

Abstract
Background
Desmoid tumors (deep fibromatoses) are clonal connective tissue malignancies that do not metastasize, but have a significant risk of local recurrence, and are associated with morbidity and occasionally mortality. Responses of desmoid patients to sorafenib on an expanded access program led us to review our experience.
Methods
After IRB approval, we reviewed data for 26 patients with desmoid tumors treated with sorafenib. Sorafenib was administered at 400 mg oral daily and adjusted for toxicity.
Findings
Sorafenib was first line therapy in 11/26 patients and the remaining 15/26 had received a median of 2 prior lines of therapy. Twenty-three of 26 patients had demonstrated evidence of progressive disease by imaging, while 3 patients had achieved maximum benefit or toxicity with chemotherapy. Sixteen of 22 (~70%) patients reported significant improvement of symptoms. At a median of 6 months (2–29) of treatment, the best RECIST 1.1 response included 6/24 (25%) patients with partial response (PR), 17/24 (70%) with stable disease and 1 with progression and death. Twelve of 13 (92%) patients evaluated by MRI had >30% decrease in T2 signal intensity, an indirect metric for increased fibrosis and loss of cellularity. Eighty percent of patients with radiological benefit had extra-abdominal desmoids.
Interpretation
Sorafenib is active against desmoid tumors. A prospective, randomized clinical trial of sorafenib against other active agents is warranted. Loss of MRI T2 signal may be a useful surrogate for defining responses, but requires validation by examination of tumor pathology.
BACKGROUND
Desmoid tumors, also known as deep fibromatoses (DT/DF), are fibroblastic neoplasms that arise from musculoaponeurotic stromal elements(1), containing small bundles of spindle cells with rare mitoses in an abundant stroma. DT/DF arises most commonly in the extremity, abdominal cavity (root of the mesentery), retroperitoneum and abdominal wall(2). DT/DF are often considered benign due to their lack of metastatic potential. However, DT/DF cause significant morbidity by infiltrating or exerting mass effects on vital structures (3, 4). Mortality from DT/DF is occasionally observed owing to the local aggressiveness of some tumors, typically in the mesentery(4).
If DT/DF remain indolent, they may be observed (3, 4). For patients with symptoms or progressive disease, primary treatment is surgical resection with wide margins. Radiation therapy (RT) is used in some patients with high risk features, or in recurrent or surgically unresectable disease (5, 6). Morbidity from multi-modal treatments remains high. Based on case reports and small series, non-steroidal anti-inflammatory agents and anti-estrogens are employed when surgery and radiation are unable to achieve local control. DT/DF have variable expression of estrogen receptor alpha (ER) and uniform expression of ER-β, however, receptor status does not correlate with clinical outcome(7).
Cytotoxic chemotherapy is usually reserved for symptomatic or progressive disease not amenable to surgery or radiation(8, 9). Anthracyclines are active when used as single agents or in combinations(10–12). Other active systemic regimens include vinca alkaloid-methotrexate combinations(13), single agent dacarbazine or temozolomide or combinations employing more than one of these agents. In our previously published series, hormonal therapy and anthracyclines (typically pegylated liposomal doxorubicin) were the most effective regimens, while other agents were less effective(8, 9).
Kinase directed therapy has also been employed against desmoids tumors. In a Phase II study with imatinib, 3 PR in 51 patients (6%) were observed after 18 or more months of treatment(14). The response rate was lower than that from smaller multicenter studies(15, 16). Sorafenib (BAY-43-9006) is a multi-targeted oral tyrosine kinase inhibitor. After we observed DT/DF patients who received sorafenib through an expanded access program who attained clinical and radiological benefit (RECIST PR and loss of MRI T2 signal intensity), we retrospectively assessed our experience with sorafenib in DT/DF patients.
METHODS
Patient selection
Following institutional IRB approval (waiver WA0209-04), we identified and reviewed medical records of 26 patients from February, 2008 to October, 2010 who received sorafenib for DT/DF in our clinic. We collected the following data: age at the date of diagnosis, presentation status (primary or recurrent), gender, presence of Gardner syndrome, primary site, primary size, radiological appearance (diffuse versus nodular), number and type of surgeries, use of radiation therapy, lines, duration and response on prior therapies (hormonal, cytotoxic and tyrosine kinase inhibitors), reason for treatment discontinuation, time to progression, documentation of progression before initiation of sorafenib, dose and toxicities of sorafenib, overall survival and status.
Imaging Techniques
Radiological measurements were made by a single board certified radiologist (R.A.L.) with experience evaluating soft tissue tumors by MRI or CT. The radiologist was blinded to patient treatment. Modalities employed in evaluating this heterogeneous group of patients include: 1) MRI with gadolinium for tumors of the extremities, chest wall, or neck (13 patients, 52 scans), 2) CT with intravenous contrast (7 patients, 26 scans) or oral contrast only (4 patients, 13 scans) for tumors involving mesentery, abdominal wall or thorax.
T2-weighted MRI values were assessed by two methods. In the first method, the radiologist drew the largest electronic Region of Interest (ROI) staying within the boundaries of each tumor at its greatest cross-sectional diameter and calculated a ratio between this value (C1) and adjacent skeletal muscle (M1). These measurements were repeated at the same location of the tumor and adjacent muscle on subsequent post-treatment examinations (C2 and M2, respectively). A normalized decrease of T2 signal intensity of 30% (i.e. [C2/M2]/[C1/M1]) was arbitrarily defined as significant. In the second method, the same radiologist visually estimated the percentage of the entire tumor on all sections that was darker than muscle on T2 weighted images, in 10% increments (0–100%); this estimate was repeated for all follow-up studies.
Role of the funding source
Grant and philanthropic support was used to support the costs of the collection and analysis of data in this retrospective analysis.
FINDINGS
Patient Characteristics
The study population is summarized in Table 1. There were 17 females and 9 males in this cohort. Median age at diagnosis and presentation was 29.5 years (range 17–57) and 31 years (range 20–59), respectively. At initial diagnosis, 16 patients underwent surgical resection and 10 patients had surgically unresectable disease, or disease only resectable by amputation. At the time of presentation, all 26 patients had unresectable disease or disease only removable by amputation (7 with multifocal disease). Seven patients had antecedent surgery and developed DT/DF at the surgical site within a median of 36 months (range 12–132); 4/7 had a history of FAP and underwent prophylactic colectomy. 3/7 (non-FAP) patients had hemicolectomy for rectal adenocarcinoma, node dissection for invasive breast cancer and breast implant for congenital breast hypoplasia. One patient had pregnancy-associated DT/DF.
Table 1.
Characteristics of 26 patients on study
| Patient and Tumor Characteristics | Prior Treatment Characteristics | |||
|---|---|---|---|---|
| Sex | ||||
| Male | 9 | Initial diagnosis: | Patients | |
| Female | 17 | Unresectable at diagnosis | 10 | |
| Recurrence following surgery | 16 | |||
| Age (yrs) | Recurrence after 2nd surgery | 3 | ||
| At diagnosis | 29.5 | |||
| At presentation | 31 | At presentation: unresectable or resectable by amputation | 26 | |
| Location: | Adjuvant Radiation | 4 | ||
| Intra-Abdominal | 12 | |||
| Extremity | 6 | Systemic therapy | 15 | |
| Trunk/Chest wall | 6 | NSAIDS | Not analyzed | |
| Head and neck | 2 | Hormone | 8 (best response, SD) | |
| Liposomal Doxorubicin | 8 | Best response: 4 SD**. | ||
| Characteristics (radiological) | Doxorubicin | 4 | ||
| Multifocal | 7 | Decarbazine | 4 | |
| Diffuse | 20 | Methotrexate | 2 | |
| Nodular | 6 | Cyclophosphamide | 2 | |
| Imatinib or Sunitinib | 6 (1 pt with minor response***) | |||
| Size | ||||
| < 5 cm (T1) | 4 | Sorafenib | ||
| 5 – 10 cm (T2) | 11 | Progression on imaging | 23 pts | |
| > 10 cm (T3) | 11 | Maximum benefit on prior therapy | 3 pts | |
| First line sorafenib | 12 pts | |||
| Risk factors | Prior therapy (median of 2) | 14 pts | ||
| FAP | 4 | |||
| Prior Surgery* | 7 | Duration of Sorafenib (median) | ||
| - Surgery for FAP | − 4/7 | - Entire cohort (months) | 6.4 (1 – 29) | |
| - Surgery for other | − 3/7 | - As 1st line (months) | 4.6 (1 – 29) | |
| Pregnancy | 1 | - As 2nd line (months) | 12 (2 – 26) | |
surgery for other reasons before diagnosis of desmoid,
SD: Stable Disease by RECIST,
minor response: decrease by 10 – 29% by RECIST.
Tumor characteristics
The histological diagnosis was confirmed at our institution in 24/26 patients. Primary sites included: abdomen/pelvis (12), extremity (6), trunk/chest wall (6) and head and neck (2). Radiological appearance of the tumor was nodular (6) or diffuse/infiltrative (20). The primary tumor size was <5 cm (4), 5–10 cm (11) and >10 cm (11).
Treatment characteristics
Local therapy
At initial diagnosis, sixteen patients underwent surgery for resectable disease. Median time to recurrence after surgery was 24 months (range 1–120). Three patients underwent a second surgery at recurrence and subsequently had unresectable disease after 10, 24 and 36 months. Four of 26 patients received adjuvant radiation following primary resection. One patient had adjuvant radiation and at recurrence had neoadjuvant radiation to the same site followed by adjuvant brachytherapy. One patient received adjuvant breast radiation for breast cancer; the DT/DF was found in the axilla following an axillary dissection.
Systemic therapy
We do not report the use of non-steroidal anti-inflammatory agents due to poor documentation, inability to evaluate compliance and the recognized low response rate. Prior systemic therapy included hormonal manipulation, chemotherapy and tyrosine kinase inhibitors. Fifteen of 26 patients received a median of 2 prior systemic treatments (range 1–5) while 11/26 patients had sorafenib as first-line treatment. Of the 15 patients who received prior systemic therapy, 8 received hormonal therapy for a median of 8 months (range 1–35) with tamoxifen (6), leuprolide (1) and medroxyprogesterone (1) before progression. Best response to hormonal treatment included stable disease and a minor response in a single patient after 6 months of therapy. Eleven of 15 patients received a median of 2 lines (range 1–3) of cytotoxic chemotherapy for a median of 7 months (range 3–42), consisting of single agents or combinations involving doxorubicin (4), liposomal pegylated doxorubicin (8), DTIC (4), methotrexate (2) and cyclophosphamide (2). Best response to chemotherapy included 4 patients with minor response and 5 patients with SD. Of these, four patients experienced long periods of observation (26, 42, 44, 84 months) following doxorubicin or liposomal pegylated doxorubicin administered for a median of 20 cycles (~20 months). Toxicity with anthracyclines included congestive heart failure, alveolar bone loss in the jaw, mucositis, alopecia and fatigue. Tyrosine kinase inhibitors other than sorafenib had been employed in 6/14 patients for a median of 3 months (range 1–7) with 1 patient intolerant of the drug, 4 patients with PD and 1 patient with minor radiologic response as best outcome.
Sorafenib administration
Sorafenib was initiated when progression was noted on imaging in 23/26 patients; 3 patients had RECIST stable disease on chemotherapy, but experienced worsening pain. Sorafenib was started in 15/26 patients after a median of 2 lines of prior systemic therapy administered for a median of 5 months. Sorafenib was initiated as first line therapy in 11/26 patients after a median of 13 months (range 2–42) from diagnosis. At the time of this report, 1st and 2nd line patients received sorafenib for a median of 4.6 (1–31) and 12 (2–26) months, respectively. Sorafenib was 1st line in 20% and 40% of patients with abdominal and extra-abdominal tumors, respectively. Sorafenib was started at a maximum dose of 400 mg daily and decreased for toxicity. No patient was treated at 400 mg twice daily; the dose approved in renal cell or hepatocellular carcinoma. The median dose of sorafenib was 200 mg daily. Some patients required further dose reductions to 200 mg every other day while others tolerated alternating doses of 400 mg and 200 mg daily (300 mg/day). Toxicities are described in Table 2 and include hand-foot syndrome, fatigue, skin rash, trichodynia, hypertension, mild alopecia and diarrhea. Side effects were well controlled with dose adjustments and inclusion of anti-diarrhea and anti-hypertensive drugs.
Table 2.
Clinical benefit, radiological responses and toxicities associated with sorafenib
| Pt | Location | Initial Symptoms | Clinical Benefit on sorafenib | Best RECIST response to sorafenib | Best T2 MRI signal change (from baseline)* | Visual approximation of “darker than muscle”. ** | Sorafenib Toxicities |
|---|---|---|---|---|---|---|---|
| MRI scans with and without gadolinium (extremity, trunk, head and neck) | |||||||
| 01 (P) | UE | Pain and discomfort at tumor site. Fore-quarter amputation only option. | Complete resolution of symptoms in 2 months (partial relief within 1 week). | −42% | −47% | 80% | Fatigue (gr 1), HF (gr 2), diarrhea (gr 1) and easy bruisability. All unresolved. |
| 02 (F) | H&N | Pain and compromised ROM of neck. | Some improvement in 4 months, complete resolution in 15 mo. | −42% | −35% | 90% | Fatigue (gr 3), HF (gr 2), GI (gr 1): all resolved completely. |
| 03 (F) | LE | Difficult to walk due to pain | Complete resolution of symptoms | −40% | −47% | 50% | Diarrhea (gr 1): resolved. |
| 04 (F) | CW/T | Shooting pain and L- ROM | Significant decrease within 1 month. Improved ROM. | −36% | −90% | 90% | Mild alopecia, Diarrhea (gr 2), HF (gr 2): unresolved. |
| 05 (P) | UE | Pain and swelling. Limited ROM. | Mild improvement after 12 months. | −31% | −80% | 90% | Occasional diarrhea, HF (resolving). |
| 06 (P) | UE | Pain, fullness and limited ROM | Significant improvement in all symptoms in 2 weeks. | −28% | −40% | 30% | Hypersensitivity and rash of soles/scalp: resolved. |
| 07 (P) | LE | Swelling of entire leg and difficulty ambulating. Pruritus at tumor site. | Significant decrease in swelling and improved ambulation. No pruritus. | −27% | −60% | 50% | HF: Grade 2 (resolved to Gr 1). Mild alopecia. |
| 08 (F) | CW/T | Pain requiring narcotics | Significant decrease in pain and softer mass. Off narcotics | −15% | −45% | 30% | Rash and HTN: resolved. |
| 09 (P) | H&N | Pain in neck | Pain and ROM improved within 1 week. | −13% | −30% | 20% | HF (gr 2), HTN (gr 2) and slight alopecia: all resolved. |
| 10 (P) | UE | Tightness over axilla and pain | Worsening pain on therapy. | −11% | −50% | 60% | HF (gr 3, ongoing), fatigue, and rash: resolved |
| 11(F) | RP/PS | Dull ache | Mild improvement | −3% | −57% | 20% | Rash (gr 3), HF (gr 1): all improved. |
| 12 (F) | UE | Significant pain and stiffness | Significant decrease in 2 weeks. | −1% | −5% | 10% | Fatigue, H&F syndrome and fatigue (all resolved) |
| 13 (P) | LE | Severe shooting pain in leg. | Slight decrease in pain. | +4% | −75% | 90% | None |
| CT scans of chest, abdomen and pelvis with or without contrast | |||||||
| 14 (P) | Abd/RP | No symptoms | No symptoms | −30% | Diarrhea (gr 2) related to surgery. | ||
| 15 (P) | CW/T | Dyspnea on exertion, sharp chest pain, edema, impending cardiopulmonary collapse. | Significant decrease in symptoms within 1 month. Complete symptom resolution in 3 months. | −19% | HF (gr 2), diarrhea (gr 1), fatigue (gr 1). | ||
| 16FAP (P) | Abd/RP | Diarrhea | Diarrhea resolved within 1 month | −19% | Skin rash | ||
| 17 (P) (P) | Abd/RP | Severe abd pain | Worse on treatment | −9% | None. OFF THERAPY FOR TOXICITY. | ||
| 18FAP (P) | Abd/RP | Pain from hydronephrosis | Pain relief with ureteral stent. | −8% | HF, fatigue, rash, dry skin. | ||
| 19 (F) | Abd/RP | No symptoms | No symptoms | −8% | Fatigue, nausea, vomiting (grade 1, resolved) | ||
| 20 (F) | Abd/RP | Intermittent abdominal pain | Worsening of symptoms | −7% | Unilateral blurry vision, scotomas. OFF THERAPY FOR TOXICITY | ||
| 21 (P) | Abd/RP | No symptoms | No symptoms | −3% | HF, diarrhea, nausea | ||
| 22 (P) | Abd/RP | No symptoms | No symptoms | +11% | Dry hands | ||
| 23FAP (P) | Abd/RP | Abd pain, distension | Worsening and pain. POD death. | +17% | OFF THERAPY FOR PROGRESSION AND DEATH | ||
| 24 (P) | Abd/RP | Intermittent abd fullness | No improvement in symptoms. | +18% | H&F syndrome, hypertension. OFF THERAPY FOR NO BENEFIT. | ||
| No radiological images since initiation of therapy | |||||||
| 25 (F) | Abd/RP | Abdominal fullness | Abdominal fullness | No scan | HTN: Gr 3. OFF THERAPY FOR TOXICITY | ||
| 26FAP (F) | RP/PS | Significant back pain | Immediate relief within 1 week. Softness on palpation. | No scan | Rash, dry skin, N/V: grade 1 | ||
LEGEND: mo: months, L-ROM: Limited range of motion. LE: lower extremity, UE: upper extremity, CW/T: Chest wall or trunk, H&N: head and neck, Abd/RP: abdomen or retroperitoneum, Abd: abdominal, RP/PS: retroperitoneal and para-spinal, GI: includes: nausea, vomiting and/or diarrhea. HF: Hand-Foot syndrome;
Methods section for calculation of signal decrease.
Decrease in T2 signal as estimated by a single radiologist blinded to treatment.
FAP: familial adenomatous polyposis. (F) First line sorafenib, (P) Prior therapies.
Clinical Outcome on Sorafenib
Clinical outcome of sorafenib therapy is detailed in Table 2 along with duration, type of response and drug toxicities. Median follow up on sorafenib was 6 months (range: 2–29). Sixteen of 22 (~70%) patients reported subjective decrease in pain and analgesic use after initiation of sorafenib; these data were not quantitated with a validated pain scale. Some examples of clinical benefit include: (1) A 21 year old male (Table 2, Pt 1, and Figure 1) had tumor extending from axilla to brachial artery, median and ulnar nerve which resulted in pain, swelling and loss of mobility. Forequarter amputation remained the only option to render the patient disease-free. Within 2 months of sorafenib the patient experienced a RECIST PR and complete resolution of symptoms; (2) a 24 year old female (Table 2, Pt 15) had a large desmoid tumor displacing her mediastinum and impending cardiopulmonary collapse despite other therapy. Within one week of sorafenib she had dramatic improvement in orthopnea, dyspnea and lower extremity edema; (3) a 21 year old woman (Table 2, Pt 9) with inoperable cervical spine mass presented with pain and compromised mobility. Therapy did not result in change in tumor size; however there was loss of T2 signal and pain relief. Notably, no symptomatic benefit was reported by patients with intra-abdominal desmoids. Three patients stopped sorafenib within 2 months due to abdominal pain, uncontrolled hypertension or visual disturbances. Neither cardiac toxicity nor bleeding was observed.
Figure 1.
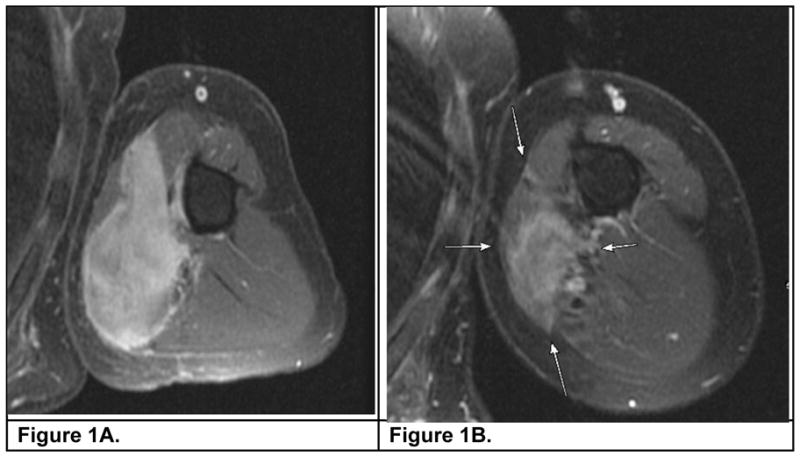
An MRI (with contrast, T2 weighted) of the upper arm depicting a desmoid tumor encasing a neurovascular bundle before (1A) and after six months (1B) of sorafenib.
Reinstitution of therapy
A patient was treated for 18 months and experienced a RECIST PR and clinical benefit. Following 6 months of observation she experienced local progression. Restarting sorafenib resulted in pain relief and tumor shrinking. Similar findings have been noted in two other patients, raising the question of response durability after treatment discontinuation.
Another patient attained SD with sorafenib after progressing on several lines of chemotherapy. Sorafenib was switched to sunitinib due to toxicity, resulting in disease progression. Re-initiation of sorafenib resulted in pain relief and disease stabilization.
Radiological response assessment
RECIST 1.1
Twenty four of 26 patients were evaluable for response. CT or MRI was obtained at a median interval of 4 months. Responses by RECIST 1.1 were: 0/14 complete responses, 6/24 partial responses (25%), 17/24 (70%) with stable disease and 1 patient with progressive disease. Seven patients experienced minor response (defined as 10–29% decrease). Figure 2 depicts RECIST responses in 24 evaluable patients. Bi-dimensional WHO size measurements were highly correlated (R2=0.92) with RECIST (figure not shown). RECIST PR was achieved at a median of 10 months and a minor response (i.e. reduction > 10% by RECIST) was noted at a median of 4 months of starting therapy. PR and SD were mostly seen in extra-abdominal tumors (p=0.03, t-test) and there was no difference in between those who received sorafenib as first-line or second-line (p = 0.9, t-test).
Figure 2.

Waterfall plot of best radiological outcome by size (RECIST 1.1) for individual patients along with duration of response (months) and type of imaging noted below each patient column.
MRI Signal Changes
13 patients were evaluable by MRI (Figure 3). T2 signal changes were quantitatively and qualitatively described (see Methods). We defined a 30% loss of T2 signal relative to muscle as a response. We anecdotally noted that patients with changes in T2 signal had described symptomatic improvement; however, this was not correlated with a formal quality of life scale. T2 changes were seen in 12/13 (~90%) patients who had an MRI. T2 signal change was seen in 100% of patients with a RECIST PR. Median time to T2 signal loss (>30%) was 5 months for those who obtained a PR (median 47% decrease) and 3.6 months for those achieving SD as best result. In this small data set, the sensitivity and specificity of T2 signal loss as a predictor of RECIST PR was 100% and 12%, respectively. T2 signal loss has a positive predictive value and negative predictive value of 42% and 100%, respectively.
Figure 3.

Waterfall plot of best radiological outcome by size (RECIST 1.1) of patients evaluated by MRI, duration of response (months) and change in T2 signal intensity (gray columns).
Survival
The 26 patients were followed for a median of 50 months (3–209 months) from initial diagnosis. Median follow-up from the start of sorafenib was 6 months (1–29). Median time to progression was not reached. Five patients progressed despite sorafenib, defined as increasing tumor size (2), symptoms (1) or drug intolerance (2). Twenty-five of 26 patients are alive with disease.
INTERPRETATION
DT/DF have a highly variable clinical presentation and natural history. There are no standard first-line systemic therapies in DT/DF as only few agents have been examined prospectively and there are no randomized studies. In this cohort, median PFS rates were not reached. Only one patient (with Gardner syndrome) had progressive disease and died. Since desmoid tumors may have a relatively indolent course, we submit that overt radiological response rate and symptomatic improvement may be better primary endpoints for outcome than PFS for clinical trials. Any response must also take into account the occasional patient with spontaneous regressions or delayed regressions from prior therapy.
The most striking aspect of this study is the rapid clinical benefit seen in 16/22 (~70%) of symptomatic patients. Clinical improvement was typically noted within 2 weeks of starting sorafenib; we had not observed such clinical benefit with imatinib. It is notable that radiological benefit (RECIST CR+PR+SD) were most common in extremity DT/DF rather than intra-abdominal tumors (p=0.03, t-test). There was no difference in radiological benefit when sorafenib was first-line or second-line treatment (p=0.9). These findings suggest the response of DT/DF to sorafenib is a function of their biology, i.e. APC mutation (Gardner syndrome) for intra-abdominal desmoids(17, 18) versus β-catenin mutation commonly observed in other DT/DF(19). Conversely, it is difficult to ascribe anti-angiogenic effects of sorafenib as directly responsible for the observed benefit. To date, there are no biomarkers that inform natural outcome or clinical benefit with therapies. Tumor or serum levels of KIT, PDGFR, PDGF-AA, PDGF-BB, and CTNNB1 or APC mutation status have not correlated with responses to TKI therapy(14–16). Research is underway to elucidate why non-abdominal desmoids respond more frequently to sorafenib than mesenteric desmoids, which may also point to the potential durability of response once treatment has been interrupted.
The optimal imaging modality to evaluate DT/DF remains undefined. Six of 24 patients experienced a RECIST PR and 7/24 had minor tumor shrinkage (10–29%) that represent a response using the alternative response criteria developed by Choi for GIST(20). Focusing more on the MRI characteristics of desmoids, studies that evaluated soft tissue tumors noted that differences in MRI characteristics are related to the ratio of fibroblasts to collagen(21). We observed that sorafenib leads to T2 signal loss in 12/13 (~90%) patients, which is suggestive of a shift in fibroblast to collagen ratio. T2 signal loss was observed in all patients with a RECIST PR. Whether T2 signal represents a biological effect is a question for prospective trials with endpoints evaluating quality of life, pre- and post-treatment pathology and time to relapse.
Quantitating MRI T2 signal intensity may be a novel radiographic metric in DT/DF. Stacchiotti et al showed that T1 signal on contrast enhanced MRI may be a novel radiologic marker of response in high grade sarcomas treated with neoadjuvant chemotherapy/radiation(22). In contrast to computed tomography, in which Hounsfield units may be used as a metric of tissue density, MRI T2 signal is a unitless number with no intrinsic meaning except for comparative purposes. In attempting to develop a reproducible metric for clinical improvement we normalized the signal intensity of lesions by using normal muscle as a reference standard. In this analysis, MRI scans were not performed with identical parameters, which are expected to cause variation in the measured T2 signal intensity, even with a reference standard. In addition, we used a region of interest (ROI) on one representative section which introduces sampling error. In these patients, the changes in signal were homogenous when they did occur, and, thus, our measurements were arguably representative of the entire lesion. The above limitations argue for a prospective study to further evaluate this metric.
Despite our attempt to capture all treated patients, the retrospective nature of this study exposes it to selection bias, as well as possible lead-time bias related to variable imaging intervals. Nonetheless, the relative rapidity of the clinical and radiological responses was striking and appeared to be in excess of what has been reported with other systemic agents, at least as pertains to desmoids not associated with Gardner syndrome. Our clinical observations are supportive of a prospective, randomized trial comparing sorafenib to other active agents, and hopefully provide an otherwise underappreciated means to manage disease in difficult anatomic locations
Acknowledgments
Funding: National Cancer Institute (NCI) grants: CA47179, CA148260 and P30 CA008748 supplement. Additional funding from: Cycle for Survival (private philanthropic) and American Society of Clinical Oncology (ASCO) Merit Award (MMG). Robert G Maki served on an advisory board for Bayer Pharmaceuticals, which supplies sorafenib through its Resources for Expert Assistance and Care Helpline (REACH) program. David D’Adamo receives research funding from Bayer Pharmaceuticals. All other authors report no conflict of interest.
We acknowledge the support of Murray F. Brennan for useful discussions on this topic.
Footnotes
CONTRIBUTIONS
Mrinal M Gounder: conception, design, collection and assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript.
Robert A Lefkowitz: collection and assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript.
Mary Louise Keohan: collection and assembly of data, final approval of manuscript.
David R D’Adamo: collection and assembly of data, final approval of manuscript.
Meera Hameed: collection and assembly of data, data analysis and interpretation, final approval of manuscript.
Cristina R Antonescu: collection and assembly of data, data analysis and interpretation, final approval of manuscript.
Samuel Singer: collection and assembly of data, final approval of manuscript.
Katherine Stout: collection and assembly of data, final approval of manuscript.
Linda Ahn: collection and assembly of data, final approval of manuscript.
Robert G Maki: conception, design, collection and assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript, administrative support, financial support.
References
- 1.Fletcher CDMUK, Mertens F. Pathology and genetics of tumours of soft tissue and bone. Lyon: IARC Press; 2002. [Google Scholar]
- 2.de Bree E, Keus R, Melissas J, Tsiftsis D, van Coevorden F. Desmoid tumors: need for an individualized approach. Expert Rev Anticancer Ther. 2009;9:525–35. doi: 10.1586/era.09.9. [DOI] [PubMed] [Google Scholar]
- 3.Lewis JJ, Boland PJ, Leung DH, Woodruff JM, Brennan MF. The enigma of desmoid tumors. Ann Surg. 1999;229:866–72. doi: 10.1097/00000658-199906000-00014. discussion 72–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith AJ, Lewis JJ, Merchant NB, Leung DH, Woodruff JM, Brennan MF. Surgical management of intra-abdominal desmoid tumours. Br J Surg. 2000;87:608–13. doi: 10.1046/j.1365-2168.2000.01400.x. [DOI] [PubMed] [Google Scholar]
- 5.Ballo MT, Zagars GK, Pollack A, Pisters PW, Pollack RA. Desmoid tumor: prognostic factors and outcome after surgery, radiation therapy, or combined surgery and radiation therapy. J Clin Oncol. 1999;17:158–67. doi: 10.1200/JCO.1999.17.1.158. [DOI] [PubMed] [Google Scholar]
- 6.Baumert BG, Spahr MO, Von Hochstetter A, Beauvois S, Landmann C, Fridrich K, et al. The impact of radiotherapy in the treatment of desmoid tumours. An international survey of 110 patients. A study of the Rare Cancer Network. Radiat Oncol. 2007;2:12. doi: 10.1186/1748-717X-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deyrup AT, Tretiakova M, Montag AG. Estrogen receptor-beta expression in extraabdominal fibromatoses: an analysis of 40 cases. Cancer. 2006;106:208–13. doi: 10.1002/cncr.21553. [DOI] [PubMed] [Google Scholar]
- 8.Constantinidou A, Jones RL, Scurr M, Al-Muderis O, Judson I. Pegylated liposomal doxorubicin, an effective, well-tolerated treatment for refractory aggressive fibromatosis. Eur J Cancer. 2009;45:2930–4. doi: 10.1016/j.ejca.2009.08.016. [DOI] [PubMed] [Google Scholar]
- 9.Pires de Camargo V, Keohan ML, D’Adamo DR, Antonescu CR, Brennan MF, Singer S, et al. Clinical outcomes of systemic therapy for patients with deep fibromatosis (desmoid tumor) Cancer. 2010;116:2258–65. doi: 10.1002/cncr.25089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Patel SR, Evans HL, Benjamin RS. Combination chemotherapy in adult desmoid tumors. Cancer. 1993;72:3244–7. doi: 10.1002/1097-0142(19931201)72:11<3244::aid-cncr2820721118>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 11.Gega M, Yanagi H, Yoshikawa R, Noda M, Ikeuchi H, Tsukamoto K, et al. Successful chemotherapeutic modality of doxorubicin plus dacarbazine for the treatment of desmoid tumors in association with familial adenomatous polyposis. J Clin Oncol. 2006;24:102–5. doi: 10.1200/JCO.2005.02.1923. [DOI] [PubMed] [Google Scholar]
- 12.Patel SR, Benjamin RS. Desmoid tumors respond to chemotherapy: defying the dogma in oncology. J Clin Oncol. 2006;24:11–2. doi: 10.1200/JCO.2005.03.6566. [DOI] [PubMed] [Google Scholar]
- 13.Weiss AJ, Horowitz S, Lackman RD. Therapy of desmoid tumors and fibromatosis using vinorelbine. Am J Clin Oncol. 1999;22:193–5. doi: 10.1097/00000421-199904000-00020. [DOI] [PubMed] [Google Scholar]
- 14.Chugh R, Wathen JK, Patel SR, Maki RG, Meyers PA, Schuetze SM, et al. Efficacy of imatinib in aggressive fibromatosis: Results of a phase II multicenter Sarcoma Alliance for Research through Collaboration (SARC) Trial. Clin Cancer Res. 2010;16:4884–91. doi: 10.1158/1078-0432.CCR-10-1177. [DOI] [PubMed] [Google Scholar]
- 15.Heinrich MC, Joensuu H, Demetri GD, Corless CL, Apperley J, Fletcher JA, et al. Phase II, open-label study evaluating the activity of imatinib in treating life-threatening malignancies known to be associated with imatinib-sensitive tyrosine kinases. Clin Cancer Res. 2008;14:2717–25. doi: 10.1158/1078-0432.CCR-07-4575. [DOI] [PubMed] [Google Scholar]
- 16.Heinrich MC, McArthur GA, Demetri GD, Joensuu H, Bono P, Herrmann R, et al. Clinical and molecular studies of the effect of imatinib on advanced aggressive fibromatosis (desmoid tumor) J Clin Oncol. 2006;24:1195–203. doi: 10.1200/JCO.2005.04.0717. [DOI] [PubMed] [Google Scholar]
- 17.Alman BA, Li C, Pajerski ME, Diaz-Cano S, Wolfe HJ. Increased beta-catenin protein and somatic APC mutations in sporadic aggressive fibromatoses (desmoid tumors) Am J Pathol. 1997;151:329–34. [PMC free article] [PubMed] [Google Scholar]
- 18.Davies DR, Armstrong JG, Thakker N, Horner K, Guy SP, Clancy T, et al. Severe Gardner syndrome in families with mutations restricted to a specific region of the APC gene. Am J Hum Genet. 1995;57:1151–8. [PMC free article] [PubMed] [Google Scholar]
- 19.Amary MF, Pauwels P, Meulemans E, Roemen GM, Islam L, Idowu B, et al. Detection of beta-catenin mutations in paraffin-embedded sporadic desmoid-type fibromatosis by mutation-specific restriction enzyme digestion (MSRED): an ancillary diagnostic tool. Am J Surg Pathol. 2007;31:1299–309. doi: 10.1097/PAS.0b013e31802f581a. [DOI] [PubMed] [Google Scholar]
- 20.Choi H, Charnsangavej C, Faria SC, Macapinlac HA, Burgess MA, Patel SR, et al. Correlation of computed tomography and positron emission tomography in patients with metastatic gastrointestinal stromal tumor treated at a single institution with imatinib mesylate: proposal of new computed tomography response criteria. J Clin Oncol. 2007;25:1753–9. doi: 10.1200/JCO.2006.07.3049. [DOI] [PubMed] [Google Scholar]
- 21.Sundaram M, McGuire MH, Schajowicz F. Soft-tissue masses: histologic basis for decreased signal (short T2) on T2-weighted MR images. AJR Am J Roentgenol. 1987;148:1247–50. doi: 10.2214/ajr.148.6.1247. [DOI] [PubMed] [Google Scholar]
- 22.Stacchiotti S, Collini P, Messina A, Morosi C, Barisella M, Bertulli R, et al. High-grade soft-tissue sarcomas: tumor response assessment--pilot study to assess the correlation between radiologic and pathologic response by using RECIST and Choi criteria. Radiology. 2009;251:447–56. doi: 10.1148/radiol.2512081403. [DOI] [PubMed] [Google Scholar]