

Abstract
Despite interest in malic enzyme(ME)s in insulin cells, mitochondrial malic enzyme (ME2) has only been studied with estimates of mRNA or with mRNA knockdown. Because an mRNA’s level does not necessarily reflect the level of its cognate enzyme, we designed a simple spectrophotometric enzyme assay to measure ME2 activity of insulin cells by utilizing the distinct kinetic properties of ME2. Mitochondrial ME2 uses either NAD or NADP as a cofactor, has a high Km for malate and is allosterically activated by fumarate and inhibited by ATP. Cytosolic ME (ME1) and the other mitochondrial ME (ME3) use only NADP as a cofactor and have lower Kms for malate. The assay easily showed for the first time that substantial ME2 activity is present in pancreatic islets of humans, rats and mice and INS-1 832/13 cells. ME2’s presence was confirmed with immunoblotting. There was no evidence that ME3 is present in these tissues.
Keywords: Mitochondrial NAD(P) malic enzyme, ME2, malate, fumarate activation, ATP inhibition, pancreatic islets, spectrophotometric enzyme assay procedure
INTRODUCTION
Malic enzymes catalyze the oxidative decarboxylation of malate to produce pyruvate and carbon dioxide with the reduction of NAD(P) to NAD(P)H. There are three malic enzyme isoforms present in various amounts in organs of mammals, one cytosolic isoform (ME1) and two mitochondrial isoforms (ME2 and ME3). ME1 (EC 1.1.1.40) and ME3 (EC 1.1.1.40) are specific for NADP as a pyridine nucleotide cofactor and have low Kms for malate. ME2 (EC 1.1.1.38) has a dual cofactor specificity for NAD and NADP, but has a higher affinity for NAD. In addition, distinct from ME1 and ME3, ME2 has a high Km for malate, is allosterically activated by fumarate and is moderately sensitive to inhibition by ATP [1-5]. Although the malic enzyme reaction is reversible, the flux of at least the cytosolic malic enzyme reaction is thought to be only in the direction of pyruvate formation in vivo [6].
In 1995 our laboratory first suggested the feasibility of a pyruvate malate shuttle involving ME1 in the cytosol and pyruvate carboxylase in the mitochondria in rat pancreatic islets and proposed that at least one role of the shuttle was to export NADPH equivalents from the mitochondria to the cytosol to promote insulin secretion [7]. In support of this concept other laboratories using 13C-NMR found evidence of pyruvate cycling involving (a) malic enzyme(s) and pyruvate carboxylase in glucose-stimulated clonal beta cells [8-10]. However, subsequently we found evidence against our own hypothesis for the necessary involvement of ME1 in insulin secretion when we failed to detect ME1 enzyme activity in pancreatic islets isolated from several normal mouse strains [11]. In agreement with this idea, there exists the Mod-1 mouse that possesses a naturally occurring null mutation for the Me1 gene such that it has no ME1 in any tissue of the body, yet is phenotypically normal [12]. In even more support for the non-requirement of ME1 for insulin secretion we crossed the Mod-1 mouse with a mouse that lacks cytosolic glycerol phosphate dehydrogenase to derive the mmgg mouse that lacks both enzymes in all tissues. This mouse also has normal blood glucose and insulin levels and glucose-stimulated insulin release from its pancreatic islets is normal [13].
Recently the role of ME1 and mitochondrial malic enzyme (ME2) in insulin secretion has become controversial with contradictory effects on insulin secretion in studies of siRNA knockdown of the mRNAs that encode the enzymes in insulin secreting tissues [14-17] and conflicting reports on ME1 levels in mouse islets from different laboratories. ME1 enzyme activity is easy to measure with spectrophotometric assays and is readily detectable in the cytosol of human and rat pancreatic islets and insulin cell lines [7, 11, 13]. Except for one recent report that ME1 activity is present in mouse islets [15], ME1 activity has been found to be absent in mouse islets [11, 13, 18-20]. However, the presence or absence of assayable ME2 enzyme activity or ME2 protein in any insulin-secreting tissue has never been reported. Pongratz et al [14] reported in 2007 that siRNA knockdown of Me1 mRNA and enzyme activity in INS-1 cells inhibited both glucose- and leucine-plus-glutamine-stimulated insulin release and that knockdown of Me2 mRNA inhibited only leucine-plus-glutamine-induced insulin release. They did not demonstrate ME2 activity with a spectrophotometric enzyme assay, but did demonstrate its presence in a preparation of isolated INS-1 mitochondria with an elegant approach in which heavy isotope tracers and NMR analysis were used. Recently, Ronnebaum et al [17] reported that knockdown of either Me1 or Me2 mRNA in rat pancreatic islets did not inhibit glucose-stimulated insulin release, but that knockdown of Me1 mRNA or Me2 mRNA did inhibit glucose-stimulated insulin release in INS-1 cells. However, they noted that the lower insulin release in the INS-1 cells with the knocked down Me2 mRNA was associated with suboptimal cell density due to a decreased rate of cell growth. They did not report measurements of levels of enzyme activity or protein of either malic enzyme [17]. Although malic enzymes were not the primary emphasis of a study by Li et al [21] of islets of a mouse with a knocked out sulfonylurea receptor, they reported the presence of Me1, Me2 and Me3 mRNAs in islets of this mouse and a normal mouse. In their article they raised the valid point that the enzyme assay conditions we used to measure malic enzyme activity when we failed to detect malic enzyme activity in cytosol and mitochondrial fractions of mouse islets reported in reference 11 would not detect ME2 activity because we used a low concentration of malate in our enzyme reaction mixture that would permit detection of primarily ME1. However, Li et al [21] did not report measurements of enzyme activity or protein of any malic enzyme.
The presence in a tissue of an mRNA that encodes an enzyme is not sufficient evidence for the presence of the enzyme, the actual functional unit of metabolism. PCR methods used to measure mRNAs are so sensitive that contamination of pancreatic islets with even a few non-islet cells can cause false positives. Furthermore, the presence of an mRNA does not accurately reflect the level of translation of the protein it encodes and even whether the protein is present in a tissue at all (Examples are briefly noted in the Results and Discussion section.). In view of the interest in the role of malic enzymes in insulin secretion it would be beneficial to have a procedure for quantifying ME2 activity in insulin-secreting tissues that is as convenient as the spectrophotometric assay used to measure ME1 in the cytosol of tissues. The distinct kinetic properties of ME2 have been discerned with purified preparations of ME2 [1-4]. Taking advantage of these properties enabled us to develop a simple spectrophotometric enzyme assay for measuring ME2 enzyme activity in crude whole-cell homogenates and mitochondrial fractions of pancreatic islets and clonal insulinoma cell lines. Although ME2 is two to three times more active in the presence of NAD than NADP [1, 4], NAD cannot be used to measure ME2 activity in crude tissue preparations because malate dehydrogenase activity, which is usually 100-1000 fold the level of malic enzyme activity in most tissues, including pancreatic islets, would be detected. Since ME2 shows a significant enzyme rate in the presence of NADP [4], we measured ME2 enzyme activity with NADP as the cofactor. The differentiation of ME2 activity from ME1 activity was made possible by measuring ME1 activity in the presence of 1 mM malate, a saturating concentration of malate in respect to the Vmax rate for ME1 (and also ME3 if ME3 was present) and that gives a negligible rate for ME2. ME2 activity was measured in the presence of 10 mM malate plus fumarate, a condition that gives a maximal rate for ME2. When ME2 activity was measured in whole-cell homogenates, the activity in the presence of the low concentration of malate was subtracted from the higher activity in the presence of the high malate plus fumarate to give the rate attributable to ME2. When a mitochondrial fraction was the enzyme source, it mattered little whether the enzyme rate in the presence of the low concentration of malate was subtracted from the enzyme rate in the presence of high malate plus fumarate because the rate in the presence of low malate was negligible compared to the rate in the presence of the high concentration of malate. Adding ATP to the enzyme reaction mixture and observing inhibition of the enzyme rate confirmed that the activity measured was that of ME2. Use of the assay indicates for the first time that assayable ME2 enzyme activity is present in pancreatic islets of the human, rat and mouse and the INS-1 832/13 cell line. In addition, the presence of ME2 protein in these tissues was demonstrated with immunoblots with anti-ME2 antibody.
EXPERIMENTAL PROCEDURES
Materials
Chemicals, anti-human ME2 antibody (Catalog Number HPA008880) and anti-beta actin antibody were from Sigma-Aldrich Corporation (St. Louis, MO). Sprague-Dawley rats and ICR(CD-4) and NSA(CF-1) mice were from Harlan Sprague-Dawley (Madison, WI). C57BL/6 mice were from the Jackson Laboratories (Bar Harbor, MA). Pancreatic islets were isolated as previously described [7, 11, 13, 22, 23]. Human pancreatic islets were from Prodo Laboratories Inc. and the Sharp-Lacy Research Institute, Irvine, CA, sponsored by the Juvenile Diabetes Research Foundation and the University of Wisconsin Islet Cell Resource Center, Madison, WI sponsored by the Islet Cell Resource Consortium.
Subcellular fractionation
Monolayers of INS-1 832/13 cells were released with trypsin and EDTA from four 150 mm tissue culture plates, washed twice with PBS and homogenized in 2 ml of 220 mM mannitol, 70 mM sucrose, 5 mM potassium Hepes buffer, pH 7.5 (KMSH) containing 1 mM dithiothreitol and protease inhibitor cocktail (ThermoScientific product no 78415) using 40 strokes in a Potter Elvehjem homogenizer (1.4 cm diameter Teflon pestle; 1.9 cm inner diameter × 12 cm mortar). Homogenization and subsequent steps were at 4°. A portion of the whole-cell homogenate was saved and the remainder was centrifuged at 600 × g for 10 min. The resulting supernatant fraction was centrifuged at 10,000 × g for 10 min to give the mitochondrial pellet. The mitochondrial pellet was suspended in 300 μl of the KMSH, dithiothreitol and protease inhibitor solution and stored frozen at -20° until used. The postmitochondrial supernatant fraction was centrifuged at 20,000 × g for 20 min to give a supernatant fraction used as cytosol. The yield of mitochondria was calculated by comparing the total amount of the activity of the exclusively mitochondrial enzyme glutamate dehydrogenase in the mitochondria to that in the whole cell homogenate. The amount of mitochondrial breakage was estimated from the amount of glutamate dehydrogenase activity in the cytosol. Subcellular fractionation of pancreatic islets to prepare a whole cell homogenate, cytosol and mitochondria was performed similarly to that for INS-1 832/13 cells except that homogenization was performed in 200-400 μl of KMSH in a microfuge test tube and the post-600-x-g-x-10 min supernatant fraction was centrifuged at 5,500 × g for 10 min to give the mitochondrial pellet as previously described [7, 11, 13, 23].
Enzyme assays
Cytosolic malic enzyme (ME1) (but also ME3 if present) activity was estimated spectrophotometrically by monitoring NADPH formation at 340 nm in an enzyme reaction mixture containing 1.0 mM malate, 0.3 mM NADP, 6 mM MgCl2, 1 mM DTT and 50 mM Tris-chloride buffer, pH 7.6 at 37° as previously described [11, 13]. Mitochondrial NAD(P) (ME2) malic enzyme activity was measured in 1 ml of a reaction mixture containing 0.3 mM NADP, 1 mM dithiothreitol, 10 mM MgCl2 and 50 mM Tris-chloride buffer, pH 7.4, and 10-30 μl of a source of enzyme (whole-cell homogenate or mitochondria). The formation of NADPH was monitored spectrophotometrically at 340 nm and 37°. Background rates were measured with no added substrate and again with 1 mM malate. The ME2 enzyme reaction was started with the addition of fumarate (5 mM), an activator of ME2, and adding malate to increase its concentration to 11 mM. Enzyme rates were also measured in the presence of 2 mM ATP, an inhibitor of ME2. Mitochondrial malic enzyme rates were measured with various orders of addition of fumarate, malate and ATP to the enzyme reaction mixture as shown in Table 1. When the enzyme activity in whole-cell homogenates was measured, the enzyme rate in the presence of 1 mM malate attributable to cytosolic malic enzyme (ME1) was subtracted from the total rate in the presence of fumarate plus 11 mM malate to give the rate attributable to mitochondrial NAD(P) malic enzyme (ME2). Because the activity of ME2 in the presence of 1 mM malate and no activator is slightly > 0, this slightly underestimates the activity of ME2 in a whole cell homogenate (See Results and Discussion). In a slightly different ME2 assay developed after all the studies reported herein were completed, an alternate approach was sometimes used in which the enzyme rate and the two background rates were measured simultaneously (instead of sequentially) in duplicate or quadruplicate companion wells containing 200 μl of the ME2 reaction mixture described above and 4-8 μl of enzyme source in a 96-well plate monitored in a Molecular Devices SpectraMax M2 Microtiter Plate Reader. The contents of the wells were identical except that the ME2 enzyme rate wells contained 10 mM malate plus 5 mM fumarate. The background rate wells contained either no malate and no fumarate or 1 mM malate and no fumarate. The background rates were subtracted from the rate with fumarate and high malate to give the rate attributable to ME2. In addition, 1.2 mM NADP was used as this gives an enzyme rate 20%-30% higher than with 0.3 mM NADP. Glutamate dehydrogenase enzyme activity was measured as previously described [23].
Table 1. Activation by fumarate and inhibition by ATP of ME2 malic enzyme assayed in a mitochondrial extract of INS-1 832/13 cells.
An extract of mitochondria was incubated in a reaction mixture in the presence of a saturating level of the enzyme substrate, malate (10 mM), and with or without the activator fumarate (5 mM) or inhibitor ATP (2 mM) added in different sequences. Sequential additions to a single cuvette are separated from the additions to other cuvettes by spaces between lines. Results show the individual rates or the mean ± SE of three replicate measurements.
| Additions (Order of addition) |
Malic Enzyme Rate (nmol NADPH formed/min/mg protein) |
|---|---|
| Fumarate (1) | 0 (3) |
| Malate (2) | 19.3 ± 1.6 (3) |
|
| |
| Malate (1) | 10.6 |
| Fumarate (2) | 19.1 |
| ATP (3) | 9.5 |
|
| |
| Malate (1) | 10.5 |
| ATP (2) | 3.2 |
| Fumarate (3) | 11.4 |
Immunoblotting
Immunoblotting was performed as previously described [23].
RESULTS AND DISCUSSION
Importance of estimating enzyme activity
Measurements of enzyme activity provide quantitative estimates of the levels of the functional units of metabolic pathways. The presence of an mRNA that encodes an enzyme is often cited as evidence for the presence of the enzyme in a tissue, but detecting an mRNA does not reflect the level of the cognate enzyme or even whether the enzyme is present. However, if an mRNA is not present, its cognate enzyme is unlikely to be present. Two examples of reports of the presence of mRNAs in rat pancreatic islets that encode proteins that are not present are of those that encode the liver isoform of pyruvate kinase and cytosolic phosphoenolpyruvate carboxykinase. Although the presence of these two mRNAs is frequently reported in pancreatic islets or clonal beta cells, the enzyme activity and immunoreactivity of pyruvate kinase in pancreatic islets are that of the M2 isoform [24-26] and phosphoenolpyruvate carboxykinase enzyme activity is not present in pancreatic islets [22, 27]. (Since phosphoenolpyruvate carboxykinase and the liver isoform of pyruvate kinase are found in gluconeogenic tissues and the beta cell does not require or possess the capacity for gluconeogenesis [28], there is no need for the beta cell to possess these two enzymes.). Besides being the most quantitative, measurements of enzyme activity show that the enzyme protein is functional. In the case of siRNA experiments, truncated mRNAs can be produced and translated into truncated proteins [29]. The truncated mRNAs may be erroneously assumed to reflect the level of full length mRNAs depending on the PCR primers used in the amplification and measurement of the mRNA level and also truncated proteins can be detected in immunoblots [29].
Mitochondrial malic enzyme ME2 vs ME3 in beta cells
Although ME2 uses both NAD and NADP as pyridine nucleotide cofactors and is more active in the presence of NAD than in the presence of NADP, ME2 activity is one-third to one-half as active in the presence of a saturating concentration of NADP compared to a saturating concentration of NAD [1, 4]. Since an enzyme reaction mixture containing NAD would detect malate dehydrogenase, which is 100 - 1,000 times more active than any malic enzyme in beta cells [30, 31] as is typical of many tissues [32], it is necessary to use NADP as the cofactor when measuring ME2 activity. ME3 uses exclusively NADP as a cofactor and, thus, an assay mixture containing NADP would detect ME3 if ME3 was present. However, Pongratz et al [14] reported that the level of Me3 mRNA in rat pancreatic islets and INS-1 cells is very low compared to the levels of Me1 and Me2 mRNAs and our data from INS-1 832/13 cells agree with their data (The ratio of mRNAs encoding ME1/ME2/ME3 was 10: 6: 2 as judged from mRNA microarrays (NimbleGen, Madison, WI), L.J. Brown and M.J. MacDonald, unpublished observation.). Li et al [21] also showed that Me3 mRNA is much lower than Me1 mRNA and Me2 mRNA in mouse islets. In mouse islets we found that the relative levels of mRNAs that encode malic enzymes are Me2 >> Me1 > Me3. Perhaps more definitively, in our hands when Me2 mRNA was knocked down 85-95% in the cell lines we derived from the INS-1 832/13 cell line, there was no longer detectable malic enzyme activity in mitochondria from these cell lines and no ME2 protein seen in immunoblots (L.J. Brown, M.J. Longacre, M.A. Kendrick and M.J. MacDonald, unpublished observation). This is consistent with ME2 being the only mitochondrial malic enzyme in this cell line. In addition, in mitochondria of islets from humans, rats and INS-1 cells, as well as in whole-cell homogenates of mouse islets that contain no ME1 activity (11, 13, 18-20), we found that the enzyme rate in the presence of 1 mM malate in the malic enzyme reaction mixture (that should measure ME3 activity if present) was not significantly different from the background rate with no substrate. Collectively, these data suggest that if ME3 is present at all in islets of the mouse, rat or human or INS-1 832/13 cells, its level is very low.
ME2 activity in the INS-1 832/13 cell line and pancreatic islets of three genera
ME1 and ME3 use only NADP and are maximally active at a low concentration of malate [1]. In crude homogenates of whole cells an estimate of ME2 activity was obtained by measuring malic enzyme activity in the cuvette in the presence of NADP and 1 mM malate and then adding fumarate and increasing the malate concentration to 11 mM. The enzyme activity in the presence of 1 mM malate was then subtracted from the enzyme activity in the presence of fumarate plus 11 mM malate to give the enzyme activity attributable to ME2. Since, as mentioned above, isolated mitochondria from our INS-1 832/13 cell lines in which ME2 mRNA was knocked down 85-95% showed no enzyme rate in the presence of 1 mM malate, this indicates there was no cytosolic malic enzyme contaminating the mitochondrial preparation and very little or no ME3 present. In mitochondria from the INS-1 832/13 cell line, which was the parent cell line for the knockdown experiments, as well as the cell line studied in the current work, the enzyme rate in the presence of 1 mM malate equaled 7-9% of the rate in the presence of fumarate and 11 mM malate (data not shown). This suggests that this slow rate could be due to ME2. Thus, subtracting the enzyme rate in the presence of 1 mM malate from the total enzyme rate might underestimate ME2 activity by at most 10%. Since ME2 is inhibited by ATP [1, 4] adding ATP to the reaction mixture gives further confirmation that the enzyme activity measured is indeed from ME2.
Table 1 shows that mitochondria of INS-1 832/13 cells possess a malic enzyme activity with the properties of ME2 with activation by fumarate and inhibition by ATP. Inhibition by ATP was greater in the absence of fumarate as is consistent with reports with purified ME2 [1, 4]. Table 2 shows that the specific enzyme activity of ME2 in mitochondria of INS-1 832/13 cells is comparable to the specific enzyme activity of ME1 in the cytosol of these cells and that about 35% of total cell malic enzyme activity is attributable to ME2. Table 3 shows that whole-cell homogenates and/or mitochondrial preparations of rat, human and mouse pancreatic islets possess ME2 activity. As mentioned above, our studies indicate that the pancreatic islets and INS-1 cells possess little or no ME3 enzyme activity. However, since ME3, like ME1, is maximally active in the presence of a low concentration of malate [1], subtracting the activity in the presence of 1 mM malate when assaying a crude whole-cell homogenate of the islets would also give activity attributable to ME2, even if ME3 were present. Malic enzyme activity in the pancreatic islet and INS-1 whole-cell homogenates and mitochondria assayed in the presence of high malate and fumarate was inhibited by ATP (Table 3), consistent with the idea that ME2 was the enzyme activity detected. We observed previously that mouse islets possessed no ME1 enzyme activity (malic enzyme activity in the cytosol fraction in the presence of 1 mM malate) [11]. Mouse whole-cell homogenate ME2 activity appeared to be higher than in homogenates of whole cells of islets from rats or humans (Table 3). Whether the apparent higher level of ME2 is a compensation for the lack of ME1 in mouse islets will require further investigation. Immunoblotting with an anti-ME2 antibody confirmed that ME2 was present in pancreatic islets of humans, the Sprague Dawley rat and the NSA and C57BL6 mouse strains and INS-1 832/13 cells (Figure 1).
Table 2. Activities of ME1 and ME2 in INS-1 832/13 cells.
Enzyme activities were measured and yields of mitochondria were calculated as described under Experimental Procedures.
| Specific Enzyme Activity of Malic Enzymes Based on Cytosol or Mitochondria Protein | |
|---|---|
| ME1 in cytosol | 29.1 ± 1.6 nmol NADPH/min/mg cytosol proteina |
| ME2 in mitochondria | 20.5 ± 0.4 nmol NADPH/min/mg mitochondrial proteinb |
| Specific Enzyme Activities Based on Cell Protein (nmol NADPH/min/mg total cell protein) | % of Total | |
|---|---|---|
| ME1 + ME2 (Measured in homogenate) |
18.7 ± 0.9 | 100% |
| ME1 (Calculated from measurement in cytosol)a |
8.7 ± 0.4 | 47% |
| ME1 (Calculated from measurement in whole cell homogenate)a |
12 ± 0.1 | 64% |
| ME2 (Calculated from activity in mitochondria and yield of mitochondria)b |
6.6 ± 0.08 | 35% |
| ME2 (Calculated by subtracting ME1 activitya from total ME activityb in homogenate) |
6.7 ± 0.9 | 36% |
Activity measured in presence of 1 mM malate
Activity measured in presence of 10 mM malate plus 5 mM fumarate.
Table 3. Mitochondrial NAD(P) malic enzyme ME2 activity is present in INS-1 832/13 cells and pancreatic islets of the mouse, rat and human.
Mice were the ICR and NSA strains. ME2 activity was measured as described under Experimental Procedures without or with ATP (2 mM). Results are the mean with the number of preparations of enzyme source in parentheses and SE (when N ≥ 3).
| Enzyme Source | ME2 Specific Enzyme Activity (nmol NADPH/min/mg protein) |
|---|---|
| INS-1 832/13 homogenate + ATP |
6.7 ± 0.9 (3)a 2.3 ± 0.2 (3)a |
| INS-1 832/13 mitochondria + ATP |
25 ± 3.9 (5) 4.8 ± 2.6 (3) |
| Mouse islet whole cell homogenate + ATP |
18 ± 3.6 (3) 5.1 ± 0.9 (3) |
| Rat islet whole cell homogenate + ATP |
5.4 ± 1.1 (5) 0 (2) |
| Rat islet mitochondria + ATP |
12 (1) 0 (1) |
| Human islet whole cell homogenate + ATP |
4.7 ± 0.22 (7) 0.8 ± 0.5 (7) |
| Human islet mitochondria | 18 ± 3.6 (3) |
Rate is extrapolated to basis of whole cell protein even though measured in mitochondrial fraction.
Figure 1. ME2 is present in human, rat and mouse pancreatic islets and INS-1 832/13 cells.
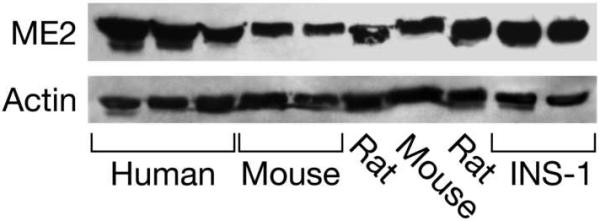
Immunoblot of islets from individual humans and individual batches of islets from Sprague-Dawley rats or mice (lanes 4 and 7, the NSA strain; lane 5, the C57BL6 strain) and INS-1 832/13 cells with 20 μg protein/lane probed with anti-human ME2 antibody. The blot was additionally probed with anti-beta actin antibody to show equal loading of protein across lanes.
ACKNOWLEDGEMENTS
This work was supported by NIH grant DK28348 and the Oscar C. Rennebohm Foundation. The authors thank Mary Rabaglia for supplying C57BL/6 mouse islets.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Lin RC, Davis EJ. J. Biol. Chem. 1974;249:3867–3875. [PubMed] [Google Scholar]
- [2].Mandella RD, Sauer LA. J. Biol. Chem. 1975;250:5877–5884. [PubMed] [Google Scholar]
- [3].Nagel WO, Sauer LA. J. Biol. Chem. 1982;257:12405–12411. [PubMed] [Google Scholar]
- [4].Moreadith RW, Lehninger AL. J. Biol. Chem. 1984;259:6222–6227. [PubMed] [Google Scholar]
- [5].Chang GG, Tong L. Biochemistry. 2003;42:12721–12733. doi: 10.1021/bi035251+. [DOI] [PubMed] [Google Scholar]
- [6].Veech RL, Eggleston LV, Krebs HA. Biochem. J. 1969;115:606–619. doi: 10.1042/bj1150609a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].MacDonald MJ. J. Biol. Chem. 1995;270:20051–20058. [PubMed] [Google Scholar]
- [8].Lu D, Mulder H, Zhao P, Burgess SC, Jensen MV, Kamzolova S, Newgard CB, Sherry AD. Proc. Natl. Acad. Sci. 2002;99:2708–2713. doi: 10.1073/pnas.052005699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Cline GW, Lepine RL, Papas KK, Kibbey RG, Shulman GI. J. Biol. Chem. 2004;279:44370–44375. doi: 10.1074/jbc.M311842200. [DOI] [PubMed] [Google Scholar]
- [10].Simpson NE, Khokhlova N, Oca-Cossio JA, Constantinidis I. Diabetologia. 2006;49:1338–1348. doi: 10.1007/s00125-006-0216-5. [DOI] [PubMed] [Google Scholar]
- [11].MacDonald MJ. Am. J. Physiol. Endo. Metab. 2002;283:E302–E310. doi: 10.1152/ajpendo.00041.2002. [DOI] [PubMed] [Google Scholar]
- [12].Lee CY, Lee SM, Lewis S, Johnson FM. Biochemistry. 1980;19:5098–5103. doi: 10.1021/bi00563a025. [DOI] [PubMed] [Google Scholar]
- [13].MacDonald MJ, Marshall LK. Mol. Cell. Biochem. 2001;220:117–125. doi: 10.1023/a:1010821821921. [DOI] [PubMed] [Google Scholar]
- [14].Pongratz RL, Kibbey RG, Shulman GI, Cline GW. J. Biol. Chem. 2007;282:200–207. doi: 10.1074/jbc.M602954200. [DOI] [PubMed] [Google Scholar]
- [15].Xu J, Han J, Long YS, Lock J, Weir GC, Epstein PN, Liu YQ. Diabetologia. 2008;51:2281–2289. doi: 10.1007/s00125-008-1155-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Guay C, Madiraju SR, Aumais A, Joly E, Prentki M. J. Biol. Chem. 2007;282:35657–35665. doi: 10.1074/jbc.M707294200. [DOI] [PubMed] [Google Scholar]
- [17].Ronnebaum SM, Jensen MV, Hohmeier HE, Burgess SC, Zhou YP, Qian S, MacNeil D, Howard A, Thornberry N, Ilkayava O, Lu D, Sherry AD, Newgard CB. J. Biol. Chem. 2008;283:28909–28917. doi: 10.1074/jbc.M804665200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ashcroft SJ, Randle PJ. Biochem. J. 1970;119:5–15. doi: 10.1042/bj1190005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Heart E, Yaney GC, Corkey RF, Schultz V, Luc E, Liu L, Deeney JT, Shirihai O, Tornheim K, Smith PJ, Corkey BE. Biochem. J. 2007;403:197–205. doi: 10.1042/BJ20061209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Heart E, Cline GW, Collis LP, Pongratz RL, Gray JP, Smith PJS. Am. J. Physiol. Endocrinol. Metab. 2009 March 17;296:E1354–E1367. doi: 10.1152/ajpendo.90836.2008. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Li C, Nissim I, Chen P, Buettger C, Najafi H, Daikhin Y, Nissim I, Collins HW, Yudkoff M, Stanley CA, Matchinsky FM. J. Biol. Chem. 2008;283:17238–17249. doi: 10.1074/jbc.M709235200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].MacDonald MJ, Chang CM. Diabetes. 1985;34:246–250. doi: 10.2337/diab.34.3.246. [DOI] [PubMed] [Google Scholar]
- [23].MacDonald MJ, Smith AD, III, Hasan NM, Sabat G, Fahein LA. J. Biol. Chem. 2007;282:30596–30606. doi: 10.1074/jbc.M702732200. [DOI] [PubMed] [Google Scholar]
- [24].MacDonald MJ, Chang CM. Mol. Cell. Biochem. 1985;68:115–120. doi: 10.1007/BF00219375. [DOI] [PubMed] [Google Scholar]
- [25].MacDonald MJ. Diabetologia. 1995;38:125. doi: 10.1007/BF02369367. [DOI] [PubMed] [Google Scholar]
- [26].Chatterton TA, Reynolds CH, Lazarus NR, Pogson CI. Biochem. J. 1982;204:605–608. doi: 10.1042/bj2040605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hedeskov CJ, Capito K, Thams P. Biochim. Biophys. Acta. 1984;791:37–44. doi: 10.1016/0167-4838(84)90278-4. [DOI] [PubMed] [Google Scholar]
- [28].MacDonald MJ, McKenzie DI, Walker TM, Kaysen JH. Horm. Metab. Res. 1992;24:158–160. doi: 10.1055/s-2007-1003284. [DOI] [PubMed] [Google Scholar]
- [29].Gu W, Cochrane M, Leggatt GR, Payne E, Choyce A, Zhou F, Tindle R, McMillan NA. Proc. Natl. Acad. Sci. U.S.A. 2009 May 4; doi: 10.1073/pnas.0812085106. Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].MacDonald MJ, Efendic S, Ostenson CG. Diabetes. 1996;45:886–890. doi: 10.2337/diab.45.7.886. [DOI] [PubMed] [Google Scholar]
- [31].MacDonald MJ, Longacre MJ, Langberg EC, Tibell A, Kendrick MA, Fukao T, Ostenson CG. Diabetologia. 2009;52:1087–1091. doi: 10.1007/s00125-009-1319-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Teller JK, Fahien LA, Davis JW. J. Biol. Chem. 1992;267:10423–10432. [PubMed] [Google Scholar]