

Abstract
Drugs of abuse such as cocaine induce long-term synaptic plasticity in the reward circuitry, which underlies the formation of drug-associated memories and addictive behavior. We reported previously that repeated cocaine exposure in vivo facilitates long-term potentiation (LTP) in dopamine neurons of the ventral tegmental area (VTA) by reducing the strength of GABAergic inhibition and that endocannabinoid-dependent long-term depression at inhibitory synapses (I-LTD) constitutes a mechanism for cocaine-induced reduction of GABAergic inhibition. The present study investigated the downstream signaling mechanisms and functional consequences of I-LTD in the VTA in the rat. Extracellular signal-regulated kinase (ERK) signaling has been implicated in long-term synaptic plasticity, associative learning, and drug addiction. We tested the hypothesis that VTA ERK activity is required for I-LTD and cocaine-induced long-term synaptic plasticity and behavioral effects. We show that the activation of receptors required for I-LTD increased ERK1/2 phosphorylation and inhibitors of ERK activation blocked I-LTD. We further demonstrate that ERK mediates cocaine-induced reduction of GABAergic inhibition and facilitation of LTP induction. Finally, we show that cocaine conditioned place preference (CPP) training (15 mg/kg; four pairings) increased ERK1/2 phosphorylation in the VTA, while bilateral intra-VTA injections of a CB1 antagonist or an inhibitor of ERK activation attenuated ERK1/2 phosphorylation and the acquisition, but not the expression, of CPP to cocaine. Our study has identified the CB1 and ERK signaling cascade as a key mediator of several forms of cocaine-induced synaptic plasticity and provided evidence linking long-term synaptic plasticity in the VTA to rewarding effects of cocaine.
Introduction
The endocannabinoid (eCB) system is critically involved in behavioral effects of many drugs of abuse, including cocaine (Gerdeman et al., 2003). A primary site of the interaction between addictive drugs and the cannabinoid receptor (CB1) is the ventral tegmental area (VTA) (Lupica and Riegel, 2005). An important function of eCBs is to mediate short-term and long-term depression (LTD) at excitatory and inhibitory synapses (I-LTD) (Gerdeman et al., 2002; Marsicano et al., 2002; Robbe et al., 2002; Chevaleyre and Castillo, 2003). Repeated cocaine exposure in vivo reduces GABAergic inhibition to VTA dopamine neurons by inducing I-LTD-like synaptic modification (Liu et al., 2005; Pan et al., 2008a). eCB-LTD and I-LTD represent a widespread and fundamental mechanism by which synaptic strength and behavior can be regulated (Gerdeman et al., 2003; Heifets and Castillo, 2009). A common property for this type of LTD/I-LTD is that CB1 antagonists block the induction of CB1-LTD/I-LTD, but cannot reverse the established LTD/I-LTD when applied after LTD induction (Chevaleyre and Castillo, 2003; Ronesi and Lovinger, 2005; Pan et al., 2008a), suggesting that other downstream signaling mechanisms must be recruited to produce the lasting synaptic depression.
Extracellular signal-regulated kinase (ERK) may represent a candidate downstream mechanism that mediates I-LTD in the VTA. CB1 receptor agonists increased phosphorylated and active ERK in cell lines (Bouaboula et al., 1995; Wartmann et al., 1995) and the hippocampus (Derkinderen et al., 2003; Jiang et al., 2005). Cocaine-induced ERK phosphorylation in the striatum was CB1 receptor dependent (Corbillé et al., 2007). ERK has been implicated in both LTP (English and Sweatt, 1996, 1997) and LTD (Gallagher et al., 2004; Grueter et al., 2006; Kellogg et al., 2009). We tested the hypothesis that ERK is activated downstream of CB1 receptors and mediates the eCB-dependent I-LTD in the VTA. An important consequence of eCB-I-LTD is to prime excitatory synapses for LTP induction (Chevaleyre and Castillo, 2003, 2004; Zhu and Lovinger, 2007). We also examined whether the CB1 and ERK signaling cascade mediates cocaine-induced reduction of GABAergic inhibition and facilitation of LTP in VTA dopamine neurons (Liu et al., 2005; Pan et al., 2008a). ERK plays a critical role in associative learning (Atkins et al., 1998; Sweatt, 2004) and cocaine addiction (Lu et al., 2005, 2006; Grueter et al., 2006; Girault et al., 2007). Cocaine exposure increased ERK phosphorylation in the VTA (Berhow et al., 1996) and other brain regions (Valjent et al., 2000; Lu et al., 2005), and systemic or local administration of ERK inhibitors blocked cocaine-seeking behaviors (Lu et al., 2006; Girault et al., 2007). However, it remains largely unknown whether the CB1 or ERK activity in the VTA affects behavioral responses of cocaine. We investigated whether the VTA CB1 and ERK signaling cascade is required for the development of conditioned place preference (CPP) to cocaine. Here, we provide evidence that the VTA CB1 and ERK signaling cascade is required for I-LTD, cocaine-induced reduction of GABAergic inhibition, facilitation of LTP induction, and the acquisition of CPP to cocaine.
Materials and Methods
Animals
Male Sprague Dawley rats (Charles River) were used for brain slice electrophysiology Western blotting (Fig. 2) (P18–P30), immunohistochemistry, Western blotting (Figs. 6, 7), and behavior experiments (300–350 g). All experimental procedures were approved by the Institutional Animal Care and Use Committee of the Medical College of Wisconsin.
Figure 2.

The CB1, D2, or group I mGluR agonists increased ERK1/2 phosphorylation but had no significant effect on p-38 phosphorylation in the VTA. A, VTA slices were treated for 10 min with vehicle (0.01% DMSO), the CB1 receptor agonist WIN 55212-2 (2 μm), the CB1 receptor antagonist AM251 (2 μm), or MEK inhibitor U0126 (20 μm). Quantitative group data (top) and representative Western blots (bottom) of phosphorylated ERK1/2 (p-ERK) in VTA homogenates are shown (n = 5 for each group; **p < 0.01, ***p < 0.001). B, Group I mGluR agonist DHPG (50 μm; 10 min) increased ERK1/2 phosphorylation; this effect was attenuated by AM251 and was abolished by U0126 (n = 4 for each group; **p < 0.01, ***p < 0.001). C, D2 receptor agonist quinpirole (10 μm; 10 min) increased ERK1/2 phosphorylation; this effect was not affected by AM251 but was abolished by U0126 (n = 5 for each group; **p < 0.01, ***p < 0.001). D, WIN 55212-2, DHPG, and quinpirole had no significant effects on p-p38 levels in VTA homogenates (n = 4 for each group; p > 0.05). For each group, four to five slices were prepared from three to four rats. Error bars indicate SEM.
Figure 6.

Intra-VTA infusions of the CB1 receptor antagonist AM251 or MEK inhibitor U0126 during the conditioning phase attenuated the acquisition of CPP to cocaine. A, Timeline of drug treatment and behavioral paradigm. Groups of rats received saline and cocaine place conditioning once daily for 8 d. Vehicle (Veh, 0.5 μl), AM251 (5 μg), or U0126 (0.1 μg) was bilaterally infused into the VTA 20 min before each cocaine or saline pairing. B, Pretest indicates that rats did not exhibit significant baseline bias in place preference in all groups (n = 6–9; p > 0.05). C, Intra-VTA infusions of AM251 or U0126 significantly attenuated CPP in cocaine-conditioned rats but did not affect CPP scores in saline-conditioned rats (n = 6–9; *p < 0.05, ***p < 0.001). D, ERK1/2 phosphorylation in the VTA was significantly increased in cocaine-conditioned rats compared with that in saline-conditioned rats. Intra-VTA infusions of AM251 or U0126 during the conditioning phase significantly decreased ERK1/2 phosphorylation (n = 4 for each group; *p < 0.01, ***p < 0.001). Western blotting was performed on VTA samples collected ∼1 h after the CPP tests. Error bars indicate SEM.
Figure 7.
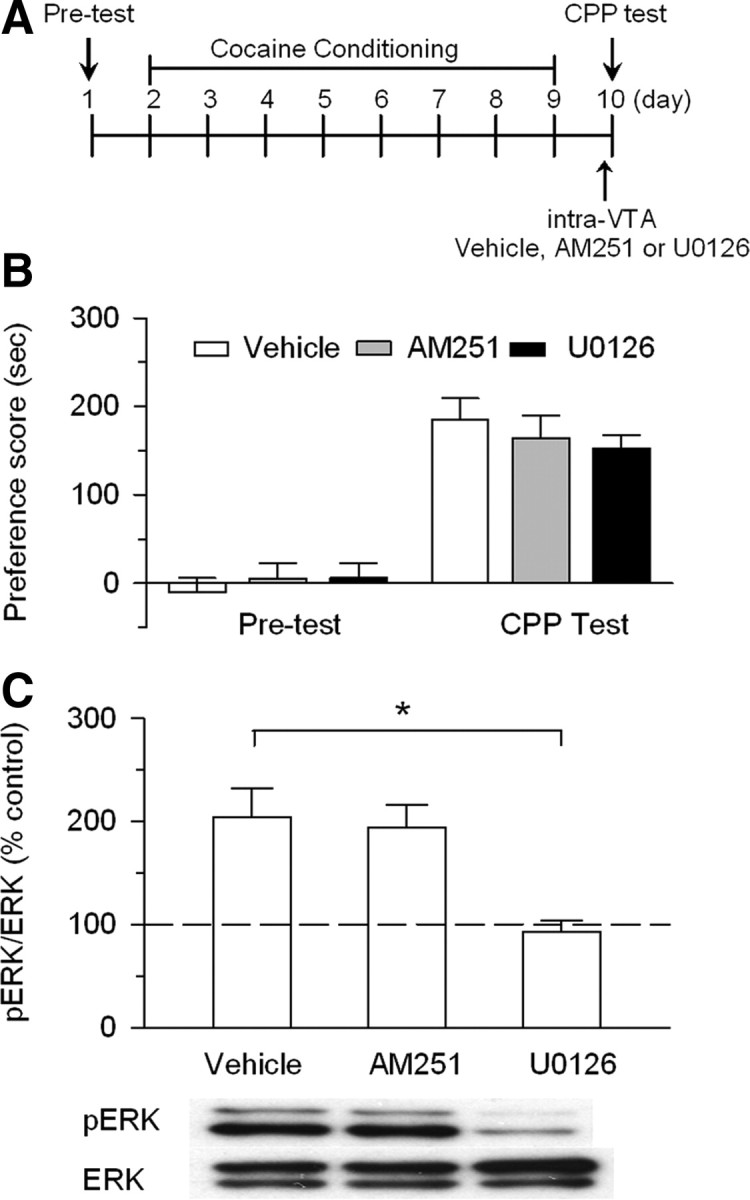
Intra-VTA infusions of AM251 or U0126 20 min before CPP test did not affect the expression of CPP to cocaine. A, Timeline of drug treatment and behavioral paradigm. Rats received only cocaine place conditioning. Vehicle (0.5 μl), AM251 (5 μg), or U0126 (0.1 μg) was bilaterally infused into the VTA 20 min before the CPP test. B, Cocaine conditioning induced significant increase in CPP scores compared with pretest. Intra-VTA infusion of AM251 or U0126 before the CPP test did not significantly affect CPP scores (n = 7–9; p > 0.05). C, Intra-VTA AM251 did not affect ERK1/2 phosphorylation in the VTA, whereas intra-VTA U0126 blocked ERK1/2 phosphorylation (n = 4 for each group; *p < 0.05). Western blotting was performed on VTA samples collected ∼1 h after the CPP tests. Error bars indicate SEM.
Brain slice preparation
Midbrain slices (250 μm) from male Sprague Dawley rats (P18–P30) were prepared as described previously (Pan et al., 2008a). In some experiments, rats were given daily intraperitoneal injection of either saline (0.9% NaCl; 1 ml/kg) or cocaine (15 mg/kg) for 5–7 d. The effectiveness of the cocaine treatment was shown by the sensitization of locomotor activity in an open field box (Liu et al., 2005). Subgroups of these rats were also given intraperitoneal injection of vehicle or one of the following receptor antagonists 20 min before each cocaine or saline injection. These include CB1 receptor antagonist 1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-(1-piperidyl)pyrazole-3-carboxamide (AM251) (2 mg/kg) and mitogen-activated protein kinase (MAPK)/ERK kinase (MEK) inhibitor α-[amino[(4-aminophenyl)thio]methylene]-2-(trifluoromethyl)benzeneacetonitrile (SL327) (50 mg/kg). Drug doses are based on previous reports (Atkins et al., 1998; Derkinderen et al., 2003; Thiemann et al., 2008). For rats that received intraperitoneal saline or cocaine injection, midbrain slices were prepared 24 h after the last cocaine or saline injection. The slices were incubated in oxygenated artificial CSF (ACSF) containing the following (in mm): 125 NaCl, 3 KCl, 2.5 CaCl2, 1 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 10 glucose. The ACSF was saturated with 95% O2 and 5% CO2. After recovery for at least 1 h at room temperature, a slice was transferred to the recording chamber and superfused with ACSF.
Electrophysiology
Whole-cell recordings were made using patch-clamp amplifiers (Multiclamp 700B) under infrared-differential interference contrast microscopy. Data acquisition and analysis were performed using DigiData 1440A digitizer and analysis software pClamp 10 (Molecular Devices). Signals were filtered at 2 kHz and sampled at 10 kHz. Dopamine neurons were identified by the presence of large Ih currents, rhythmic firing at low frequency, and prominent afterhyperpolarization (Johnson and North, 1992; Jones and Kauer, 1999; Liu et al., 2005). It has been shown that these electrophysiological characteristics do not exclusively belong to dopamine neurons (Margolis et al., 2006; Zhang et al., 2010). Putative dopamine neurons in our study may contain a small number of nondopamine neurons, which should be randomly distributed in different experimental groups.
For recording of evoked IPSCs, recordings were made in the presence of glutamate receptor antagonists CNQX (20 μm) and AP-5 (50 μm) and neurons were voltage clamped at −70 mV. Electrical stimulation was delivered by a bipolar tungsten stimulation electrode (WPI) that was placed ∼150 μm rostral to the recorded neuron, using square pulses with duration of 50 μs and frequency of 0.05–0.1 Hz. Glass pipette was filled with a solution containing the following (in mm): 100 K-gluconate, 50 KCl, 10 HEPES, 0.2 EGTA, 2 MgCl2, 4 Mg-ATP, 0.3 Na2GTP, and 10 Na2-phosphocreatine at pH 7.2 (with KOH). For recording of maximal IPSCs, neurons were voltage clamped at −20 mV and 100 mm K-gluconate and 50 mm KCl were replaced with Cs-gluconate (145 mm) and CsCl (5 mm). Maximal IPSCs were elicited by gradually increasing the stimulation intensity to recruit saturating IPSCs (Huang et al., 1999; Liu et al., 2005) and performed blind to the treatment history of the rat. For LTP experiments, neurons were current clamped at −70 mV and the internal solution contained the following (in mm): 142 potassium gluconate, 8 KCl, 10 HEPES, 0.2 EGTA, 2 MgCl2, 4 Mg-ATP, 0.3 Na2GTP, and 10 Na2-phosphocreatine, pH 7.2 (with KOH). The spike-timing protocol for LTP induction consisted of 20 bursts of EPSP–spike pairs, with each burst consisting of five paired stimuli delivered at 100 ms intervals (10 Hz) and an interburst interval of ∼5 s (Liu et al., 2005). The postsynaptic spikes were evoked by injection of depolarizing current pulses (1–2 nA; 2–3 ms), with the onset of EPSPs preceding the peak of postsynaptic spikes by ∼5 ms. EPSPs were evoked at 0.1 Hz before and after LTP induction. Series resistance (15–25 MΩ) was monitored throughout the recordings, and data were discarded if the resistance changed by >20%. All recordings were performed at 32 ± 1°C by using an automatic temperature controller (Warner Instruments).
Immunohistochemistry
Immunohistochemical procedures were based on published studies with minor modifications (Valjent et al., 2004). Twenty-four hours after the last cocaine or saline treatment, rats were deeply anesthetized with intraperitoneal injection of sodium pentobarbital (60 mg/kg; Sigma-Aldrich) and perfused through the aorta with 4% paraformaldehyde in 0.1 m sodium PBS with 4% sucrose, pH 7.4. After perfusion, brains were removed and fixed in the same fixative overnight at 4°C. The brains were then cryoprotected in increasing concentrations of sucrose (10, 20, and 30%) in 0.1 m PBS at 4°C, frozen on dry ice, and stored at −80°C until use. Coronal sections containing VTA were cut at 30 μm thickness with a cryostat. After H2O2 treatment and rinsing three times in PBS, free-floating sections were blocked for 1 h at room temperature with blocking solution (1% bovine serum albumin, 5% normal goat serum, and 1% Triton X-100 in 0.1 m PBS, pH 7.4). Sections were then incubated with 1:350 phosphorylated ERK (p-ERK) antibody (anti-phospho Thr202-Tyr204 ERK1/2; Cell Signaling Technology) at 4°C for 48 h. After rinsing three times, 5 min each, in PBS, sections were incubated in the secondary antibodies: 1:100 goat anti-rabbit IgG-HRP (Bio-Rad) for 2 h at room temperature. After rinsing twice with PBS and once with 0.1 m ammonium phosphate buffer (APB), pH 7.0, immunoreactivity was detected with 0.05% DAB–0.004% H2O2 in APB for 5 min, and the reaction was stopped with APB for 5 min and then rinsed in PBS three times, dehydrated, and coverslipped. Control sections were processed with a nonimmune serum in place of the primary antibodies. Sections were analyzed by using a Zeiss Axioimager Z1 microscope. For each animal, every fourth VTA section was examined and the mean counts of p-ERK-positive neurons in the VTA were determined manually in one side of the brain. The number of p-ERK-positive cells was normalized to the area. The boundary of VTA was determined with the aid of rat brain atlas (Paxinos and Watson, 1998).
Western blotting
The extent of ERK or p38 MAPK activation was determined by Western blotting with protein and phosphoprotein-specific antibodies (Thr202/Tyr204 for p-ERK, and Thr180/Tyr182 for p-38 MAPK) (Cell Signaling Technology). In experiments shown in Figure 2, VTA slices were maintained in a static incubation chamber in oxygenated ACSF at 32 ± 1°C. Slices were treated with agonists and/or antagonists for 10 min, and the VTA was dissected and then homogenized in 0.2 ml of lysis buffer, pH 7.6, containing 50 mm Tris-acetate, 50 mm NaF, 10 mm EDTA, 10 mm EGTA, 0.01% Triton X, protease inhibitors, and protein phosphatase inhibitors I and II (Sigma-Aldrich). In experiments shown in Figures 6 and 7, rats were anesthetized with isoflurane and rapidly decapitated at the specified times after CPP testing. The brains were immediately removed and placed in oxygenated ACSF at 4°C. Midbrain slices were prepared, and VTA was dissected bilaterally, and then homogenized in lysis buffer. All above samples were centrifuged at 10,000 × g for 10 min. The supernatant was subjected to SDS-PAGE. Total protein concentrations were assayed using Bio-Rad protein assay kit (Bio-Rad Laboratories).The concentration of protein is calibrated to 1 μg/μl with 2× loading buffer containing 0.1 m Tris-HCl, pH 6.8, 4% (w/v) SDS, 20% (v/v) glycerol, 10% (v/v) 2-mercaptoethanol, and 0.04% (w/v) bromophenol blue. Samples (10 μg of protein per sample) were separated in 10% SDS-PAGE. Following the transfer, blots were blocked in solution containing 5% (w/v) milk and 0.1% (v/v) Tween 20 in Tris-buffered saline (TBS-T) for 2 h at room temperature, and incubated with antibody for phosphorylated ERK (1:1000 dilution; Cell Signaling) overnight at 4°C. Blots were then washed several times with TBS-T and probed with horseradish peroxidase-conjugated secondary antibody for 2 h at room temperature before being developed using ECL immunoblotting detection system. The immunoblots were then stripped at 60°C for 30 min with stripping buffer containing 62.5 mm Tris-HCl, pH 6.8, 2% (w/v) SDS, 100 mm 2-mercaptoethanol, and reprobed with an antibody recognizing ERK (1:1000 dilution; Cell Signaling).
Animal surgery and intra-VTA injections
Rats were anesthetized with ketamine and xylazine (90 + 10 mg/kg, i.p.) and placed in a stereotaxic device (David Kopf Instruments). Bilateral guide cannulae (26 gauge; Plastics One) were implanted 1 mm above the VTA using the following stereotaxic coordinates (from bregma, anteroposterior, −5.2 mm; mediolateral, ±1.0 mm; dorsoventral, −7.0 mm) (Paxinos and Watson, 1998). Obturators were placed in the guide cannulae such that they extended 1 mm beyond them and were left there at all times except during microinjections. After 7–8 d of recovery, animals were trained for CPP experiments. Immediately before a conditioning or test session, the obturator was removed from one of the guide cannulae, and a stainless-steel injector tube (30 gauge; Plastics One) was inserted to a depth 1 mm beyond the end of the guide cannula. The injector tube was connected through polyethylene tubing to a 10 μl Hamilton microsyringe. AM251, 1,4-diamino-2,3-dicyano-1,4-bis(2-aminophenylthio)butadiene (U0126), or corresponding vehicle (0.5 μl per side) was manually injected at a rate of 0.5 μl over 60 s. The injector was kept in the guide cannula for an additional 60 s to ensure adequate diffusion from the injector tip. A similar microinjection was made on the contralateral side. Twenty minutes after the intra-VTA microinjection, rats were intraperitoneally injected with saline (0.9% NaCl; 1 ml/kg) or cocaine (15 mg/kg) and were placed in the conditioning environments immediately after injection. Cocaine hydrochloride for intraperitoneal injection was dissolved in saline (0.9% NaCl). SL327, U0126, and AM251 for intraperitoneal or intra-VTA injections were first dissolved in DMSO, and then diluted in saline solution to get final concentration of DMSO to 12.5%. The same concentration of diluted DMSO was used as vehicle controls.
Conditioned place preference
A three-chamber CPP apparatus was used for CPP test (MED Associates). Two sliding doors separated the central chamber from two side chambers, which have distinct wall colors (white or black) and flooring (bar or grid). Animal movement and times spent in each chamber were measured by computer-interfaced infrared photobeams. CPP protocol consisted of the following sessions: (1) pretest (day 1): animals were allowed to explore both sides of chambers for 20 min and time spent in each side was recorded. Most of the rats (∼90%) did not show side preference before drug injections and stayed approximately the same time in each chamber. Rats showing unconditioned side preference (staying 180 s longer than other side) were excluded. (2) Conditioning (day 2–9): place conditioning was conducted over a period of 8 d using an unbiased procedure. Rats were randomly assigned to cocaine or saline conditioning groups. Within each drug treatment group, the drug-paired chamber was assigned in a counterbalanced order.
Cocaine conditioning.
Rats received cocaine injection (15 mg/kg, i.p.) on days 2, 4, 6, and 8, and were immediately confined to one chamber for 20 min. On days 3, 5, 7, and 9, rats received saline injection (0.9% NaCl; 1 ml/kg, i.p.) and were immediately confined to the opposite chamber for 20 min.
Saline conditioning.
Rats received daily saline injection (0.9% NaCl; 1 ml/kg, i.p.) and were immediately confined to one chamber for 20 min on days 3, 5, 7, and 9, and were confined to the opposite chamber for 20 min on days 2, 4, 6, and 8. Subgroups of rats also received bilateral intra-VTA infusions of vehicle, AM251, or U0126 20 min before each intraperitoneal cocaine or saline injection (see Fig. 6). (3) CPP test (day 10): All of the animals were allowed to explore ad libitum for 20 min between the two sides and time spent on each side is recorded. Subgroups of rats also received bilateral intra-VTA infusions of vehicle, AM251, or U0126 20 min before CPP test (see Fig. 7).
Histological verification of VTA cannula placements
After completion of the CPP experiments, VTA cannula placements were anatomically verified. The animals were anesthetized with pentobarbital (60 mg/kg, i.p.) and then perfused with PBS followed by 4% paraformaldehyde. Brains were cut into 40 μm sections and stained with cresyl violet then examined with light microscopy. Based on the stereotaxic atlas of Paxinos and Watson (1998), 11 rats with misplaced cannulae were excluded from behavioral analysis.
Chemicals
Cocaine hydrochloride, CNQX-Na2, AP-5, picrotoxin, and all other common chemicals were obtained from Sigma-Aldrich. Other agonists and antagonists were obtained from Tocris Bioscience.
Specific experiments
Experiment 1: effects of MEK and p38 inhibitors on eCB-dependent I-LTD VTA dopamine neurons.
We prepared midbrain slices from a total of 37 male Sprague Dawley rats (P18–P30) and examined the effects of bath application of CB1, ERK, and p38 blockers on I-LTD in VTA dopamine neurons. Drugs applied into slices included the CB1 receptor antagonist AM251 (2 μm), MEK inhibitors SL327 (10 μm) and U0126 (20 μm), U0124 (20 μm, an inactive analog of U0126), and p38 MAPK inhibitors 4-[4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-1H-imidazol-5-yl]pyridine (SB 203580) (10 μm) and 4-[4-(4-fluorophenyl)-5-(4-pyridinyl)-1H-imidazol-2-yl]phenol (SB 202190) on (10 μm). The concentrations of the inhibitors were based on previous studies (Atkins et al., 1998; Favata et al., 1998; Pan et al., 2008a). For each control or drug application group, six to eight neurons were recorded in six to eight slices that were prepared from four to six rats.
Experiment 2: effects of CB1, D2, and group I mGluR agonists on p-ERK1/2 and p-p38 levels in the VTA.
Midbrain slices were prepared as described in experiment 1. Slices were incubated with one of the following agonists, (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-napthalenylmethanone (WIN 55212-2) (2 μm), quinpirole (10 μm), or (RS)-3,5-dihydroxyphenylglycine (DHPG) (50 μm), alone or with vehicle, AM251 (2 μm), or U0126 (20 μm). Ten minutes later, the VTA was dissected and then were homogenized. Western blotting was performed using antibodies against ERK, p-ERK, p-38, or p-p38. For each experimental group, four to five slices were prepared from three to four rats.
Experiment 3: effects of intraperitoneal injection of AM251 or SL327 on cocaine-induced reduction of GABAergic inhibition and facilitation of LTP induction.
Rats (18–23 d of age) were given daily intraperitoneal injections with either saline or cocaine (15 mg/kg) for 5–7 d. Immediately after saline or cocaine injection, rats were placed on an open field box (45 cm length × 35 cm wide × 30 cm deep) for 15 min. The effectiveness of the cocaine treatment was shown by the sensitization of locomotor activity, as described in our previous study (Liu et al., 2005). Vehicle (12.5% DMSO in saline), AM251 (2 mg/kg), or SL327 (50 mg/kg) was intraperitoneally injected 20 min before each intraperitoneal saline or cocaine injection. The doses of AM251 and SL327 were based on previous studies (Atkins et al., 1998; Rutkowska, 2004). Midbrain slices were prepared ∼24 h after the last saline or cocaine injection. Whole-cell recordings were made from VTA dopamine neurons, and maximal IPSCs, miniature IPSCs (mIPSCs), and LTP were recorded. In each group of maximal IPSC and mIPSC experiments, 8–12 neurons were recorded in 8–12 slices, which were prepared from four to six rats. In each group of LTP experiments, six to nine neurons were recorded in six to nine slices from four to five rats.
Experiment 4: effects of intraperitoneal injection of AM251 or SL327 on cocaine-induced phosphorylated ERK immunoreactivity in the VTA.
We used six groups of rats (300–350 g; n = 4 each group; total number, 24) to determine whether repeated cocaine exposure increased ERK activation in the VTA and whether CB1 receptor activation contributed to the ERK activation. Rats were given daily intraperitoneal injections with either saline or cocaine (15 mg/kg) for 6 d, similar to those described in experiment 3. Vehicle (12.5% DMSO in saline), AM251 (2 mg/kg), or SL327 (50 mg/kg) was intraperitoneally injected 20 min before each intraperitoneal saline or cocaine injection. Rats were killed ∼24 h after the last saline or cocaine injection. VTA sections were immunostained with p-ERK antibody, and p-ERK-positive neurons were counted and analyzed.
Experiment 5: effects of intra-VTA infusion of AM251 or U0126 on the acquisition of cocaine CPP and ERK phosphorylation.
We used six groups of rats (300–350 g; n = 6–9 each group; total number, 47) to determine the role of CB1 receptor and ERK1/2 in the acquisition of cocaine CPP (see Fig. 6A). Cocaine or saline place conditioning was conducted once daily for 8 d, as described above. Vehicle (0.5 μl per side), AM251 (5 μg; 0.5 μl per side), or U0126 (0.1 μg; 0.5 μl per side) was bilaterally infused into the VTA via preimplanted cannulae 20 min before cocaine or saline pairing with a particular chamber. The doses of AM251 and U0126 were based on previous studies (Lu et al., 2005; Xi et al., 2006). Twenty-four hours after the last pairing, CPP was tested without any drug or vehicle administration (see Fig. 6A). Approximately 1 h after CPP tests, rats were killed and midbrain slices were prepared (n = 4 rats each group). VTA was dissected bilaterally and then homogenized in lysis buffer. Western blotting was performed using antibodies against p-ERK and ERK.
Experiment 6: effects of intra-VTA infusion of AM251 or U0126 on the expression of cocaine CPP and ERK phosphorylation.
We used three groups of rats (n = 7–9 each group; total number, 25) to determine the role of CB1 receptor and ERK1/2 in the expression of cocaine CPP (see Fig. 7A). Rats received cocaine conditioning but no intra-VTA infusion was made during place conditioning. Twenty minutes before the CPP test, vehicle (0.5 μl per side), AM251 (5 μg; 0.5 μl per side), or U0126 (0.1 μg; 0.5 μl per side) was bilaterally infused into the VTA and their effects on the expression of cocaine CPP was tested. Approximately 1 h after the CPP tests, rats were killed (n = 4 rats each group), and VTA was dissected bilaterally and then homogenized in lysis buffer. Western blotting was performed using antibodies against p-ERK and ERK.
Statistics
Data are presented as the mean ± SEM. LTDs are calculated as follows: 100 × (mean amplitude of IPSCs during the final 10 min of recording/mean amplitude of baseline IPSCs). LTPs are calculated as follows: 100 × (mean amplitude of EPSPs during the final 10 min of recording/mean amplitude of baseline EPSPs). The CPP score was defined as the time spent in the cocaine-paired chamber minus the time spent in the saline-paired chamber. Immunoblots were analyzed by densitometry using ImageJ (http://rsb.info.nih.gov/ij/index.html). Data sets were compared with either Student's t test, one-way or two-way ANOVA followed by Tukey's post hoc analysis. Results were considered to be significant at p < 0.05.
Results
Experiment 1: effects of ERK and p38 inhibitors on eCB-dependent I-LTD in VTA dopamine neurons
VTA dopamine neurons receive GABAergic inhibitory inputs mainly from local interneurons (Kalivas et al., 1993; Xia et al., 2011). Electron microscopy studies suggest that CB1 receptors are expressed on inhibitory and excitatory axonal terminals (Mátyás et al., 2008). We have shown previously that application of 10 Hz stimulation for 5 min in the presence of cocaine (3 μm) induced I-LTD in VTA dopamine neurons in midbrain slices and this I-LTD was blocked by the CB1 receptor antagonist AM251 (Pan et al., 2008a). Replicating these findings, we found that the combination of cocaine application (3 μm) with 10 Hz stimulation induced I-LTD in VTA dopamine neurons (66.2 ± 7.5% of baseline; n = 6), and this I-LTD was blocked by the CB1 receptor antagonist AM251 (2 μm; 92.6 ± 6.7% of baseline; n = 7; t(11) = 2.6; p < 0.05 vs control) (Fig. 1A).
Figure 1.

ERK, but not p38 MAPK, is required for eCB-dependent I-LTD in VTA dopamine neurons. A, The presence of cocaine (3 μm) 3 min before and 5 min during the 10 Hz stimulation [indicated by arrow (↑)] induced I-LTD in VTA dopamine neurons (n = 6). Bath application of the CB1 receptor antagonist AM251 (2 μm) blocked I-LTD (n = 7; p < 0.05 vs vehicle). Top, Sample IPSCs in the VTA before (indicated by “1”) and after (indicated by “2”) the 10 Hz stimulation. B, Bath application of MEK inhibitor SL327 (10 μm; n = 7) blocked I-LTD (p < 0.05 vs vehicle). C, MEK inhibitor U0126 (20 μm; n = 8) blocked I-LTD, whereas U0124 (20 μm; n = 7), the inactive analog of U0126, had no significant effect on I-LTD (p < 0.05 vs U0126). D, Bath application of p38 MAPK inhibitor SB 203580 (10 μm; n = 8) or SB 202190 (10 μm; n = 6) had no significant effects on I-LTD (p > 0.05 vs vehicle). For each group, six to eight neurons were recorded in six to eight slices that were prepared from four to six rats.
Previous studies indicate that the activation of CB1 receptors is necessary for the induction, but not the maintenance of eCB-mediated LTD or I-LTD (Gerdeman et al., 2002; Chevaleyre and Castillo, 2003; Ronesi et al., 2004; Pan et al., 2008a), suggesting that other downstream signaling molecule(s) must be recruited to produce the long-lasting depression. We tested the hypothesis that ERK is activated downstream of the CB1 receptor and is required for the eCB-dependent I-LTD. ERK is phosphorylated and activated by MAPK/ERK kinase (MEK) (Nakielny et al., 1992). If ERK is activated downstream of the CB1 receptor and mediates I-LTD in the VTA, I-LTD should be blocked by MEK inhibitors. We found that bath application of selective MEK inhibitor SL327 (10 μm) blocked I-LTD (92.2 ± 5.7% of baseline; n = 7; t(12) = 2.4; p < 0.05 vs control) (Fig. 1B). Application of another MEK inhibitor, U0126 (20 μm) also blocked I-LTD (91.0 ± 6.3% of baseline; n = 8; t(13) = 2.7; p < 0.05) (Fig. 1C). In contrast, U0124 (20 μm), an inactive analog of U0126, had no significant effect on I-LTD (65.9 ± 6.9% of baseline; n = 7; t(11) = 0.03; p > 0.05 vs control) (Fig. 1C). These results suggest that ERK is required for the eCB-dependent I-LTD in the VTA.
CB1 receptor agonists increased p38 MAPK phosphorylation and activation in the hippocampus (Derkinderen et al., 2001). p38 MAPK is required for mGluR-dependent LTD in hippocampal slices (Bolshakov et al., 2000; Rush et al., 2002). To test whether p38 MAPK is required for the eCB-dependent I-LTD in the VTA, we examined the effect of selective p38 MAPK inhibitors SB 203580 and SB 202190 on I-LTD. We found that I-LTD was not significantly affected by 10 μm SB 203580 (74.1 ± 6.8% of baseline; n = 8; t(12) = 0.8; p > 0.05 vs control) or 10 μm SB 202190 (73.2 ± 6% of baseline; n = 6; t(10) = 0.7; p > 0.05 vs control) (Fig. 1D). These results indicate that p38 MAPK is not required for eCB-dependent I-LTD in the VTA.
Experiment 2: effects of CB1, D2, and group I mGluR agonists on p-ERK1/2 and p-p38 levels in the VTA
To test whether activation of the CB1 receptor could trigger ERK activation in the VTA, we examined the effect of the CB1 receptor agonist WIN 55212-2 on ERK1/2 phosphorylation in isolated VTA slices, using phosphospecific antibodies that recognize the dually phosphorylated (Thr202/Tyr204) and active ERK. In hippocampal cell culture or slices, CB1-induced ERK phosphorylation reaches peak value in 5–15 min (Derkinderen et al., 2003; Jiang et al., 2005). It is likely that ERK phosphorylation in the VTA follows similar time course. Incubation of the VTA slices with 2 μm WIN 55212-2 for 10 min increased ERK phosphorylation (both ERK1 and ERK2 combined) as measured by Western blots of homogenates from the VTA (F(1,24) = 11.1; p < 0.01; Tukey's post hoc test, vehicle vs WIN 55212-2, p < 0.001; n = 5) (Fig. 2A). The WIN 55212-2-induced increase in the phosphorylated ERK1/2 was blocked by 2 μm AM251 or 20 μm U0126 (F(2,24) = 6.8; p < 0.01; vehicle vs AM251, p < 0.01; n = 5; vehicle vs U0126, p < 0.001; n = 5) (Fig. 2A). These results suggest that the CB1 receptor agonist induces ERK phosphorylation and activation in the VTA.
We have shown previously that both group I mGluRs or D2 dopamine receptors are also required for eCB-dependent I-LTD in the VTA since I-LTD was blocked by antagonists to either group I mGluRs or D2 dopamine receptors (Pan et al., 2008a). We examined whether group I mGluR agonist DHPG can activate ERK in the VTA. Incubation of VTA slices with 50 μm DHPG for 10 min increased ERK1/2 phosphorylation (F(1,18) = 37.1; p < 0.001; vehicle vs DHPG, p < 0.001; n = 4) (Fig. 2B), while AM251 (2 μm) or U0126 (20 μm) treatment attenuated or blocked DHPG-induced ERK1/2 phosphorylation (F(2,18) = 12.1; p < 0.001; vehicle vs AM251, p < 0.01, n = 4; vehicle vs U0126, p < 0.001, n = 4) (Fig. 2B). These results are consistent with our previous findings that group I mGluR activation induces eCB release to activate CB1 receptors, leading to I-LTD induction in the VTA (Pan et al., 2008a). We next examined whether activation of D2 dopamine receptors can activate ERK in the VTA. Incubation of the VTA slices with 10 μm D2 receptor agonist quinpirole for 10 min also increased phosphorylated ERK1/2 (F(1,24) = 24.1; p < 0.001; vehicle vs quinpirole, p < 0.01, n = 5) (Fig. 2C), while AM251 (2 μm) or U0126 (20 μm) treatment had main effect on quinpirole-induced ERK1/2 phosphorylation (F(2,24) = 7.7; p < 0.01) and significant WIN 55212-2 by treatment interaction (F(2,24) = 5.3; p < 0.05). However, Tukey's post hoc tests indicate that the quinpirole-induced increase in the phosphorylated ERK1/2 was not affected by AM251 (p > 0.05; n = 5) (Fig. 2C), suggesting that the CB1 receptor is not an intermediary for D2 receptor-induced ERK1/2 activation. In contrast, the D2 receptor-induced ERK1/2 activation was blocked by MEK/ERK inhibitor U0126 (p < 0.001; n = 5) (Fig. 2C).
We investigated whether agonists to CB1, group I mGluR, and D2 receptors induce p-38 MAPK phosphorylation and activation in the VTA. Incubation of the VTA slices with WIN 55212-2 (2 μm), DHPG (50 μm), or quinpirole (10 μm) for 10 min had no significant effects on phosphorylated p38 (p-p38) levels as measured by Western blots of VTA homogenates (F(3,12) = 0.4; p > 0.05) (Fig. 2D). Thus, the activation of the receptors required for I-LTD induction resulted in ERK1/2, but not p-38, phosphorylation and activation in the VTA.
Experiment 3: effects of intraperitoneal injection of AM251 or SL327 on cocaine-induced reduction of GABAergic inhibition and facilitation of LTP induction
We have shown that daily intraperitoneal injection of cocaine to rats for 5–7 d reduces the mean amplitude of maximal IPSCs and mIPSCs in VTA dopamine neurons of midbrain slices, indicating that repeated cocaine exposure in vivo reduces the strength of GABAergic inhibition to VTA dopamine neurons (Liu et al., 2005). We have further demonstrated that repeated cocaine exposure in vivo reduces the strength of GABAergic inhibition in dopamine neurons by inducing an I-LTD-like synaptic plasticity (Pan et al., 2008a). Having shown that the CB1 receptor antagonist AM251 or MEK inhibitor SL327 blocked I-LTD, we next determined whether intraperitoneal injection of AM251 (2 mg/kg) or SL327 (50 mg/kg) blocked cocaine-induced reduction of GABAergic inhibition. Consistent with our previous studies (Liu et al., 2005; Pan et al., 2008a), we found that the mean amplitude of maximal IPSCs was significantly decreased in cocaine-treated rats compared with saline-treated rats (F(1,47) = 5.8; p < 0.05; Tukey's post hoc test, saline vs cocaine, p < 0.01) (Fig. 3A). Pretreatment with AM251 or SL327 had no significant effects on the mean amplitude of maximal IPSCs in saline-injected rats, but blocked the decreases in the mean amplitude of maximal IPSCs in cocaine-injected rats (F(2,47) = 5.7; p < 0.01; vehicle vs AM251, p < 0.01; vehicle vs U0126, p < 0.01) (Fig. 3A). Similarly, pretreatment with AM251 or SL327 had no significant effects on the mean amplitude of mIPSCs in saline-injected rats, but blocked cocaine-induced decreases in the mean amplitude of mIPSCs in cocaine-injected rats (F(2,48) = 3.5; p < 0.05; vehicle vs AM251, p < 0.01; vehicle vs U0126, p < 0.01) (Fig. 3B). Thus, in vivo administration of AM251 or SL327 blocked cocaine-induced reduction of GABAergic inhibition in VTA dopamine neurons. These results demonstrate that pharmacological inhibitors of I-LTD also blocked cocaine-induced reduction of GABAergic inhibition and provide further evidence that I-LTD represents a cellular mechanism for cocaine-induced reduction of GABAergic inhibition.
Figure 3.

Effects of in vivo pretreatment (intraperitoneal) with antagonists to receptors involved in I-LTD on cocaine-induced decrease in the amplitude of maximal IPSCs and mIPSCs in dopamine neurons in midbrain slices, which were prepared ∼24 h after the last saline or cocaine injection. A, Top, Sample IPSCs in response to stimuli of incremental intensities (20–140 μA). The neurons were voltage clamped at −20 mV. Bottom, Repeated intermittent intraperitoneal injections of cocaine for 5–7 d reduced the mean amplitude of maximal IPSCs compared with time-matched saline injections. Pretreatment (intraperitoneal) with CB1 receptor antagonist AM251 (2 mg/kg, i.p.) or MEK inhibitor SL327 (50 mg/kg, i.p.) 20 min before each intraperitoneal cocaine injection abolished cocaine-induced reduction of the mean amplitude of maximal IPSCs (n = 8–12 for each bar; **p < 0.01). B, Same as in A, except that mIPSCs were recorded at a holding potential of −70 mV (n = 7–10 for each bar; **p < 0.01). Sample mIPSCs from saline or cocaine-treated groups are shown on the top. In each group, 8–12 neurons were recorded in 8–12 slices, which were prepared from four to six rats. Error bars indicate SEM.
We have shown previously that LTP in VTA dopamine neurons is tightly controlled by GABAergic inhibition and repeated cocaine exposure in vivo for 5–7 d facilitates LTP induction by reducing GABAergic inhibition (Liu et al., 2005). We next determined whether intraperitoneal injection of AM251 (2 mg/kg) or SL327 (50 mg/kg) blocked cocaine-induced facilitation of LTP induction. EPSPs were evoked by stimulating excitatory inputs and VTA dopamine neurons were current clamped at −70 mV, the reversal potential of IPSCs. GABAA receptor blocker picrotoxin was absent in the ACSF. Under this condition, the EPSPs were abolished by the AMPA receptor antagonist CNQX (20 μm) and the NMDA receptor antagonist d-AP5 (50 μm) (n = 3) (data not shown), indicating that they are mediated by the activation of glutamate receptors. To induce LTP, we used a spike-timing protocol consisting of bursts of EPSP–spike pairs, with the onset of EPSPs preceding the peak of the postsynaptic spike by ∼5 ms (Liu et al., 2005). We found that repetitive EPSP–spike pairing induced a long-lasting increase in the amplitude of EPSPs (LTP) in VTA dopamine neurons in slices in vehicle plus cocaine-treated rats, but not from vehicle plus saline-treated rats (t(13) = 2.7; p < 0.05) (Fig. 4A,G). The same EPSP–spike pairing did not induce LTP in slices in AM251 plus cocaine-treated rats, nor did it induce LTP in SL327 plus cocaine-treated rats (Fig. 4B,C,G). These results suggest that pretreatment of the rats with CB1 receptor antagonist AM251 or MEK inhibitor SL327 blocked cocaine-induced facilitation of LTP induction. As control experiments, we found that LTP was not induced in AM251 plus saline-treated rats or SL327 plus saline-treated rats (Fig. 4B,C,G).
Figure 4.

Effects of in vivo pretreatment (intraperitoneal) of AM251 or SL327 on cocaine-induced facilitation of LTP induction in VTA dopamine neurons. A, The EPSP–spike pairing protocol induced LTP in midbrain slices prepared from rats treated with vehicle plus cocaine for 5–7 d (n = 7), but not from rats treated with vehicle plus saline for 5–7 d (n = 8). The arrow indicates application of the LTP induction protocol. Experiments in A–C were performed in the absence of picrotoxin. B, C, Pretreatment (intraperitoneal) with CB1 receptor antagonist AM251 (2 mg/kg, i.p.) or MEK inhibitor SL327 (50 mg/kg, i.p.) 20 min before each intraperitoneal cocaine injection abolished cocaine-induced facilitation of LTP induction (n = 7–9). The same drug pretreatment had no significant effects on EPSPs in saline-treated rats (n = 7–8). D–F, The experiments are same in A–C, except that picrotoxin (50 μm) was included in the ACSF to block GABAergic inhibition (n = 6–9). G, H, Summary of LTP induction under various conditions in the absence (G) or presence (H) of picrotoxin. EPSP amplitude (percentage) was calculated as follows: 100 × (mean amplitude of EPSPs during the final 10 min of recording/mean amplitude of baseline EPSPs). *p < 0.05 compared with vehicle control. In each group, six to nine neurons were recorded in six to nine slices from four to five rats. Error bars indicate SEM.
The above experiments have shown that in vivo administration of AM251 or SL327 blocked cocaine-induced facilitation of LTP induction in the VTA. Because LTP induction in VTA dopamine neurons is tightly controlled by GABAergic inhibition (Liu et al., 2005), one possibility is that the blockade of cocaine-induced reduction of GABAergic inhibition may explain the effects of AM251 or SL327 on LTP induction. To test this possibility, we repeated the above LTP experiments in the presence of GABAA receptor antagonist picrotoxin. In the continuous presence of picrotoxin (50 μm), LTP with similar magnitude was induced in dopamine neurons in slices from vehicle plus cocaine-treated rats or vehicle plus saline-treated rats (t(13) = 0.4; p > 0.05) (Fig. 4D,H). Notably, pretreatment (intraperitoneal) of rats with AM251 (2 mg/kg) or SL327 (50 mg/kg) 20 min before each intraperitoneal saline or cocaine injection had no significant effect on LTP induced in picrotoxin (AM251, t(12) = 0.3, p > 0.05; SL327, t(13) = 0.4, p > 0.05) (Fig. 4E,F,H). Thus, blockade of GABAergic inhibition with picrotoxin abrogated the effects of AM251 and SL327 on LTP, suggesting that AM251 or SL327 blocked cocaine-induced facilitation of LTP induction by altering the strength of GABAergic inhibition to dopamine neurons.
Experiment 4: effects of intraperitoneal injection of AM251 or SL327 on cocaine-induced p-ERK immunoreactivity in the VTA
We first examined whether repeated cocaine exposure affected ERK phosphorylation in the VTA. We found that daily intraperitoneal injection of cocaine (15 mg/kg) to rats for 6 d significantly increased the number of p-ERK-positive neurons in the VTA compared with time-matched saline injection (F(1,18) = 89.5; p < 0.001) (Fig. 5A–C). Single cocaine exposure did not significantly change the number of p-ERK-positive cells in the VTA (data not shown). High-magnification view of VTA sections indicates that soma and dendrites of VTA neurons were labeled with p-ERK (Fig. 5B, bottom). However, we cannot exclude the possibility that axonal terminals in the VTA were p-ERK-immunoreactive due to the limit of resolution. Next, we examined whether the CB1 receptor was involved in cocaine-induced increase in ERK phosphorylation. Intraperitoneal injection of AM251 (2 mg/kg) or SL327 (50 mg/kg) 20 min before each cocaine injection significantly attenuated cocaine-induced increase in the number of ERK-positive neurons in the VTA (F(2,18) = 43.66; p < 0.001) (Fig. 5A–C). Thus, CB1 receptor activation contributes to cocaine-induced increase in the number of p-ERK-positive neurons in the VTA.
Figure 5.

The CB1 receptor activation contributes to cocaine-induced activation of ERK in the VTA. Immunostaining of activated ERK was performed using a phosphospecific antibody reacting with p-ERK. A, Intraperitoneal injection of AM251 (2 mg/kg) or SL327 (50 mg/kg) 20 min before each saline injection did not significantly affected the number of p-ERK-positive cells in the VTA. B, Repeated daily cocaine injections (15 mg/kg, i.p.; 6 d) increased p-ERK-positive cells in the VTA compared with repeated saline injections. Intraperitoneal injection of AM251 or SL327 20 min before each cocaine injection significantly attenuated cocaine-induced increase in the number of ERK-positive cells. Bottom, High-magnification view indicates that soma and dendrites of VTA neurons were labeled with p-ERK. Scale bars, 20 μm. C, Summarized data of the number of p-ERK-positive cells under various experimental conditions (4 rats each group; ***p < 0.001). Error bars indicate SEM.
Experiment 5: effects of intra-VTA infusion of AM251 or U0126 on the acquisition of cocaine CPP and ERK phosphorylation
Intra-VTA infusion of MEK inhibitor PD98059 [2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-4-one] impaired the development of locomotor sensitization to cocaine (Pierce et al., 1999). However, it remains unexplored whether intra-VTA infusion of CB1 receptor antagonists or MEK inhibitors affects CPP to cocaine. We examined whether intra-VTA infusion of AM251 or U0126 during the conditioning phase affected CPP to cocaine. The timeline for baseline preference test (pretest), cocaine or saline conditioning, intra-VTA infusion, and CPP test is described in Figure 6A. During pretest, rats did not exhibit significant differences in the time spent in each chamber (p > 0.05), indicating that there was no unconditioned place preference (Fig. 6B). Then, cocaine or saline place conditioning was conducted once daily for 8 d. Vehicle (0.5 μl per side), AM251 (5 μg; 0.5 μl per side), or U0126 (0.1 μg; 0.5 μl per side) was bilaterally infused into the VTA via preimplanted cannulae 20 min before cocaine or saline pairing with a particular chamber. Twenty-four hours after the last pairing, CPP was tested without any drug or vehicle administration (Fig. 6A). A two-way ANOVA revealed that cocaine conditioning and intra-VTA drug treatments had significant main effects on CPP scores (cocaine: F(1,41) = 28.68, p < 0.001; intra-VTA infusion: F(2,41) = 8.72, p < 0.001; cocaine by intra-VTA infusion interaction: F(2,41) = 10.62, p < 0.001) (Fig. 6C). Tukey's post hoc tests showed that intra-VTA infusion of AM251 or U0126 produced significant decreases in CPP score in cocaine-conditioned rats (p < 0.001) but did not affect CPP score in saline-conditioned rats (p > 0.05). The latter finding indicates that the attenuation of CPP to cocaine by intra-VTA AM251 or U0126 was not because of an aversive effect of AM251 or U0126. Interestingly, cocaine-conditioned rats infused with U0126 had a significantly lower CPP score compared with those infused with AM251 (p < 0.05). These results indicate that MEK or CB1 receptor blockade during the acquisition phase of the CPP paradigm attenuated cocaine-induced place preference, and U0126 produced greater attenuation of cocaine CPP than AM251.
We determined whether the acquisition of CPP to cocaine was associated with activation of ERK in the VTA and whether AM251 or U0126 affected ERK activation. We used Western blotting to detect phosphorylated, activated ERK (English and Sweatt, 1997). Approximately 1 h after the CPP tests, rats were killed and VTA was dissected bilaterally from midbrain slices. Western blotting was performed using antibodies against p-ERK and ERK. We found that VTA ERK phosphorylation was significantly increased in cocaine-conditioned rats compared with that of saline-conditioned rats; intra-VTA infusion of AM251 or U0126 during the conditioning phase significantly attenuated ERK phosphorylation levels in cocaine-conditioned rats (cocaine: F(1,18) = 31.45, p < 0.001; intra-VTA infusion: F(2,18) = 9.35, p < 0.01; cocaine by intra-VTA infusion interaction: F(2,18) = 6.41, p < 0.01) (Fig. 6D). These results indicate that the acquisition of CPP to cocaine is accompanied by an increase in ERK phosphorylation and activation in the VTA, and eCB signaling contributes to cocaine-induced ERK activation.
Experiment 6: effects of intra-VTA infusion of AM251 or U0126 on the expression of cocaine CPP and ERK phosphorylation
We examined whether the blockade of the CB1 receptor and MEK in the VTA during the test phase affected the expression of CPP to cocaine (Fig. 7A). Pretest indicated rats did not show side preference before drug injections (Fig. 7B). Rats received cocaine conditioning but no intra-VTA infusion was made during place conditioning. Twenty minutes before the CPP test, vehicle (0.5 μl per side), AM251 (5 μg; 0.5 μl per side), or U0126 (0.1 μg; 0.5 μl per side) was bilaterally infused into the VTA. A one-way ANOVA indicates that neither AM251 nor U0126 significantly affected CPP to cocaine (F(2, 22) = 0.634; p > 0.05). Together, these results indicate that eCB or ERK activity is required for the acquisition, but not the expression of CPP to cocaine.
We determined whether intra-VTA infusion of AM251 or U0126 20 min before the CPP test affected ERK phosphorylation in the VTA in cocaine-conditioned rats. Western blotting was performed on VTA samples collected ∼1 h after the CPP tests. We found that intra-VTA drug treatment significantly altered ERK phosphorylation (F(2,9) = 8.39; p < 0.01). Tukey's post hoc test indicates that AM251 had no significant effect on ERK phosphorylation compared with intra-VTA vehicle (p > 0.05), whereas intra-VTA U0126 significantly decreased ERK phosphorylation (p < 0.05). These results indicate that once ERK was activated following cocaine conditioning, CB1 receptor blockade no longer affected ERK activation.
Figure 8 shows the locations of the cannula tips from the experiments in Figures 6 and 7. These sites are representative of those used for intra-VTA infusions. A total of 83 rats were used for intra-VTA infusion and CPP test; 11 rats with cannula tips outside of the VTA were excluded from analysis.
Figure 8.
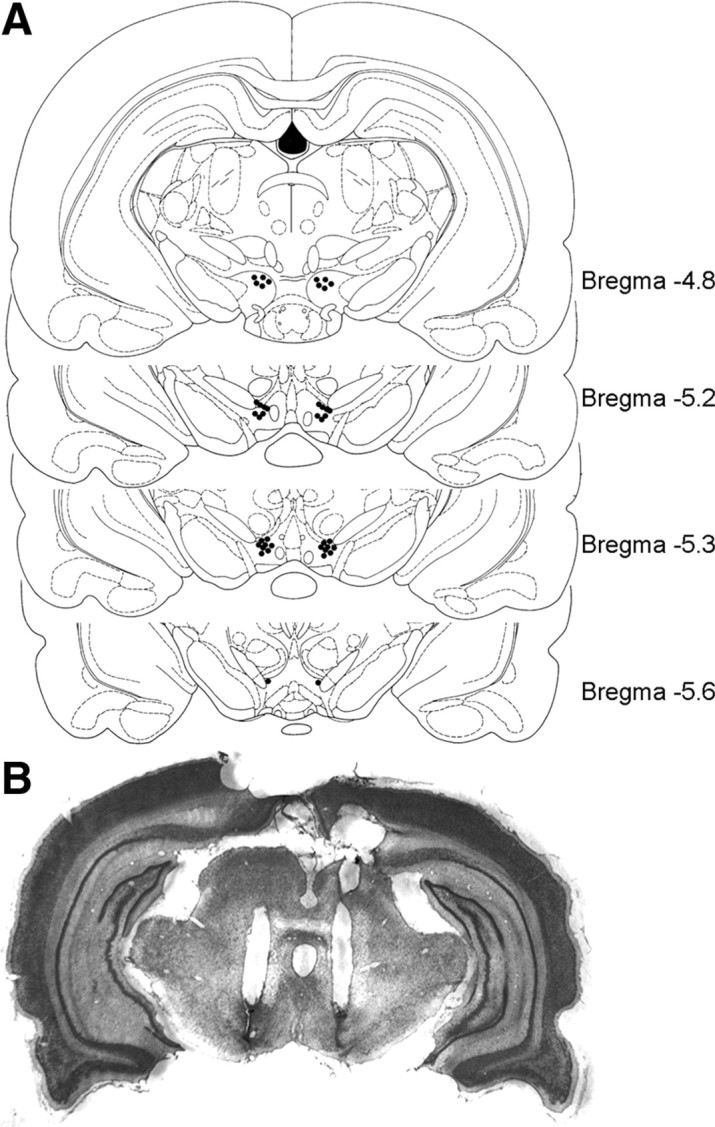
Histological verification of VTA cannula placements. A, Locations of intra-VTA infusion sites from the behavioral data summarized in Figures 6 and 7. For clarity, representative samples of bilateral cannula placements are shown. B, Cresyl violet-stained coronal section of typical bilateral intra-VTA cannula placements.
Discussion
Here, we demonstrate that ERK is required for eCB-dependent I-LTD in VTA dopamine neurons. Furthermore, the VTA CB1 receptor and ERK signaling cascade mediates cocaine-induced reduction of GABAergic inhibition, facilitation of LTP induction, and the acquisition of cocaine CPP. These findings have identified key signaling mechanisms linking cocaine-induced long-term synaptic plasticity in the VTA to the rewarding effects of cocaine.
The requirement of ERK for eCB-dependent I-LTD in the VTA
We provide evidence that ERK is activated downstream of CB1 receptors and is required for eCB-dependent I-LTD in VTA. MEK inhibitors U0126 and SL327 blocked I-LTD, whereas U0124, an inactive analog of U0126, did not affect I-LTD. We have shown previously that the activation of D2 dopamine receptors, group I mGluRs, and CB1 receptors is required for I-LTD induction (Pan et al., 2008a). We showed that incubation of VTA slices with D2 agonist quinpirole, mGluR agonist DHPG, or CB1 agonist WIN 55212-2 resulted in ERK phosphorylation, consistent with previous studies showing that quinpirole, DHPG, or WIN 55212-2 increase ERK phosphorylation in the striatum, hippocampus, and other brain regions (Yan et al., 1999; Brami-Cherrier et al., 2002; Derkinderen et al., 2003; Gallagher et al., 2004; Grueter et al., 2006). Together, these data support a model in which D2, CB1, and mGluRs cooperate to activate ERK to induce I-LTD in the VTA. However, the relationship between ERK activation and I-LTD is not linear since each of the CB1, D2, or mGluR antagonists almost completely blocked I-LTD (Pan et al., 2008a).
It remains to be determined how the activation of D2, mGluR, and CB1 receptors leads to ERK activation. Either D2 or CB1 receptor activation inhibits adenylyl cyclase, resulting in decreased cAMP/protein kinase A (PKA) activity (Neve et al., 2004; Howlett, 2005). cAMP can either stimulate or inhibit the ERK pathway via different signaling mechanisms (Roberson et al., 1999; Stork and Schmitt, 2002). CB1 agonists activate ERK via a decrease in cAMP/PKA activity in cell lines and in hippocampal slices (Davis et al., 2003; Derkinderen et al., 2003). We and others have shown that cAMP/PKA signaling is required for eCB-dependent LTD/I-LTD (Chevaleyre et al., 2007; Mato et al., 2008; Pan et al., 2008b; Chiu et al., 2010). The decreased cAMP/PKA activity might constitute a mechanism for CB1 and D2-induced ERK activation in the VTA. New protein synthesis is required for eCB-LTD in the striatum (Yin et al., 2006). ERK regulates new protein synthesis in responses to neuronal activity (Kelleher et al., 2004). We speculate that ERK is “upstream” of protein synthesis, which may be required for eCB-dependent I-LTD in the VTA.
The CB1 and ERK signaling cascade mediates cocaine-induced reduction of GABAergic inhibition and facilitation of LTP induction
Our previous studies suggest that repeated intraperitoneal cocaine injection for 5–7 d reduces the strength of GABAergic inhibition to dopamine neurons by inducing an I-LTD-like synaptic plasticity in vivo (Pan et al., 2008a). The present study extended these findings by showing that intraperitoneal injection of CB1 antagonist AM251 or MEK inhibitor U0126 prevented cocaine-induced decrease in the amplitude of maximal IPSCs and mIPSCs. An important consequence of cocaine-induced reduction of GABAergic inhibition is to prime excitatory synapses for LTP induction (Liu et al., 2005). We found that in vivo pretreatment of AM251 or SL327 also prevented cocaine-induced facilitation of LTP induced in the absence of GABAA receptor blocker picrotoxin but had no significant effect on LTP induced in the presence of picrotoxin. These results suggest that the observed effects of AM251 and SL327 on LTP can be attributable to their blockade of cocaine-induced reduction of GABAergic inhibition. Together, our data suggest that the CB1 receptor–ERK signaling cascade mediates I-LTD, cocaine-induced reduction of GABAergic inhibition, and facilitation of LTP induction.
The following caveats should be considered in the interpretation of the present data. First, how cocaine exposure affects spike-timing LTP at VTA dopamine neurons still remains controversial, although the consensus is that such LTP can be induced in the presence of picrotoxin in naive or saline-treated animals (Liu et al., 2005; Argilli et al., 2008; Luu and Malenka, 2008). Second, recent studies indicate that midbrain dopamine neurons are heterogenous in terms of coding and target projection. They encode both reward-predicting and aversive stimuli (Ungless et al., 2004; Matsumoto and Hikosaka, 2009) and project to different targets (Lammel et al., 2008). However, the present study did not distinguish different dopamine neuronal populations. Finally, the electrophysiological recordings were made in slices prepared from 18- to 30-d-old rats because the slice quality is better in young animals. Thus, the functional relevance of our findings should be interpreted with caution.
Repeated cocaine injections increased the number of p-ERK-positive cells, and intraperitoneal injections of AM251 or SL327 attenuated the observed effects. Cocaine-induced increase in ERK activation in the striatum was significantly attenuated in CB1 receptor knock-out mice (Corbillé et al., 2007). Thus, CB1 receptor activation contributes to cocaine-induced ERK activation in the VTA and striatum. Interestingly, these studies showed the soma and dendrites of VTA and striatal neurons were labeled with p-ERK. Furthermore, intraperitoneal injection of the CB1 agonist Δ9-tetrahydrocannabinol significantly increased p-ERK immunoreactivity in the soma and dendrites of hippocampal CA1 pyramidal neurons and neurons in other brain regions (Valjent et al., 2004). It is known that CB1 receptors are expressed on inhibitory presynaptic axonal terminals in the VTA (Mátyás et al., 2008) and hippocampus (Katona et al., 1999; Tsou et al., 1999). These data suggest that stimulation of the CB1 receptor increased ERK activation through indirect mechanisms. We speculate that the CB1 receptor-mediated disinhibition and associated increase in neuronal activity may contribute to ERK activation. We have shown previously that both presynaptic ad postsynaptic mechanisms contribute to I-LTD induction (Pan et al., 2008a). The increased p-ERK at the soma and dendrites may constitute a putative postsynaptic mechanism for I-TLD.
Contributions of VTA CB1 and ERK1/2 signaling cascade to cocaine CPP
Both the eCB system and ERK are critically involved in cocaine addiction (Gerdeman et al., 2003; Lupica and Riegel, 2005; Lu et al., 2006; Girault et al., 2007). Cocaine exposure increased ERK1/2 phosphorylation in the VTA (Berhow et al., 1996) and nucleus accumbens (Valjent et al., 2000, 2004; Corbillé et al., 2007). Systemic or local infusions of CB1 antagonists or MEK inhibitors into the nucleus accumbens attenuated or blocked cocaine-induced addictive behaviors (Valjent et al., 2000; Miller and Marshall, 2005; Xi et al., 2006; Wiskerke et al., 2008; Orio et al., 2009). ERK signaling in the central amygdala is critical to incubation of cocaine craving (Lu et al., 2005). However, there are relatively few studies that investigate the role of the CB1 receptor or ERK activity in the VTA in the behavioral effects of cocaine. Intra-VTA injections of a MEK inhibitor blocked cocaine-induced behavioral sensitization without affecting acute locomotor activity (Pierce et al., 1999) and reversed the potentiation of cue-induced cocaine seeking that was induced by intra-VTA injections of brain- or glia-derived neurotrophic factor (Lu et al., 2004, 2009). The present study showed that intra-VTA injections of AM251 or U0126 before each place conditioning attenuated or blocked the acquisition of CPP to cocaine. Possible mechanisms for the observed effects include decreased rewarding effects of cocaine or decreased ability to learn the association between environmental cues and rewarding effects of cocaine. Drugs of abuse usurp the neural mechanisms normally subserving reward-related learning (Hyman et al., 2006). ERK is required for contextual fear conditioning, a form of associative learning (Atkins et al., 1998). Long-term synaptic plasticity in the reward circuit of the brain may represent a putative cellular model for this reinforcement learning (Kauer, 2004). We have shown that both CB1 receptor and ERK are required for cocaine-induced facilitation of LTP induction in the VTA. We speculate that the decreased ability of associative learning might explain why intra-VTA AM251 or U0126 attenuated the acquisition of CPP to cocaine.
However, we found that intra-VTA injection of AM251 or U0126 20 min before CPP test did not affect the expression of CPP to cocaine. Although the CB1 receptor and ERK activity in the VTA are required for the establishment of association between environmental cues and rewarding effects of cocaine, they are not necessary for maintenance of cue-associated memories once such association is established. The CB1 receptor and ERK may activate other downstream signaling mechanisms, including the expression of immediate-early genes, to maintain cocaine-seeking behavior during the expression of CPP (Valjent et al., 2006). Alternatively, other brain regions such as the nucleus accumbens may be recruited subsequently to maintain CPP to cocaine (Miller and Marshall, 2005; Valjent et al., 2005).
Cocaine-induced locomotor sensitization and increase in ERK phosphorylation in the striatum were CB1 receptor dependent (Corbillé et al., 2007). We found that repeated cocaine exposure increased ERK phosphorylation in the VTA, and this effect was partially blocked by AM251. In addition, ERK is activated downstream of the CB1 receptor and is required for I-LTD. Together, these results revealed that the CB1 receptor and ERK function as a signaling pair that mediates long-term synaptic plasticity in the VTA and rewarding effects of cocaine.
Footnotes
This work was supported by National Institutes of Health Grant DA024741 and by Advancing a Healthier Wisconsin.
References
- Argilli E, Sibley DR, Malenka RC, England PM, Bonci A. Mechanism and time course of cocaine-induced long-term potentiation in the ventral tegmental area. J Neurosci. 2008;28:9092–9100. doi: 10.1523/JNEUROSCI.1001-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD. The MAPK cascade is required for mammalian associative learning. Nat Neurosci. 1998;1:602–609. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- Berhow MT, Hiroi N, Nestler EJ. Regulation of ERK (extracellular signal-regulated kinase), part of the neurotrophin signal transduction cascade, in the rat mesolimbic dopamine system by chronic exposure to morphine or cocaine. J Neurosci. 1996;16:4707–4715. doi: 10.1523/JNEUROSCI.16-15-04707.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolshakov VY, Carboni L, Cobb MH, Siegelbaum SA, Belardetti F. Dual MAP kinase pathways mediate opposing forms of long-term plasticity at CA3-CA1 synapses. Nat Neurosci. 2000;3:1107–1112. doi: 10.1038/80624. [DOI] [PubMed] [Google Scholar]
- Bouaboula M, Poinot-Chazel C, Bourrié B, Canat X, Calandra B, Rinaldi-Carmona M, Le Fur G, Casellas P. Activation of mitogen-activated protein kinases by stimulation of the central cannabinoid receptor CB1. Biochem J. 1995;312:637–641. doi: 10.1042/bj3120637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brami-Cherrier K, Valjent E, Garcia M, Pagès C, Hipskind RA, Caboche J. Dopamine induces a PI3-kinase-independent activation of Akt in striatal neurons: a new route to cAMP response element-binding protein phosphorylation. J Neurosci. 2002;22:8911–8921. doi: 10.1523/JNEUROSCI.22-20-08911.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron. 2003;38:461–472. doi: 10.1016/s0896-6273(03)00235-6. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Endocannabinoid-mediated metaplasticity in the hippocampus. Neuron. 2004;43:871–881. doi: 10.1016/j.neuron.2004.08.036. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Heifets BD, Kaeser PS, Südhof TC, Purpura DP, Castillo PE. Endocannabinoid-mediated long-term plasticity requires cAMP/PKA signaling and RIM1alpha. Neuron. 2007;54:801–812. doi: 10.1016/j.neuron.2007.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu CQ, Puente N, Grandes P, Castillo PE. Dopaminergic modulation of endocannabinoid-mediated plasticity at GABAergic synapses in the prefrontal cortex. J Neurosci. 2010;30:7236–7248. doi: 10.1523/JNEUROSCI.0736-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbillé AG, Valjent E, Marsicano G, Ledent C, Lutz B, Hervé D, Girault JA. Role of cannabinoid type 1 receptors in locomotor activity and striatal signaling in response to psychostimulants. J Neurosci. 2007;27:6937–6947. doi: 10.1523/JNEUROSCI.3936-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MI, Ronesi J, Lovinger DM. A predominant role for inhibition of the adenylate cyclase/protein kinase A pathway in ERK activation by cannabinoid receptor 1 in N1E-115 neuroblastoma cells. J Biol Chem. 2003;278:48973–48980. doi: 10.1074/jbc.M305697200. [DOI] [PubMed] [Google Scholar]
- Derkinderen P, Ledent C, Parmentier M, Girault JA. Cannabinoids activate p38 mitogen-activated protein kinases through CB1 receptors in hippocampus. J Neurochem. 2001;77:957–960. doi: 10.1046/j.1471-4159.2001.00333.x. [DOI] [PubMed] [Google Scholar]
- Derkinderen P, Valjent E, Toutant M, Corvol JC, Enslen H, Ledent C, Trzaskos J, Caboche J, Girault JA. Regulation of extracellular signal-regulated kinase by cannabinoids in hippocampus. J Neurosci. 2003;23:2371–2382. doi: 10.1523/JNEUROSCI.23-06-02371.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English JD, Sweatt JD. Activation of p42 mitogen-activated protein kinase in hippocampal long term potentiation. J Biol Chem. 1996;271:24329–24332. doi: 10.1074/jbc.271.40.24329. [DOI] [PubMed] [Google Scholar]
- English JD, Sweatt JD. A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J Biol Chem. 1997;272:19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- Gallagher SM, Daly CA, Bear MF, Huber KM. Extracellular signal-regulated protein kinase activation is required for metabotropic glutamate receptor-dependent long-term depression in hippocampal area CA1. J Neurosci. 2004;24:4859–4864. doi: 10.1523/JNEUROSCI.5407-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdeman GL, Ronesi J, Lovinger DM. Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci. 2002;5:446–451. doi: 10.1038/nn832. [DOI] [PubMed] [Google Scholar]
- Gerdeman GL, Partridge JG, Lupica CR, Lovinger DM. It could be habit forming: drugs of abuse and striatal synaptic plasticity. Trends Neurosci. 2003;26:184–192. doi: 10.1016/S0166-2236(03)00065-1. [DOI] [PubMed] [Google Scholar]
- Girault JA, Valjent E, Caboche J, Hervé D. ERK2: a logical AND gate critical for drug-induced plasticity? Curr Opin Pharmacol. 2007;7:77–85. doi: 10.1016/j.coph.2006.08.012. [DOI] [PubMed] [Google Scholar]
- Grueter BA, Gosnell HB, Olsen CM, Schramm-Sapyta NL, Nekrasova T, Landreth GE, Winder DG. Extracellular-signal regulated kinase 1-dependent metabotropic glutamate receptor 5-induced long-term depression in the bed nucleus of the stria terminalis is disrupted by cocaine administration. J Neurosci. 2006;26:3210–3219. doi: 10.1523/JNEUROSCI.0170-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heifets BD, Castillo PE. Endocannabinoid signaling and long-term synaptic plasticity. Annu Rev Physiol. 2009;71:283–306. doi: 10.1146/annurev.physiol.010908.163149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett AC. Cannabinoid receptor signaling. Handb Exp Pharmacol. 2005;2005:53–79. doi: 10.1007/3-540-26573-2_2. [DOI] [PubMed] [Google Scholar]
- Huang ZJ, Kirkwood A, Pizzorusso T, Porciatti V, Morales B, Bear MF, Maffei L, Tonegawa S. BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell. 1999;98:739–755. doi: 10.1016/s0092-8674(00)81509-3. [DOI] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- Jiang W, Zhang Y, Xiao L, Van Cleemput J, Ji SP, Bai G, Zhang X. Cannabinoids promote embryonic and adult hippocampus neurogenesis and produce anxiolytic- and antidepressant-like effects. J Clin Invest. 2005;115:3104–3116. doi: 10.1172/JCI25509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SW, North RA. Two types of neurone in the rat ventral tegmental area and their synaptic inputs. J Physiol. 1992;450:455–468. doi: 10.1113/jphysiol.1992.sp019136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Kauer JA. Amphetamine depresses excitatory synaptic transmission via serotonin receptors in the ventral tegmental area. J Neurosci. 1999;19:9780–9787. doi: 10.1523/JNEUROSCI.19-22-09780.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW, Churchill L, Klitenick MA. GABA and enkephalin projection from the nucleus accumbens and ventral pallidum to the ventral tegmental area. Neuroscience. 1993;57:1047–1060. doi: 10.1016/0306-4522(93)90048-k. [DOI] [PubMed] [Google Scholar]
- Katona I, Sperlágh B, Sík A, Käfalvi A, Vizi ES, Mackie K, Freund TF. Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J Neurosci. 1999;19:4544–4558. doi: 10.1523/JNEUROSCI.19-11-04544.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauer JA. Learning mechanisms in addiction: synaptic plasticity in the ventral tegmental area as a result of exposure to drugs of abuse. Annu Rev Physiol. 2004;66:447–475. doi: 10.1146/annurev.physiol.66.032102.112534. [DOI] [PubMed] [Google Scholar]
- Kelleher RJ, 3rd, Govindarajan A, Jung HY, Kang H, Tonegawa S. Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell. 2004;116:467–479. doi: 10.1016/s0092-8674(04)00115-1. [DOI] [PubMed] [Google Scholar]
- Kellogg R, Mackie K, Straiker A. Cannabinoid CB1 receptor-dependent long-term depression in autaptic excitatory neurons. J Neurophysiol. 2009;102:1160–1171. doi: 10.1152/jn.00266.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammel S, Hetzel A, Häckel O, Jones I, Liss B, Roeper J. Unique properties of mesoprefrontal neurons within a dual mesocorticolimbic dopamine system. Neuron. 2008;57:760–773. doi: 10.1016/j.neuron.2008.01.022. [DOI] [PubMed] [Google Scholar]
- Liu QS, Pu L, Poo MM. Repeated cocaine exposure in vivo facilitates LTP induction in midbrain dopamine neurons. Nature. 2005;437:1027–1031. doi: 10.1038/nature04050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Dempsey J, Liu SY, Bossert JM, Shaham Y. A single infusion of brain-derived neurotrophic factor into the ventral tegmental area induces long-lasting potentiation of cocaine seeking after withdrawal. J Neurosci. 2004;24:1604–1611. doi: 10.1523/JNEUROSCI.5124-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Hope BT, Dempsey J, Liu SY, Bossert JM, Shaham Y. Central amygdala ERK signaling pathway is critical to incubation of cocaine craving. Nat Neurosci. 2005;8:212–219. doi: 10.1038/nn1383. [DOI] [PubMed] [Google Scholar]
- Lu L, Koya E, Zhai H, Hope BT, Shaham Y. Role of ERK in cocaine addiction. Trends Neurosci. 2006;29:695–703. doi: 10.1016/j.tins.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Lu L, Wang X, Wu P, Xu C, Zhao M, Morales M, Harvey BK, Hoffer BJ, Shaham Y. Role of ventral tegmental area glial cell line-derived neurotrophic factor in incubation of cocaine craving. Biol Psychiatry. 2009;66:137–145. doi: 10.1016/j.biopsych.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupica CR, Riegel AC. Endocannabinoid release from midbrain dopamine neurons: a potential substrate for cannabinoid receptor antagonist treatment of addiction. Neuropharmacology. 2005;48:1105–1116. doi: 10.1016/j.neuropharm.2005.03.016. [DOI] [PubMed] [Google Scholar]
- Luu P, Malenka RC. Spike timing-dependent long-term potentiation in ventral tegmental area dopamine cells requires PKC. J Neurophysiol. 2008;100:533–538. doi: 10.1152/jn.01384.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Lock H, Hjelmstad GO, Fields HL. The ventral tegmental area revisited: is there an electrophysiological marker for dopaminergic neurons? J Physiol. 2006;577:907–924. doi: 10.1113/jphysiol.2006.117069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann C, Zieglgänsberger W, Di Marzo V, Lutz B. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- Mato S, Lafourcade M, Robbe D, Bakiri Y, Manzoni OJ. Role of the cyclic-AMP/PKA cascade and of P/Q-type Ca++ channels in endocannabinoid-mediated long-term depression in the nucleus accumbens. Neuropharmacology. 2008;54:87–94. doi: 10.1016/j.neuropharm.2007.04.014. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Hikosaka O. Two types of dopamine neuron distinctly convey positive and negative motivational signals. Nature. 2009;459:837–841. doi: 10.1038/nature08028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mátyás F, Urbán GM, Watanabe M, Mackie K, Zimmer A, Freund TF, Katona I. Identification of the sites of 2-arachidonoylglycerol synthesis and action imply retrograde endocannabinoid signaling at both GABAergic and glutamatergic synapses in the ventral tegmental area. Neuropharmacology. 2008;54:95–107. doi: 10.1016/j.neuropharm.2007.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CA, Marshall JF. Molecular substrates for retrieval and reconsolidation of cocaine-associated contextual memory. Neuron. 2005;47:873–884. doi: 10.1016/j.neuron.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Nakielny S, Cohen P, Wu J, Sturgill T. MAP kinase activator from insulin-stimulated skeletal muscle is a protein threonine/tyrosine kinase. EMBO J. 1992;11:2123–2129. doi: 10.1002/j.1460-2075.1992.tb05271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve KA, Seamans JK, Trantham-Davidson H. Dopamine receptor signaling. J Recept Signal Transduct Res. 2004;24:165–205. doi: 10.1081/rrs-200029981. [DOI] [PubMed] [Google Scholar]
- Orio L, Edwards S, George O, Parsons LH, Koob GF. A role for the endocannabinoid system in the increased motivation for cocaine in extended-access conditions. J Neurosci. 2009;29:4846–4857. doi: 10.1523/JNEUROSCI.0563-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B, Hillard CJ, Liu QS. Endocannabinoid signaling mediates cocaine-induced inhibitory synaptic plasticity in midbrain dopamine neurons. J Neurosci. 2008a;28:1385–1397. doi: 10.1523/JNEUROSCI.4033-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B, Hillard CJ, Liu QS. D2 dopamine receptor activation facilitates endocannabinoid-mediated long-term synaptic depression of GABAergic synaptic transmission in midbrain dopamine neurons via cAMP-protein kinase A signaling. J Neurosci. 2008b;28:14018–14030. doi: 10.1523/JNEUROSCI.4035-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. Ed 4. New York: Academic; 1998. The rat brain in stereotaxic coordinates. [DOI] [PubMed] [Google Scholar]
- Pierce RC, Pierce-Bancroft AF, Prasad BM. Neurotrophin-3 contributes to the initiation of behavioral sensitization to cocaine by activating the Ras/mitogen-activated protein kinase signal transduction cascade. J Neurosci. 1999;19:8685–8695. doi: 10.1523/JNEUROSCI.19-19-08685.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbe D, Kopf M, Remaury A, Bockaert J, Manzoni OJ. Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proc Natl Acad Sci U S A. 2002;99:8384–8388. doi: 10.1073/pnas.122149199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, English JD, Adams JP, Selcher JC, Kondratick C, Sweatt JD. The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. J Neurosci. 1999;19:4337–4348. doi: 10.1523/JNEUROSCI.19-11-04337.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronesi J, Lovinger DM. Induction of striatal long-term synaptic depression by moderate frequency activation of cortical afferents in rat. J Physiol. 2005;562:245–256. doi: 10.1113/jphysiol.2004.068460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronesi J, Gerdeman GL, Lovinger DM. Disruption of endocannabinoid release and striatal long-term depression by postsynaptic blockade of endocannabinoid membrane transport. J Neurosci. 2004;24:1673–1679. doi: 10.1523/JNEUROSCI.5214-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush AM, Wu J, Rowan MJ, Anwyl R. Group I metabotropic glutamate receptor (mGluR)-dependent long-term depression mediated via p38 mitogen-activated protein kinase is inhibited by previous high-frequency stimulation and activation of mGluRs and protein kinase C in the rat dentate gyrus in vitro. J Neurosci. 2002;22:6121–6128. doi: 10.1523/JNEUROSCI.22-14-06121.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowska M. The effect of AM 251, a cannabinoid CB1 receptor antagonist, on food intake in rats. Acta Pol Pharm. 2004;61:401–403. [PubMed] [Google Scholar]
- Stork PJ, Schmitt JM. Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol. 2002;12:258–266. doi: 10.1016/s0962-8924(02)02294-8. [DOI] [PubMed] [Google Scholar]
- Sweatt JD. Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol. 2004;14:311–317. doi: 10.1016/j.conb.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Thiemann G, Di Marzo V, Molleman A, Hasenöhrl RU. The CB1 cannabinoid receptor antagonist AM251 attenuates amphetamine-induced behavioural sensitization while causing monoamine changes in nucleus accumbens and hippocampus. Pharmacol Biochem Behav. 2008;89:384–391. doi: 10.1016/j.pbb.2008.01.010. [DOI] [PubMed] [Google Scholar]
- Tsou K, Mackie K, Sañudo-Peña MC, Walker JM. Cannabinoid CB1 receptors are localized primarily on cholecystokinin-containing GABAergic interneurons in the rat hippocampal formation. Neuroscience. 1999;93:969–975. doi: 10.1016/s0306-4522(99)00086-x. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Magill PJ, Bolam JP. Uniform inhibition of dopamine neurons in the ventral tegmental area by aversive stimuli. Science. 2004;303:2040–2042. doi: 10.1126/science.1093360. [DOI] [PubMed] [Google Scholar]
- Valjent E, Corvol JC, Pages C, Besson MJ, Maldonado R, Caboche J. Involvement of the extracellular signal-regulated kinase cascade for cocaine-rewarding properties. J Neurosci. 2000;20:8701–8709. doi: 10.1523/JNEUROSCI.20-23-08701.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Pagès C, Hervé D, Girault JA, Caboche J. Addictive and non-addictive drugs induce distinct and specific patterns of ERK activation in mouse brain. Eur J Neurosci. 2004;19:1826–1836. doi: 10.1111/j.1460-9568.2004.03278.x. [DOI] [PubMed] [Google Scholar]
- Valjent E, Pascoli V, Svenningsson P, Paul S, Enslen H, Corvol JC, Stipanovich A, Caboche J, Lombroso PJ, Nairn AC, Greengard P, Herve D, Girault JA. Regulation of a protein phosphatase cascade allows convergent dopamine and glutamate signals to activate ERK in the striatum. Proc Natl Acad Sci U S A. 2005;102:491–496. doi: 10.1073/pnas.0408305102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Aubier B, Corbillé AG, Brami-Cherrier K, Caboche J, Topilko P, Girault JA, Hervé D. Plasticity-associated gene Krox24/Zif268 is required for long-lasting behavioral effects of cocaine. J Neurosci. 2006;26:4956–4960. doi: 10.1523/JNEUROSCI.4601-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wartmann M, Campbell D, Subramanian A, Burstein SH, Davis RJ. The MAP kinase signal transduction pathway is activated by the endogenous cannabinoid anandamide. FEBS Lett. 1995;359:133–136. doi: 10.1016/0014-5793(95)00027-7. [DOI] [PubMed] [Google Scholar]
- Wiskerke J, Pattij T, Schoffelmeer AN, De Vries TJ. The role of CB1 receptors in psychostimulant addiction. Addict Biol. 2008;13:225–238. doi: 10.1111/j.1369-1600.2008.00109.x. [DOI] [PubMed] [Google Scholar]
- Xi ZX, Gilbert JG, Peng XQ, Pak AC, Li X, Gardner EL. Cannabinoid CB1 receptor antagonist AM251 inhibits cocaine-primed relapse in rats: role of glutamate in the nucleus accumbens. J Neurosci. 2006;26:8531–8536. doi: 10.1523/JNEUROSCI.0726-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, Driscoll JR, Wilbrecht L, Margolis EB, Fields HL, Hjelmstad GO. Nucleus accumbens medium spiny neurons target non-dopaminergic neurons in the ventral tegmental area. J Neurosci. 2011;31:7811–7816. doi: 10.1523/JNEUROSCI.1504-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Feng J, Fienberg AA, Greengard P. D2 dopamine receptors induce mitogen-activated protein kinase and cAMP response element-binding protein phosphorylation in neurons. Proc Natl Acad Sci U S A. 1999;96:11607–11612. doi: 10.1073/pnas.96.20.11607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HH, Davis MI, Ronesi JA, Lovinger DM. The role of protein synthesis in striatal long-term depression. J Neurosci. 2006;26:11811–11820. doi: 10.1523/JNEUROSCI.3196-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang TA, Placzek AN, Dani JA. In vitro identification and electrophysiological characterization of dopamine neurons in the ventral tegmental area. Neuropharmacology. 2010;59:431–436. doi: 10.1016/j.neuropharm.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu PJ, Lovinger DM. Persistent synaptic activity produces long-lasting enhancement of endocannabinoid modulation and alters long-term synaptic plasticity. J Neurophysiol. 2007;97:4386–4389. doi: 10.1152/jn.01228.2006. [DOI] [PubMed] [Google Scholar]