

Abstract
RNA-seq is a method for studying the transcriptome of cells or tissues by massively-parallel sequencing of tens of millions of short DNA fragments. However, the broad dynamic range of gene expression levels, which span more than five orders of magnitude, necessitates considerable over-sequencing to characterize low-abundance RNAs at sufficient depth. Here, we describe a method that enables efficient sequencing of low-abundance RNAs by normalizing or reducing the range of from the most abundant RNA species to the least abundant RNA species. This normalization is achieved using an approach that was developed for generating expressed sequence tag (EST) libraries that uses the crab duplex-specific nuclease and exploits the kinetics of DNA annealing. That is, double stranded cDNA is denatured, allowed to partially re-anneal and the most abundant species, which re-anneal most rapidly are digested with crab duplex-specific nuclease. This procedure substantially decreases the proportion of sequence reads from highly-expressed RNAs, facilitating assessment of the full spectrum of the sequence and structure of transcriptomes.
Keywords for indexing: RNA-seq, Library normalization, Crab duplex nuclease, High-throughput DNA sequencing
INTRODUCTION
This unit describes the generation of a normalized RNA-seq library for next-generation sequencing by utilizing the preference of the crab duplex nuclease (DSN) to digest double-stranded rather than single-stranded DNA (Bogdanova et al., 2008; UNIT 5.12). In this approach (Basic Protocol 1), polyadenylated RNA is used to generate a complex RNA-seq library. The library is denatured and incompletely renatured, and then digested with DSN. The kinetics of DNA annealing are such that at any given time abundant DNA molecules are more likely to have re-annealed and become double stranded, while rare molecules are more likely to remain single-stranded. Thus, preferentially digesting double-stranded DNA with DSN yields a library markedly enriched for more rare DNA species. Massively-parallel sequencing of these normalized libraries allows for efficient and comprehensive assessment of the sequence and structure of the polyadenylated transcriptome.
BASIC PROTOCOL 1
The first step in the construction of a normalized RNA-seq library is to make a high-complexity RNA-seq library (i.e. a library that includes sufficient depth of sequence from all positions of all polyadenylated RNAs). This involves starting with about 400ng polyadenylated RNA from about 20μg total RNA. (This method should also be suitable for non-polyadenylated RNA; start at step 5.) The quality of the RNA is crucial and can be determined using a Bioanalyzer (Agilent) (Schroeder et al., 2006) or agarose gel. All steps in the protocol are performed with an excess of reagents to ensure each reaction approaches completion.
The resulting high-complexity RNA-seq library is amplified to generate 500-1200 ng DNA using 19-20 bp primers corresponding to the inner sequences of the Illumina paired-end adaptors. Normalization of the RNA-seq library is then achieved by denaturing and re-annealing at 68°C, followed by treatment with the duplex-specific nuclease (DSN). The annealing temperature of the resulting amplified RNA-seq library’s adaptor sequence is ~57°C, which minimizes non-specific targeting by DSN. A second amplification and normalization is then performed. The normalized library is then amplified with the full-length Illumina paired-end primers. See Figure 1 for an overview of the library construction.
Fig. 1.
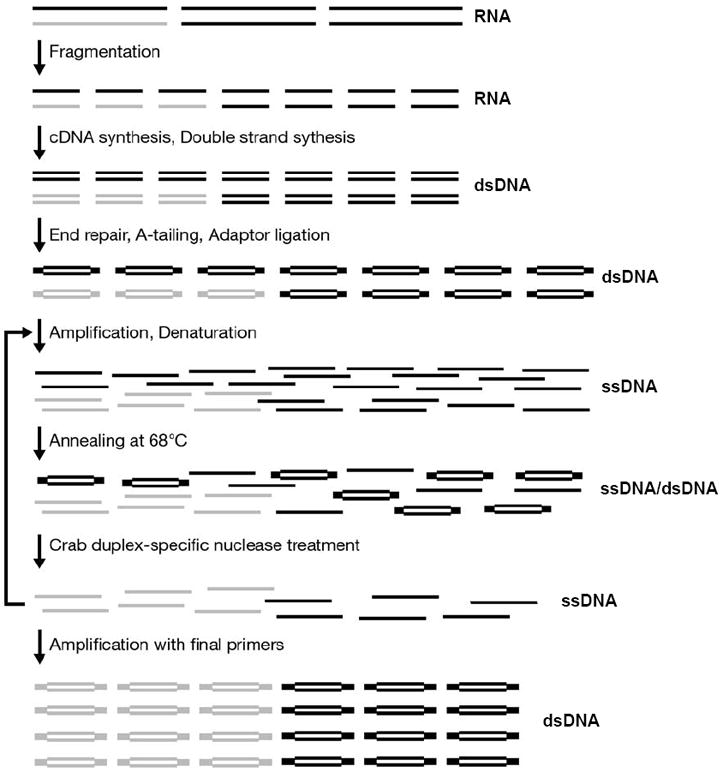
Normalized RNA-seq library construction. Illustration of the normalization process starting from a rare (grey) and abundant (black) RNA transcript.
Materials
Trizol (Invitrogen, cat#15596-018) or RNeasy kit (Qiagen, cat#74104)
Dynabeads mRNA DIRECT Kit (Invitrogen, cat#610-12)
DynaMag-2 magnet (Invitrogen, cat#123-21D)
Qubit fluorometer (Invitrogen, cat#Q32857)
Quant-iT RNA Assay Kit (Invitrogen, cat#Q32852)
5X Fragmentation buffer (Reagents and Solutions)
Glycogen (Roche, cat#10901393001)
SuperScript III Reverse Transcriptase (Invitrogen, cat#18080-044)
Second-Strand Buffer (Invitrogen, cat#10812-014)
E. coli RNAse H (Invitrogen, cat#18021071)
QIAquick PCR purification kit (Qiagen, cat#28104)
E. coli DNA Polymerase I (Invitrogen, cat#18010025)
End-It DNA End-Repair Kit (Epicentre Biotechnologies, cat#ER81050)
dATP (Roche, cat#11051440001)
Quick Ligation Kit (New England BioLabs, cat#M2200L)
50-bp ladder (New England Biolabs, cat# N3236L)
MinElute PCR Purification Kit (Qiagen, cat#28004)
SYBR Gold (cat# S11494)
QIAquick Gel Extraction Kit (Qiagen, cat#28004)
Phusion High-Fidelity DNA Polymerase (New England Biolabs, cat#F-530S)
SYBR Green I (Invitrogen, cat#S7563)
AMPure beads (Agencourt AMPure 60 mL Kit, cat#A29152)
4-20% TBE Gel (Invitrogen, cat#EC62252BOX)
Dark Reader Transilluminator (Clarechemical, cat#DR-88M)
Quant-iT dsDNA HS Assay Kit (Invitrogen, cat#Q32851)
Duplex-Specific Nuclease (Evrogen, cat#EVN-EA001-KI01)
25bp ladder (Invitrogen, cat#10597-011)
Non-Stick RNase-free Microfuge Tubes, 1.5ml (Ambion, cat#12450)
4X Hybridization buffer (Reagents and Solutions)
Oligonucleotides (Reagents and Solutions) (Integrated DNA Technologies)
Stage I: polyA selection, RNA fragmentation, cDNA and Second Strand synthesis, End repair, A-addition, Adapter ligation, Gel purification, First amplification
Vortex the oligo(dT) beads (Dynabeads) at low speed to make homogeneous. Transfer 100μl to a 1.5ml tube (non-stick tubes are used throughout the experiment). Capture the beads by placing the tube on the DynaMag-2 magnet, and wash with 50μl Lysis/Binding buffer (Dynabeads kit). Repeat capture and resuspend in 50μl Lysis/Binding buffer.
Prepare the RNA on ice (previously extracted using an RNeasy kit or Trizol and quantified) by adjusting the volume to 50μl with ddH2O. Add 50μl Lysis/Binding buffer, heat at 61°C for 2 minutes and immediately place back on ice. Transfer the RNA to the beads and mix by pipetting. Bind at room temperature for 10 minutes with gentle agitation.
- Capture the beads on the magnet and discard the supernatant. Wash twice with 200μl buffer B (Dynabeads kit). Wash once with 50μl buffer B, discard the buffer and resuspend in 20μl ddH2O. Elute the RNA from the beads by incubating at 73°C for 1-2 minutes. Immediately capture the beads on the magnet and transfer the eluate to a new tube and place on ice.It is critical not to exceed the 73°C temperature during elution to prevent extensive RNA degradation.
- Perform a second round of polyA selection by repeating steps 1-3 using the eluate from step 3 in place of total RNA in step 2 (adjust the volume to 50μl by adding 30μl of ddH2O). As in step 3, elute into 20μl of ddH2O and place the tube on ice.At this stage, the mRNA can be stored at -80°C until use.
Measure the mRNA concentration with the Quant-iT RNA Assay Kit using 1μl of the eluate. Use 400ng mRNA, raise to a volume of 16μl with ddH20, and add 4μl of 5X Fragmentation buffer. Incubate at 94°C for 2-5 minutes and place on ice.
Precipitate the fragmented RNA by adding 8μl 3M Sodium Acetate pH 5.7 and 1μl of glycogen. Mix and then add 240μl ethanol. Vortex and incubate at -20°C for 30-50 minutes. Pellet the precipitated RNA by centrifugation at ~14,000rpm at 4°C for 15 minutes. Remove the supernatant and wash the pellet with 800μl 70% ethanol. Re-centrifuge for 2 minutes, completely remove the ethanol and dry the pellet for a few minutes at room temperature.
-
Synthesize the first strand. Resuspend the RNA pellet in 30μl random hexamers (Super Script III cDNA synthesis kit) and place on ice. Add the following:
- 8μl 10mM dNTPs
- 2μl ddH2O
Mix and incubate at 65°C for 4 minutes.
Place on ice and add:- 8μl DTT
- 8μl 10X RT Buffer
- 16μl MgCl2
- 2μl RNAse OUT
- 6μl Super Script III
Incubate for 10 minutes at 25°C, 25 minutes at 42°C, 25 minutes at 50°C, and hold at 4°C.
-
Synthesize the second strand. Add as a mastermix the following (amounts per sample):
- 204μl ddH2O
- 80μl 5X Second Strand buffer
- 12μl 10mM dNTPs
- 4μl RNAse H
Mix and add 15μl DNA Polymerase I. Mix again.
Incubate at 16°C for 2.5 hours and hold at 4°C overnight. Purify the resulting second stranded product using a Qiagen PCR purification kit according to the kit’s instructions. Elute with 90μl buffer EB (Qiagen).Following this step, the product can be stored at 4°C or -20°C. -
Perform End Repair. Using the End Repair Kit (Epicentre), make a mastermix of the following (amount per sample):
- 20μl 10X End Repair Buffer
- 20μl 10mM ATP
- 20μl dNTP
- 6μl End Repair Enzyme
- 89μl ddH2O
Add 110μl of End Repair Mix to the 90μl sample eluate from step 8 and incubate at 20°C for 55 minutes and hold at 4°C. Use a Qiagen PCR column to purify the sample and elute into 80μl EB.
-
For A-tailing prepare a MasterMix of the following and add to the sample:
- 10μl NEB2 (New England Biolabs) Buffer
- 4μl 5mM dATP
- 6μl 3’-5’ KlenowExo
Incubate at 37°C for 1 hour. Use a Qiagen PCR column to purify the sample and elute into 44μl EB.A complete reaction is critical as A-tailing facilitates the addition of an adenosine overhang at the 3’ end which prevents self-ligation during the next step. - Perform Adapter Ligation. First add 6μl of 100μM PE adapter and 56μl 2X Ligation buffer to the sample and mix. Add 6μl Quick Ligase and mix well. Incubate at 24°C for 20 minutes. Use the Qiagen minElute PCR purification kit and elute into 10μl EB.Double eluting by passing the first eluate through the same column is recommended.
Add gel loading dye to the above eluate and run on a 2% agarose gel. Stain the gel with SYBR gold (1:10,000) for 10-15 minutes and visualize with the Dark Reader Transilluminator. SYBR gold offers improved sensitivity compared to ethidium bromide and the transilluminator avoids exposing the DNA to UV radiation. Extract an 100bp band size between 180-350bp (Figure 2A). Use the QIagen Gel extraction kit to extract the DNA and elute with 25μl buffer EB. (Pass the eluate through the column to double-elute.)
-
Perform real-time PCR to determine the desired number of amplification cycles. For each sample, use:
- 5μl 5X buffer (Phusion polymerase)
- 1μl of 10μM Primer 1 (Inner primers)
- 1μl of 10μM Primer 2 (Inner primers)
- 1.25μl 10mM dNTPs
- 0.4μl Phusion polymerase
- 0.25μl SYBR Green I (1:1000 diluted)
- 15.6μl ddH2O
- 0.5μl RNA-seq library
Amplify:- 1st Stage: 98°C for 30”
- 2nd Stage: 98°C for 10”; 59°C for 30”; 72°C for 30”. Repeat 24 times.
- 3rd Stage: 72°C for 5’
The optimal cycle number for the final PCR amplification is determined by selecting the point before the real-time PCR reaction saturates. That is, use the graphed fluorescence versus cycle number from the real-time PCR. The optimal cycle number is at the end of the exponential phase but before the reaction reaches a plateau.Choosing cycles within the exponential range facilitates uniform amplification of the library and minimizes amplification errors. - Use the sample generated in step 12 to create 20 replicate reactions prepared as in step 13 (substituting SYBR green with ddH2O) with the number of cycles determined in step 12. This aims at amplifying about half the sample (the remainder can be saved to be used if needed to repeat the amplification or to make a non-normalized library). Following the completion of the reaction, combine the product from all tubes. Save 1-2μl to be run on the polyacrylamide gel during step 15, and purify the remainder using 2x AMPure beads (perform 2 rounds of purification) according to the manufacturer’s instructions. Elute with 20μl ddH2O.This set-up creates enough material for normalization, while using most of the initial material for amplification. This aims at amplifying a high complexity library.
- Dilute 1μl from the purified product 1:4 with water. Use 1μl of the diluted product to estimate the library concentration using the Quant-iT dsDNA HS Assay Kit. Run 1-2μl of the diluted product on a 4-20% polyacrylamide gel (UNIT 2.5A; it is useful to run alongside a 1μl aliquot from prior to the DNA purification). Stain with SYBR Gold and visualize as in Figure 2B.It is critical that the library appears as a smear and not a discrete band.
Fig. 2.

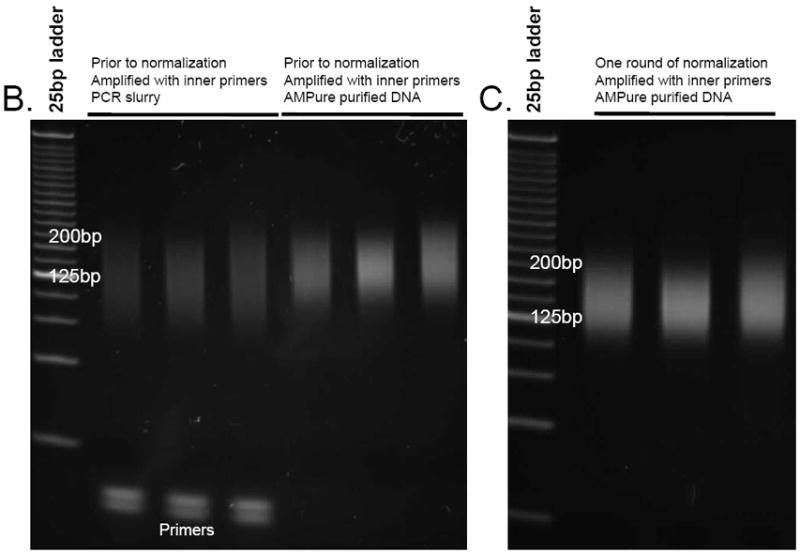
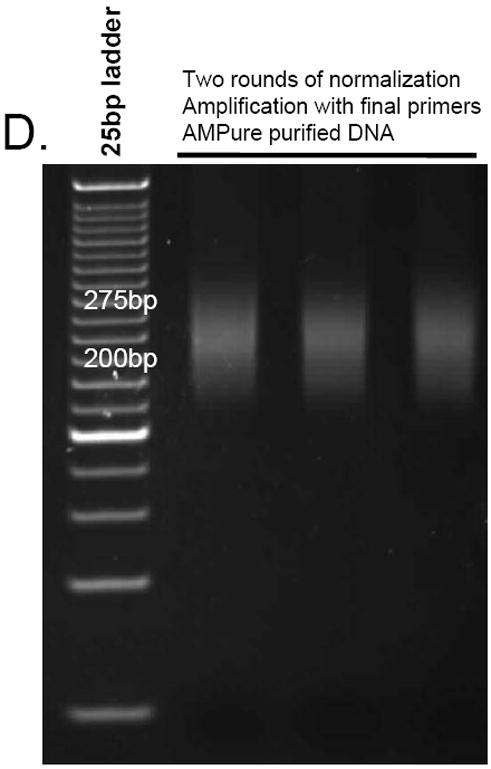
(A) Library excision from a 2% agarose gel stained with SYBR gold (step 12). Here, an 180-280bp fragment was excised. B-D. Aliquots from different stages of library preparation. (B) Amplified library prior to normalization (step 15), (C) library after one round of normalization (step 18), (D) library after two rounds of normalization amplified with the final Illumina primers (step 19)
Stage II: First normalization, Second amplification, Purification, Second normalization
-
16
Bring 500-1200ng amplified DNA to a volume of 12μl. Add 4μl of 4X Hybridization buffer (Reagents and Solutions), mix, denature at 98°C for 2 minutes and re-anneal at 68°C for 6 hours in a thermocycler.
-
17For this step always work near the thermocycler and keep the sample on the thermocycler at 68°C except when spinning down the contents of the tube. First, pre-warm the 2X Master buffer (dilute from the 10X provided in the Duplex-specific nuclease kit) on the thermocycler and add 20μl to the sample. Mix and incubate for 10 minutes. Add 3μl of DSN enzyme (reconstitute and test in accordance to the manufacturer’s instructions), mix well, quickly spin down, and incubate for 25 minutes at 68°C. Add 1μl of 125mM EDTA, mix well, quickly spin down and incubate for 5 minutes. Following this reaction, immediately place on ice and freeze if needed.It is critical that the hybridization temperature (68°C) be maintained throughout this procedure. Please note that the DSN enzyme may still be active – keep the reaction cold or frozen to avoid non-specific degradation.
-
18Follow the steps 13-17 to re-amplify the library with the inner primers and re-normalize. As template use 1μl of the product slurry from step 17. Before normalizing, run a 1 μl aliquot on a polyacrylamide gel.The library should appear as a smear and not a discrete band when run on a polyacrylamide gel (Figure 2C).
Stage III: Final amplification, Purification
-
19Use the Illumina PE primers to amplify the product from step 18. Follow the directions from Steps 13-15 to amplify the library with using 1μl of each 5μM Illumina PE primer and 1μl of the product slurry from Step 18. Purify with 1.8x beads and elute with 25μl ddH2O. Measure the concentration and assess the DNA library size as in Step 15.When running on a polyacrylamide gel, the library should appear as a smear (Figure 2D). The higher size of the smear is due to the added sequences from using the full-length paired-end primers.
-
20
Submit the library for end-sequencing at a concentration recommended by the sequencing facility.
REAGENTS AND SOLUTIONS
5X Fragmentation buffer:
200 mM Tris acetate pH 8.2
500 mM potassium acetate
150 mM magnesium acetate
4X Hybridization buffer:
200mM Hepes pH 7.5
2M NaCl
COMMENTARY
Background Information
RNA-seq is transforming our understanding of transcriptomes by revealing important information about transcript diversity, sequence, and structure. Comparison of these RNA transcriptomes between various cellular states has also facilitated study of transcript function and regulation. (Nagalakshmi et al., 2008; Sultan et al., 2008; Wang et al., 2008; Tang et al., 2009; Trapnell et al., 2010).
However, despite the improved power of next-generation sequencing technologies, the very broad dynamic range of gene expression has been an obstacle to studies of RNA transcript structural changes in low expressing genes such as transcription factors, which, even at low levels, are of profound physiologic importance. Sequencing-based strategies to assess expression profiles such as Polony Multiplex Analysis of Gene Expression (PMAGE) and Deep Sequencing Analysis of Gene Expression (DSAGE; UNIT 25B.9 and Kim et al., 2007), have demonstrated that in the cardiac left ventricle the 50 most highly-expressed genes, which consist of mitochondrial, structural, sarcomere and energy production genes, comprise greater than 40% of the polyadenylated RNAs and the most abundant RNA can comprise as much as 10% of the polyadenylated RNAs. By contrast, low-expressing RNAs are often found to comprise as few as 1 molecule per million, indicating that the range of RNA transcript expression levels spans at least five orders of magnitude. This large range impedes comprehensive characterization of transcripts expressed at low levels.
Critical parameters and Troubleshooting
An important consideration in constructing a normalized RNA-seq library is the complexity of the library prior to normalization. Lower complexity libraries can still be normalized, but will result in non-uniform sequencing of lower-expressing genes as well as a high number of sequenced duplicate reads.
Library cross-contamination is a major concern in this procedure, because such problems will be exaggerated by the normalization and PCR amplification steps. Contamination can be minimized by physically separating amplification steps from post-amplified products and washing any lab equipment that comes in direct contact with the libraries, especially any gel chambers or gel excision equipment.
RNA degradation is also a significant concern. During RNA isolation steps, significant effort is required to avoid RNA degradation. In addition to working quickly, keeping the RNA on ice and using RNAse-free glass- or plasticware, RNAse inhibitors can be added to the sample. The RNA should be handled at the indicated temperatures, as higher temperatures or prolonged incubation can increase degradation. Degradation during the polyA selection will result in loss of transcript 5’ sequences, because selection is performed using the 3’ polyA tail.
Confirm that the DSN enzyme is active within a week prior to use, per the manufacturer’s instructions. Also, see the Troubleshooting table for other critical parameters.
Anticipated results
Amplification cycles are expected to be in the range of 8-12 cycles and 12-15 cycles for the final amplification. When amplified within the exponential phase, by using real-time PCR, these products are expected to appear as a smear rather than as discrete bands, as in Figures 2B-D. The library smears resulting from the amplifications using the inner primers are expected to be smaller than the gel-extracted library size, since they only amplify the inner part of the universal adaptor sequence. Following amplification with the long Illumina final primers, the library size is expected to increase (Figure 2).
In the normalized libraries, highly-expressed RNA transcripts are expected to be decreased in proportion in the library by about 10-fold compared to their original proportion (the highest expressing gene is reduced 50-times in proportion). Lower-expressed RNA transcripts are expected to be enriched ~10-fold. Additionally, some previously undetectable RNA transcripts and some pre-mRNAs are expected to be present at non-negligible levels. Untranscribed regions of the genome should not be enriched, but may contain rare interspersed reads not exceeding 1-2 reads depth.
Time considerations
The RNA-seq library preparation takes about four days and normalization and final amplification takes about three days.
Table 1.
Oligonucleotide sequences
| Oligonucleotide | Sequence |
|---|---|
| *Illumina PE adapter 1 | 5’ Phosphate-GATCGGAAGAGCGGTTCAGCAGGAATGCCGAG |
| *Illumina PE adapter 2 | 5’ ACACTCTTTCCCTACACGACGCTCTTCCGATCT |
| Inner PE primer 1 | 5’ CACGACGCTCTTCCGATCT |
| Inner PE primer 2 | 5’ CTGAACCGCTCTTCCGATCT |
| Illumina PE primer 1 | 5’AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT |
| Illumina PE primer 2 | 5’CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT |
To prepare the adapter, resuspend each adapter oligonucleotide to 200μM concentration using water or reduced Tris-EDTA buffer. Mix equal amounts of each adaptor oligonucleotide. Using a thermocycler, anneal using the program: 4 minutes at 95°C, -0.1°C/second to 12°C, hold at 4°C. The annealed adapters can be stored at -20°C until use.
Table 2.
Troubleshooting for normalized RNA-seq library construction
| Problem | Possible Cause | Solution |
|---|---|---|
| RNA reads are not uniform | RNA degradation (The 5’ of the transcript is more susceptible since the polyA selection is done from the 3’ end) | Start with high quality RNA; use RNA with RNA Integrity Number > 8; reduce fragmentation time; begin with a single RNA sample and work quickly. |
| Excessive (>15) PCR cycles are needed for amplifying the library before the normalization step | Low starting RNA concentration | Test RNA concentration and quality at the beginning. |
| Poor library generation, perhaps due to inactive enzymes/reagents | Start with a new batch of reagents | |
| Wrong primers | Make sure that the inner primers are being used for the first amplifications | |
| A discrete band appears instead of a smear | Low complexity library or PCR cycles exceeded the linear phase | Check reactions; carefully assess the number of the real-time PCR cycles needed to amplify the library. |
| Coverage enrichment only effects moderately-expressed genes | Low complexity library | Start with a greater amount of polyA RNA. Utilize more template by setting up more amplification reactions. |
| If starting with total RNA, polyA-select prior library construction. | ||
| Gene expression profiles do not appear normalized | Inactive DSN enzyme or another problem with the DSN reaction. Also, the library may not have been sufficiently amplified. | Test DSN enzyme activity according to the manufacturer’s instructions. Assess the effect of DSN by including a no-enzyme tube (and compare overall digestion in the sample with real-time PCR). |
| Excessive number of cycles are needed to amplify the library after it was normalized (but not before normalization); if sequenced, low complexity library | DSN may have degraded the library. | Make sure that the library is well denatured. Check thermocycler quality or increase denaturing time. During normalization, do not allow the tubes to be at room temperature beyond the short time needed to spin down the contents. Always work on ice when setting up follow-up amplifications after normalization as the DSN enzyme may still be active. Also make sure the buffer concentrations used during normalization are correct. |
| Unexpected read sequences (e.g. from a different organism or unexpected genomic regions) | Contamination | Make sure the library is not contaminated with another library. Keep a separate area with equipment and reagents for work during the pre-amplification phase of the library. Use a negative control during the real-time PCR amplification and run on a gel to check if a curve is visible. |
Acknowledgments
The authors are grateful to the principal investigators Christine & Jonathan Seidman. This work was funded by grants from U54 Syscode, NHLBI Cardiac Development Consortium, and the Leducq Foundation.
References
- Bogdanov EA, Shagina I, et al. Normalizing cDNA libraries. Curr Protoc Mol Biol. 2010;Chapter 5(Unit 5 12):1–27. doi: 10.1002/0471142727.mb0512s90. [DOI] [PubMed] [Google Scholar]
- Cloonan N, Forrest AR, et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat Methods. 2008;5(7):613–9. doi: 10.1038/nmeth.1223. [DOI] [PubMed] [Google Scholar]
- Kim JB, Porreca GJ, et al. Polony multiplex analysis of gene expression (PMAGE) in mouse hypertrophic cardiomyopathy. Science. 2007;316(5830):1481–4. doi: 10.1126/science.1137325. [DOI] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–8. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Nagalakshmi U, Wang Z, et al. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008;320(5881):1344–9. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultan M, Schulz MH, et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science. 2008;321(5891):956–60. doi: 10.1126/science.1160342. [DOI] [PubMed] [Google Scholar]
- Tang F, Barbacioru C, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods. 2009;6(5):377–82. doi: 10.1038/nmeth.1315. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–5. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456(7221):470–6. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, et al. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10(1):57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhulidov PA, Bogdanova EA, et al. Simple cDNA normalization using kamchatka crab duplex-specific nuclease. Nucleic Acids Res. 2004;32(3):e37. doi: 10.1093/nar/gnh031. [DOI] [PMC free article] [PubMed] [Google Scholar]