

Abstract
Patients with end-stage renal disease (ESRD), whether on conservative, peritoneal or hemodialysis therapy, have elevated genomic damage in peripheral blood lymphocytes and an increased cancer incidence, especially of the kidney. The damage is possibly due to accumulation of uremic toxins like advanced glycation endproducts or homocysteine. However, other endogenous substances with genotoxic properties, which are increased in ESRD, could be involved, such as the blood pressure regulating hormones angiotensin II and aldosterone or the inflammatory cytokine TNF-α. This review provides an overview of genomic damage observed in ESRD patients, focuses on possible underlying causes and shows modulations of the damage by modern dialysis strategies and vitamin supplementation.
Keywords: dialysis, genotoxicity, uremic toxins
1. Introduction
In the United States of America, 13.1% of the population over 20 years were estimated to be suffering from chronic kidney disease in 2004, and in 2007, 0.18% received renal replacement therapy ([1] and United States Renal Data System, www.usrds.org). An approximate 45% increase in prevalence is expected in 2020 due to the increasing age of the population (United States Renal Data System, www.usrds.org). Similar values have been observed in other industrialized nations. Thus, an increasing number of people will receive renal replacement therapy for lengthier periods of time.
Besides causing cardiovascular and other complications, end-stage renal disease (ESRD) is further characterized by a high incidence of cancer [2,3,4]. Of the cancer types recorded in these epidemiological studies, the risk of kidney cancer is tremendously increased. Depending on the continent, it is three- to ten-times higher than that seen in the control population [2].
Several biological parameters of ESRD patients were measured to determine their burden of genomic damage. Structural DNA damage, like single and double strand breaks and alkali labile sites, detectable with one of the standard genotoxicity assays, the comet assay, was significantly increased in lymphocytes of patients under dialysis [5,6,7]. These types of DNA damage are theoretically repairable. Yet, DNA repair capacity is reduced by prolonged hemodialysis therapy [8]. Unrepaired or improperly repaired DNA lesions may have serious consequences, such as premature ageing [9,10], atherosclerosis [11] and cancer predisposition [12].
Furthermore, unrepairable genomic damage per se, such as sister chromatid exchanges and chromosome breaks, were found in lymphocytes of dialysis patients [13,14,15]. In addition to the DNA in the nucleus, the mitochondrial DNA was also shown to be affected: deletions and mutations were observed in mitochondrial DNA of muscle cells and in the easily accessible hair follicles [16,17,18]. Furthermore, the most prominent oxidative DNA lesion, the modified base 7,8-dihydro-8-oxo-guanine (8-oxodG), is increased in ESRD [19]. 8-oxodG has a high mutagenic potential and repair errors or unrepaired 8-oxodGs lead to G:C→T:A transversions, which are often found in genes altered in tumors [20].
In this review, the impact of different dialysis therapies and of supplementation with vitamins on genomic damage of dialysis patients will be discussed. Also, underlying mechanisms of the origin of the increased DNA damage in ESRD will be described.
2. Causes of Increased Genomic Damage in ESRD
2.1. Genotoxic Uremic Toxins
Several minerals and molecules accumulate in the blood of patients with ESRD, which, if not removed, may lead to death. Besides urea, which is the classical uremic toxin, over 90 other toxins were included in the encyclopedic overview of uremic retention solutes initiated by the European Uremic Toxin Work Group (EUTox). These compounds, excreted by healthy kidneys under normal conditions, are classified as low molecular weight water-soluble compounds, middle molecules and protein-bound compounds [21]. In the seven years since the first review of EUTox, over 25 additional solutes have been identified ([22] and EUTox database: http://www.nephro-leipzig.de/eutoxdb/viewtoxins.php). It is conceivable that not all compounds responsible for the uremic syndrome have yet been discovered.
Genotoxic or mutagenic properties were reported for some of the toxins in the database, e.g., hydroquinone, indoxyl sulfate, leptin, methylglyoxal, N-ε-(carboxymethyl)lysine and TNF-α [6,23,24,25,26]. Other compounds, like trihalomethanes and heterocyclic amines, not included in the EUTox database but shown to accumulate in ESRD, are known mutagens [27,28]. These toxins, as well as other, not yet recognized substances can contribute to the genomic damage in ESRD by directly causing DNA damage.
2.2. Oxidative Stress
Reactive oxygen species (ROS) perform important physiological functions, for example in immune defense or in cellular signal transduction. However, when present in high concentrations they damage cellular structures, including lipids, membranes, proteins and nucleic acids [29]. Oxidative damage to DNA comprises single and double strand breaks, DNA base modifications, abasic sites and DNA crosslinks. Cells can protect themselves against oxidative damage with antioxidants, antioxidative enzymes and repair of the lesions [30]. This balance between the formation of ROS and protective mechanisms can be destabilized in situations of excessive production of free radicals and/or of deficient antioxidant defense, resulting in oxidative stress.
In kidney disease, oxidative stress is already found in the early stages, reflected by increased levels of various biomarkers of oxidative stress [31,32]. With decline of kidney function, the oxidative stress increases [33,34]. Different sources of oxidative stress in ESRD have been identified: (1) The dialysis procedure itself causes oxidative stress, with ROS being excreted by inflammatory cells on the surface of the dialysis membranes, which depletes scavenging molecules [35,36]; (2) Indeed, a significant reduction of the glutathione based scavenging system was observed in ESRD patients, in addition to lower levels of antioxidant enzymes such as superoxide dismutase, and a vitamin C-, as well as a vitamin E-deficiency [31,37,38,39,40]; (3) The dietary restriction necessary in ESRD, with a low intake of fresh fruits and vegetables, also plays a role in the vitamin deficiency; (4) Many factors accompanying ESRD, like malnutrition or chronic volume overload, lead to a permanent state of microinflammation, which further increases ROS formation by activated neutrophils [41].
3. Influence of Different Dialysis Therapies on Genomic Damage in ESRD
Dialysis is applied to achieve the removal of toxic solutes and of fluid overload whereby the patient’s blood is in contact with dialysis fluid across a semi-permeable membrane. This membrane can be the natural peritoneal membrane in peritoneal dialysis or an artificial membrane in a hemodialysis filter used for extracorporeal dialysis. The driving force of the fluid and solute exchange is osmosis, hydrostatic pressure, and a concentration gradient leading to diffusion and convection [42]. Dialysis achieves a solute removal of approximately 10% of that of a healthy kidney and cannot replace the kidney’s endocrine functions. Therefore, dialysis patients are on a special diet and need supplementary medication.
The most commonly used modalities in renal replacement therapy are peritoneal dialysis (PD) and hemodialysis (HD). Recent studies recommend peritoneal dialysis as an initial therapy, when applicable, to better preserve the important residual renal function and prolong overall survival [43,44]. The benefits of PD for survival are maintained up to the third year, after which survival with PD and HD is the same, or even slightly lower in patients on PD [45,46]. HD is conventionally delivered thrice weekly for four hours either in a dialysis center or at home. An improved uremic state can be obtained by shorter interdialytic intervals in daily HD, achieving an increased removal of small solutes and lower plasma concentrations of various uremic toxins [47,48,49,50]. A further technique is hemodiafiltration (HDF), which combines the diffusion mechanism of HD with the convective component of hemofiltration. In this way, the clearance of both low and high molecular weight uremic toxins (up to 25 kDa) is improved [51,52].
3.1. Hemodialysis—Hemodiafiltration
A meta-analysis of 20 studies totaling 657 patients comparing convective dialysis strategies with conventional HD did not find significant differences in mortality, number of hospital admissions per year or dialysis adequacy. Due to the small number of patients included in the individual studies and the overall suboptimal quality of these studies, the convective modalities could not be evaluated definitively [53]. Not included in this analysis were the results of the large European Dialysis Outcomes and Practice Pattern Study (DOPPS), which found that patients receiving high efficiency HDF had a 35% lower mortality risk than those receiving low-flux HD [54]. Studies comparing HD and HDF observed a better clearance of middle molecules such as β2 microglobulin, leptin or complement factor D [55,56], but even small molecules like urea and phosphate were better removed by HDF [57]. This more effective removal of uremic compounds seems to be essential for the reduction of the mortality risk, since the beneficial effect persisted even after statistical adjustment for dialysis efficiency (Kt/V) [58].
HDF also had a more beneficial influence on oxidative stress parameters than did HD. Total antioxidant capacity and glutathione peroxidase activity were increased and reactive oxygen metabolites, including pentosidine, advanced oxidation protein products (AOPPs) and oxidized LDL, were decreased in patients undergoing HDF compared to HD [59,60,61,62]. In addition, the p22phox subunit of NADPH oxidase—one of the enzymes possibly partly responsible for the increased oxidative stress—was found to be less expressed in patients on HDF compared to patients on HD [62]. Last, but not least, the amount of the oxidative 8-oxodG base modification, quantified in lymphocytes after a dialysis session, was significantly lower in HDF than in HD [60], supporting the hypothesis that the HDF procedure is superior not only in eliminating molecules generating oxidative stress or produced by oxidative stress, but itself causes less oxidative stress than HD [63].
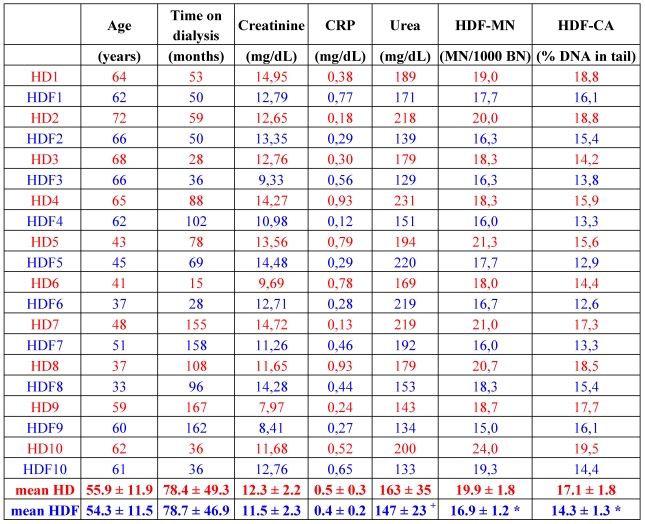
The fact that HDF better removes middle molecules, and decreases oxidative stress and also inflammation, as shown by lower C reactive protein (CRP) values of the patients [59,61], led to the assumption that patients on HDF might also show lower DNA damage. Results obtained from the comparison of the two different treatments are conflicting. One study, totaling 26 patients in the HD and 15 patients in the HDF group, did not show a difference in the DNA damage quantified with the comet assay [7]. In contrast, a yet unpublished study with 10 matched pairs of HD and HDF patients revealed less DNA damage measured by comet assay as well as a significant reduction of micronuclei in the HDF group patients (Table 1). Seven patients, who underwent a change of therapy from HD to HDF and were observed for a further eight months on HDF revealed a significant amelioration of DNA damage, as detected by comet assay, but no change in micronuclei frequency, which could be due to the rather long lifespan of lymphocytes [64]. Encompassing the observations of Vaslaki et al. [61], who found an improvement of many oxidative stress and inflammation parameters after switching HD patients to HDF for three months, HDF indeed might be more beneficial, concerning oxidative and genomic damage, than HD. To prove this, further studies have to be conducted analyzing these parameters.
Table 1.
Patient characteristics, selected plasma parameters and markers of genotoxicity of matched patients on hemodialysis (red) and hemodiafiltration (blue). HD: hemodialysis; HDF: hemodiafiltration; CRP: C reactive protein; MN: micronuclei; BN: binucleated cells; CA: comet assay; 1–10: patient numbers. Methods used to obtain these data are explained in [64]. * p < 0.001 vs. HD; + p = 0.074; paired t-test (SPSS 18).
 |
3.2. Hemodialysis—Daily Dialysis
Another possibility for improving dialysis therapy is to increase the time or the frequency of HD. The necessity to test such modalities has arisen out of the realization that no significant improvement of survival has been achieved despite technical advances in dialysis [65]. Survival of patients undergoing dialysis at one, two and five years of follow-up is 86%, 75% and 49%, respectively. Observations from the early 1980s, when Charra et al. treated patients for eight hours thrice weekly with slow flow HD and found increased survival, suggested that a dialysis session with longer duration could be more physiological and also beneficial for the patient [66,67]. Three different techniques are currently used. Short daily hemodialysis (SDHD) consists typically of 2–3.5 hours, six to seven times a week, long intermittent hemodialysis (LIHD) lasts 6–8 hours, three times a week, and nocturnal home hemodialysis (NHHD) is carried out overnight for 6–8 hours, five to seven times a week. All these dialysis modalities were studied mostly in small trials, resulting in the observation of improvements in a variety of clinical aspects and of the patients’ quality of life perception [68,69]. Additionally, a reduction in mortality between 17 to 78% was reported, as well as an increase of life expectancy of 9–15 years [70,71].
A recently published meta-analysis of 17 clinical studies analyzing the clinical effectiveness of SDHD identified possible reasons for the better outcome of patients receiving the daily treatment [72]. The clearest effect reported was the improvement of blood pressure control, resulting in a reduction of antihypertensive medication [47,73,74,75,76,77]. This is most probably achieved by a reduction of extracellular fluid excess, a lower interdialytic volume and a better control of sodium balance [72]. Erythropoetin treatment was also reduced in many patients under SDHD, explained by a better nutritional status as well as a removal of erythropoesis inhibiting uremic toxins [78,79]. Moreover, a better clearance of other uremic compounds was observed: of phosphorus [48,74,80,81,82], urea [47,48,73,74,77,82], homocysteine [83] and advanced glycation endproducts (AGEs, explained in detail below) [84]. Another beneficial aspect could be reduced inflammation as proven by lower levels of the inflammatory markers IL-6, TNF-α and CRP in SDHD patients compared to standard HD [81,85,86].
A cross-sectional study comparing patients on SDHD and standard HD therapy with regard to genomic damage, markers of microinflammation and removal of AGEs, could not detect differences in the inflammatory state [87]. Nevertheless, genomic damage assessed with the micronucleus frequency test was significantly lower under SDHD, even approaching values of the healthy control group. An explanation for this could be the better removal of uremic toxins in SDHD patients. In the same study, the improved clearance of members of the AGE group was shown, which exhibited genotoxic potential in vitro [24].
4. Effects of Vitamin Supplementation on Genomic Damage in ESRD
ESRD patients often suffer from vitamin deficiency of small water-soluble vitamins, such as vitamin C, due to a high rate of removal by dialysis and a restricted dietary intake. On the other hand, large fat-soluble vitamins, e.g., vitamin A, may be retained in the body [88]. Further reasons for the vitamin derangements in ESRD are drug interactions, which mostly affect the activity of folic acid, vitamin B6 and B12 [89]. Supplementation with vitamins in HD is therefore primarily used to compensate the deficiency. In addition, the distinct mechanisms of how the respective vitamins can ameliorate the state of health of the ESRD patients have been studied. Although the majority of investigations were conducted with supplementation of vitamin D and C, this review will now focus on studies analyzing the potential protective effects of vitamin supplementation against genomic damage in ESRD, employing vitamin E, B1, B12 and folic acid.
4.1. Vitamin E
While studies treating oxidative stress and inflammation with oral vitamin E produced conflicting results, the outcome of the four studies in dialysis patients concentrating on the effect of vitamin E supplementation on DNA damage unanimously show beneficial effects, which most probably can be ascribed to the antioxidative actions of vitamin E. Independent of the type of administration, oral or per vitamin E-coated dialyser membrane, DNA damage was observed to decrease [5,19,90,91]. In the two studies in which patients were given vitamin E orally, DNA damage was assessed by comet assay in one and by quantifying 8-oxodG by HPLC in the other [5,19], thereby measuring both unspecific structural DNA damage and a specific oxidative base modification. The same holds true for the two studies analyzing the impact of vitamin E-coated membranes, one applying the comet assay and the other quantifying 8-oxodG [90,91]. The latter study also included a crossover design, where patients changed the dialyser membrane type from standard cellulose to vitamin E-coated cellulose and vice versa. In those patients changing to the vitamin E-coated membrane, the 8-oxodG levels in leukocyte DNA significantly fell by 41% after only eight weeks, while it rose in the other group by 66% [91]. An increasing number of studies employing vitamin E-coated dialysers report favorable effects on markers of inflammation and oxidative stress, but also on blood pressure [92,93,94]. One advantage of the vitamin E-coated dialyser membrane is that the vitamin E can execute its free radical scavenging activity directly when the blood first contacts the dialyser, without activation of the inflammatory response by the membrane in the first place. Additionally, a consumption of endogenous vitamin E is prevented [91]. New vitamin E-coated dialysers based on polysulfone are being tested, which will probably be even more biocompatible than the ones coated with cellulose [92].
4.2. Vitamin B1
Beside oxidative stress generated by the activated immune cells, the formation of ROS can also be initiated by some uremic toxins, e.g., AGEs, which are a heterogeneous class of molecules formed by non-enzymatic reactions between reducing sugars and free amino groups of proteins, peptides, amino acids, lipids and also nucleic acids [95]. Best characterized are the protein modifications, including N-ε-(carboxymethyl)lysine, pentosidine and methylglyoxal-derived hydroimidazolone. The resulting adducts cannot be degraded and accumulate on long-living proteins. They are characterized by brown color, fluorescence and a tendency to polymerize. Features of the AGE-cross-linked proteins include a decreased solubility and a high resistance to proteolysis, changing the biomechanical and biochemical properties of the affected tissues [96]. AGE formation is a normal process in ageing [97]. However, in some diseases AGEs accumulate, for example in diabetes mellitus due to elevated plasma glucose levels. In ESRD, AGE accumulation results from impaired renal removal, disturbed metabolism and higher oxidative stress [98,99,100]. AGE accumulation is associated with the development of complications of diabetes (nephropathy, retinopathy and neuropathy [101]) and ESRD (dialysis-related amyloidosis, bone resorption and atherosclerosis [102,103,104]). These deleterious effects are mediated in part by the modified proteins, but also by AGE-specific receptors. The most studied receptor of AGEs, RAGE, is a multi-ligand receptor of the immunoglobulin superfamily, a signal transducing receptor modulating cellular functions [105]. Binding of AGEs to RAGE leads to oxidative stress via the induction of NADPH oxidase and to activation of proinflammatory molecules, among them NF-κB, which initiates increased expression of RAGE itself and starts a vicious circle [106,107]. These adverse effects of AGEs, together with their significantly increased plasma levels in ESRD, fulfill the criteria of uremic toxins: these compounds were classified as protein-bound uremic toxins [21].
Vitamin B1 (thiamine), in the form of the lipid-soluble thiamine precursor benfotiamine, was shown to inhibit the formation of AGEs [108]. Thiamine is required for the normal function of heart, muscle and nerves. It plays a key role in cellular energy metabolism, converting carbohydrates to energy [109], as the co-factor of the enzyme transketolase, which converts glyceraldehyde-3-phosphate and fructose-6-phosphate to intermediates of the pentose phosphate pathway [110]. Supplementation with benfotiamine in diabetic patients demonstrated a significant relief in neuropathic pain and a marked improvement in vibration perception thresholds [111]. Vitamin B1 has been shown to be decreased in many diabetic patients due to enhanced urinary excretion and malabsorption [111]. This deficiency manifests itself in reduced transketolase activities compared to healthy subjects [112]. Low transketolase is connected to neuropathies observed in approximately 25% of uremic patients and probably contributes to the unexplained cases of encephalopathy in the dialysis population [113,114,115].
Strikingly, although thiamine deficiency in dialysis patients was already observed in the early 1980s [116] and the hypothesis of a role of transketolase deficiency in uremic neuropathy exists since the 1970s [113], to date no clinical studies have analyzed the effect of thiamine supplementation on neurological problems in dialysis patients. By contrast, first clinical trials of thiamine in diabetic patients with neuropathies were conducted as early as 1992 and reported improvement of the symptoms [117]. Pharmacokinetic studies in dialysis patients were carried out, revealing the superiority of the bioavailability of benfotiamine versus thiamine [118].
Benfotiamine, therefore, was used in the first studies analyzing the impact of vitamin B1 supplementation on genomic damage of HD patients [119]. In two small consecutive prospective studies totaling 30 treated patients, the administration of 450–600 mg per day of benfotiamine for four months successfully decreased the micronucleus frequency of peripheral lymphocytes. The number of micronuclei was inversely correlated to the erythrocyte transketolase activity. Surprisingly, no effect of benfotiamine on plasma content of fluorescent AGEs could be detected. Since an intrinsic antioxidative capacity of benfotiamine unrelated to its anti-AGE effects was observed in vitro as well as in vivo [120,121], and the antioxidative plasma capacity of the benfotiamine-treated patients increased, the reduction of genomic damage can be explained with this property of benfotiamine. Possibly longer observation periods in humans are needed to reliably detect a lowering of plasma AGEs.
4.3. Folic Acid in Combination with Vitamin B12
Another uremic toxin whose formation can probably be influenced by supplementation with vitamins is homocysteine. Hyperhomocysteinemia was detected in approximately 85% of ESRD patients and persists throughout dialysis and even after renal transplantation [122]. Homocysteine is a normal intermediate in methionine metabolism, usually recycled to methionine with the involvement of folic acid and vitamin B12 [123]. Methionine is used for protein synthesis and, after conversion to S-adenosylmethionine, is an important methyl donor for transmethylation reactions, needed for example for DNA cytosine-methylation. Inhibition of DNA methyltransferases by S-adenosylhomocysteine was observed in individuals with increased homocysteine plasma levels, accompanied by hypomethylated DNA [124]. Insufficient DNA methylation renders the DNA less stable [125], possibly leading to genomic damage. Indeed, a correlation between plasma homocysteine levels and the number of micronuclei was found in humans [126].
Clinical studies analyzing the efficacy of folic acid alone or in combination with vitamin B12 (sometimes further combined with vitamin B6) on homocysteine levels in dialysis patients report inconsistent results. The plasma level of homocysteine was reduced markedly in only a few of the recently conducted trials [127,128], while there were only small changes in others [129,130]. The primary outcomes of the larger studies, all-cause mortality, cardiovascular events or cognitive function, were not ameliorated at all by the vitamin supplementations [127,131,132,133].
To date, the only study to explore the effect of supplementation with folic acid and vitamin B12 on genomic damage in dialysis patients observed a significant decline in plasma homocysteine levels, but no corresponding changes in the percentage of DNA methylation [134]. Nevertheless, genomic damage, assessed as micronucleus frequency, decreased in a therapy-length dependent manner. A possible antioxidant capacity of the supplemented vitamins could account for this beneficial effect.
5. Further Uremic Toxins with Genotoxic Properties
Apart from the above described uremic toxins with genotoxic effects, advanced glycation endproducts and homocysteine, further substances classified as uremic toxins show DNA damaging effects (Table 2). This was confirmed primarily by the comet assay, but also by detection of chromosome breaks and formation of the oxidative base modification 8-oxodG. It can be assumed that these molecules contribute to the increased genomic damage observed in dialysis patients. Since the uremic syndrome is characterized by accumulation of multiple compounds, synergistic actions of these compounds, amplifying their genotoxic potential, cannot be ruled out. Identification of additional uremic toxins and intensified research uncovering their toxicological properties will lead to an expansion of Table 2.
Table 2.
| Uremic toxin | DNA damage | References |
|---|---|---|
| Angiotensin II | 8-oxodG, micronuclei, double strand breaks, abasic sites | [135,136] |
| Hydroquinone | DNA strand breaks, micronuclei, 8-oxodG | [137] |
| Indoxyl sulfate | DNA strand breaks | [120] |
| Leptin | DNA strand breaks | [6] |
| Methylglyoxal | DNA strand breaks, 8-oxodG | [24,138] |
| N-ε-(carboxymethyl)lysine | DNA strand breaks | [24] |
| N-methyl-2-pyridone-5-carboxamide | DNA strand breaks | [139] |
| Nitrosodimethylamine | DNA strand breaks, micronuclei, known rat-liver carcinogen | [140,141,142] |
| Thiocyanate | DNA strand breaks | [143] |
| TNF-α | 8-oxodG, DNA strand breaks | [25,144] |
6. Conclusions
The possible modulation of genomic damage in ESRD by more intensive HD techniques more effectively removing uremic toxins and by supplementation with substances counteracting adverse effects of uremic toxins further supports the presumption that these compounds are mainly responsible for the formation of the DNA damage. The ability of a uremic toxin to induce the generation of oxidative stress seems to be a prerequisite for the formation of DNA damage. In addition to improving the overall condition of dialysis patients, HD techniques ensuring a more effective removal of uremic toxins, could also reduce the accumulation of genomic damage, possibly preventing the complication “malignancy” in dialysis patients.
References
- 1.Coresh J., Selvin E., Stevens L.A., Manzi J., Kusek J.W., Eggers P., Van Lente F., Levey A.S. Prevalence of chronic kidney disease in the United States. JAMA. 2007;298:2038–2047. doi: 10.1001/jama.298.17.2038. [DOI] [PubMed] [Google Scholar]
- 2.Maisonneuve P., Agodoa L., Gellert R., Stewart J.H., Buccianti G., Lowenfels A.B., Wolfe R.A., Jones E., Disney A.P., Briggs D., McCredie M., Boyle P. Cancer in patients on dialysis for end-stage renal disease: an international collaborative study. Lancet. 1999;354:93–99. doi: 10.1016/s0140-6736(99)06154-1. [DOI] [PubMed] [Google Scholar]
- 3.Stewart J.H., Buccianti G., Agodoa L., Gellert R., McCredie M.R., Lowenfels A.B., Disney A.P., Wolfe R.A., Boyle P., Maisonneuve P. Cancers of the kidney and urinary tract in patients on dialysis for end-stage renal disease: analysis of data from the United States, Europe, and Australia and New Zealand. J. Am. Soc. Nephrol. 2003;14:197–207. doi: 10.1097/01.ASN.0000039608.81046.81. [DOI] [PubMed] [Google Scholar]
- 4.Teschner M., Garte C., Rückle-Lanz H., Mader U., Stopper H., Klassen A., Heidland A. Incidence and spectrum of malignant disease among dialysis patients in North Bavaria. Dtsch. Med. Wochenschr. 2002;127:2497–2502. doi: 10.1055/s-2002-35637. [DOI] [PubMed] [Google Scholar]
- 5.Kan E., Undeger U., Bali M., Basaran N. Assessment of DNA strand breakage by the alkaline COMET assay in dialysis patients and the role of Vitamin E supplementation. Mutat. Res. 2002;520:151–159. doi: 10.1016/s1383-5718(02)00205-x. [DOI] [PubMed] [Google Scholar]
- 6.Horoz M., Bolukbas F.F., Bolukbas C., Aslan M., Koylu A.O., Gunaydin N., Selek S., Kocyigit A. The association of circulating leptin level with peripheral DNA damage in hemodialysis subjects. Clin. Biochem. 2006;39:918–922. doi: 10.1016/j.clinbiochem.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 7.Stopper H., Boullay F., Heidland A., Vienken J., Bahner U. Comet-assay analysis identifies genomic damage in lymphocytes of uremic patients. Am. J. Kidney Dis. 2001;38:296–301. doi: 10.1053/ajkd.2001.26094. [DOI] [PubMed] [Google Scholar]
- 8.Vamvakas S., Bahner U., Becker P., Steinle A., Gotz R., Heidland A. Impairment of DNA repair in the course of long-term hemodialysis and under cyclosporine immunosuppression after renal transplantation. Transplant. Proc. 1996;28:3468–3473. [PubMed] [Google Scholar]
- 9.Herbert K.E., Mistry Y., Hastings R., Poolman T., Niklason L., Williams B. Angiotensin II-mediated oxidative DNA damage accelerates cellular senescence in cultured human vascular smooth muscle cells via telomere-dependent and independent pathways. Circ. Res. 2008;102:201–208. doi: 10.1161/CIRCRESAHA.107.158626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li H., Mitchell J.R., Hasty P. DNA double-strand breaks: a potential causative factor for mammalian aging? Mech. Ageing Dev. 2008;129:416–424. doi: 10.1016/j.mad.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mercer J., Mahmoudi M., Bennett M. DNA damage, p53, apoptosis and vascular disease. Mutat. Res. 2007;621:75–86. doi: 10.1016/j.mrfmmm.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 12.Loft S., Poulsen H.E. Cancer risk and oxidative DNA damage in man. J. Mol. Med. 1996;74:297–312. doi: 10.1007/BF00207507. [DOI] [PubMed] [Google Scholar]
- 13.Buemi M., Floccari F., Costa C., Caccamo C., Belghity N., Campo S., Pernice F., Bonvissuto G., Coppolino G., Barilla A., Criseo M., Crasci E., Nostro L., Arena A. Dialysis-related genotoxicity: sister chromatid exchanges and DNA lesions in T and B lymphocytes of uremic patients. Genomic damage in patients on hemodiafiltration. Blood Purif. 2006;24:569–574. doi: 10.1159/000097080. [DOI] [PubMed] [Google Scholar]
- 14.Stopper H., Meysen T., Bockenforde A., Bahner U., Heidland A., Vamvakas S. Increased genomic damage in lymphocytes of patients before and after long-term maintenance hemodialysis therapy. Am. J. Kidney Dis. 1999;34:433–437. doi: 10.1016/s0272-6386(99)70069-7. [DOI] [PubMed] [Google Scholar]
- 15.Roth J.M., Restani R.G., Goncalves T.T., Sphor S.L., Ness A.B., Martino-Roth M.G., Garcias G.L. Genotoxicity evaluation in chronic renal patients undergoing hemodialysis and peritoneal dialysis, using the micronucleus test. Genet. Mol. Res. 2008;7:433–443. doi: 10.4238/vol7-2gmr441. [DOI] [PubMed] [Google Scholar]
- 16.Lim P.S., Ma Y.S., Cheng Y.M., Chai H., Lee C.F., Chen T.L., Wei Y.H. Mitochondrial DNA mutations and oxidative damage in skeletal muscle of patients with chronic uremia. J. Biomed. Sci. 2002;9:549–560. doi: 10.1159/000064728. [DOI] [PubMed] [Google Scholar]
- 17.Lim P.S., Cheng Y.M., Wei Y.H. Large-scale mitochondrial DNA deletions in skeletal muscle of patients with end-stage renal disease. Free Radic. Biol. Med. 2000;29:454–463. doi: 10.1016/s0891-5849(00)00334-8. [DOI] [PubMed] [Google Scholar]
- 18.Liu C.S., Ko L.Y., Lim P.S., Kao S.H., Wei Y.H. Biomarkers of DNA damage in patients with end-stage renal disease: mitochondrial DNA mutation in hair follicles. Nephrol. Dial. Transplant. 2001;16:561–565. doi: 10.1093/ndt/16.3.561. [DOI] [PubMed] [Google Scholar]
- 19.Domenici F.A., Vannucchi M.T., Jordao A.A., Jr., Meirelles M.S., Vannucchi H. DNA oxidative damage in patients with dialysis treatment. Ren. Fail. 2005;27:689–694. doi: 10.1080/08860220500242678. [DOI] [PubMed] [Google Scholar]
- 20.Nakabeppu Y., Sakumi K., Sakamoto K., Tsuchimoto D., Tsuzuki T., Nakatsu Y. Mutagenesis and carcinogenesis caused by the oxidation of nucleic acids. Biol. Chem. 2006;387:373–379. doi: 10.1515/BC.2006.050. [DOI] [PubMed] [Google Scholar]
- 21.Vanholder R., De Smet R., Glorieux G., Argiles A., Baurmeister U., Brunet P., Clark W., Cohen G., De Deyn P.P., Deppisch R., et al. Review on uremic toxins: classification, concentration, and interindividual variability. Kidney Int. 2003;63:1934–1943. doi: 10.1046/j.1523-1755.2003.00924.x. [DOI] [PubMed] [Google Scholar]
- 22.Vanholder R., Van Laecke S., Glorieux G. What is new in uremic toxicity? Pediatr. Nephrol. 2008;23:1211–1221. doi: 10.1007/s00467-008-0762-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McGregor D. Hydroquinone: an evaluation of the human risks from its carcinogenic and mutagenic properties. Crit. Rev. Toxicol. 2007;37:887–914. doi: 10.1080/10408440701638970. [DOI] [PubMed] [Google Scholar]
- 24.Schupp N., Schinzel R., Heidland A., Stopper H. Genotoxicity of advanced glycation end products: involvement of oxidative stress and of angiotensin II type 1 receptors. Ann. N. Y. Acad. Sci. 2005;1043:685–695. doi: 10.1196/annals.1333.079. [DOI] [PubMed] [Google Scholar]
- 25.Wheelhouse N.M., Chan Y.S., Gillies S.E., Caldwell H., Ross J.A., Harrison D.J., Prost S. TNF-alpha induced DNA damage in primary murine hepatocytes. Int. J. Mol. Med. 2003;12:889–894. [PubMed] [Google Scholar]
- 26.Schmid U., Stopper H., Heidland A., Schupp N. Benfotiamine exhibits direct antioxidative capacity and prevents induction of DNA damage in vitro. Diabetes Metab. Res. Rev. 2008;24:371–377. doi: 10.1002/dmrr.860. [DOI] [PubMed] [Google Scholar]
- 27.Vanholder R.C., Glorieux G., De Smet R., De Deyn P.P. Low water-soluble uremic toxins. Adv. Ren. Replace. Ther. 2003;10:257–269. doi: 10.1053/j.arrt.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 28.Bakir A., Williams R.H., Shaykh M., Dunea G., Dubin A. Biochemistry of the uremic syndrome. Adv. Clin. Chem. 1992;29:61–120. doi: 10.1016/s0065-2423(08)60222-x. [DOI] [PubMed] [Google Scholar]
- 29.Valko M., Rhodes C.J., Moncol J., Izakovic M., Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006;160:1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 30.Halliwell B. Oxidative stress, nutrition and health. Experimental strategies for optimization of nutritional antioxidant intake in humans. Free Radic. Res. 1996;25:57–74. doi: 10.3109/10715769609145656. [DOI] [PubMed] [Google Scholar]
- 31.Kao M.P., Ang D.S., Pall A., Struthers A.D. Oxidative stress in renal dysfunction: mechanisms, clinical sequelae and therapeutic options. J. Hum. Hypertens. 2010;24:1–8. doi: 10.1038/jhh.2009.70. [DOI] [PubMed] [Google Scholar]
- 32.Oberg B.P., McMenamin E., Lucas F.L., McMonagle E., Morrow J., Ikizler T.A., Himmelfarb J. Increased prevalence of oxidant stress and inflammation in patients with moderate to severe chronic kidney disease. Kidney Int. 2004;65:1009–1016. doi: 10.1111/j.1523-1755.2004.00465.x. [DOI] [PubMed] [Google Scholar]
- 33.Dounousi E., Papavasiliou E., Makedou A., Ioannou K., Katopodis K.P., Tselepis A., Siamopoulos K.C., Tsakiris D. Oxidative stress is progressively enhanced with advancing stages of CKD. Am. J. Kidney Dis. 2006;48:752–760. doi: 10.1053/j.ajkd.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 34.Terawaki H., Yoshimura K., Hasegawa T., Matsuyama Y., Negawa T., Yamada K., Matsushima M., Nakayama M., Hosoya T., Era S. Oxidative stress is enhanced in correlation with renal dysfunction: examination with the redox state of albumin. Kidney Int. 2004;66:1988–1993. doi: 10.1111/j.1523-1755.2004.00969.x. [DOI] [PubMed] [Google Scholar]
- 35.Jackson P., Loughrey C.M., Lightbody J.H., McNamee P.T., Young I.S. Effect of hemodialysis on total antioxidant capacity and serum antioxidants in patients with chronic renal failure. Clin. Chem. 1995;41:1135–1138. [PubMed] [Google Scholar]
- 36.Spittle M.A., Hoenich N.A., Handelman G.J., Adhikarla R., Homel P., Levin N.W. Oxidative stress and inflammation in hemodialysis patients. Am. J. Kidney Dis. 2001;38:1408–1413. doi: 10.1053/ajkd.2001.29280. [DOI] [PubMed] [Google Scholar]
- 37.Richard M.J., Arnaud J., Jurkovitz C., Hachache T., Meftahi H., Laporte F., Foret M., Favier A., Cordonnier D. Trace elements and lipid peroxidation abnormalities in patients with chronic renal failure. Nephron. 1991;57:10–15. doi: 10.1159/000186208. [DOI] [PubMed] [Google Scholar]
- 38.Mimic-Oka J., Simic T., Ekmescic V., Dragicevic P. Erythrocyte glutathione peroxidase and superoxide dismutase activities in different stages of chronic renal failure. Clin. Nephrol. 1995;44:44–48. [PubMed] [Google Scholar]
- 39.Vaziri N.D., Dicus M., Ho N.D., Boroujerdi-Rad L., Sindhu R.K. Oxidative stress and dysregulation of superoxide dismutase and NADPH oxidase in renal insufficiency. Kidney Int. 2003;63:179–185. doi: 10.1046/j.1523-1755.2003.00702.x. [DOI] [PubMed] [Google Scholar]
- 40.Locatelli F., Canaud B., Eckardt K.U., Stenvinkel P., Wanner C., Zoccali C. Oxidative stress in end-stage renal disease: an emerging threat to patient outcome. Nephrol. Dial. Transplant. 2003;18:1272–1280. doi: 10.1093/ndt/gfg074. [DOI] [PubMed] [Google Scholar]
- 41.Cachofeiro V., Goicochea M., de Vinuesa S.G., Oubina P., Lahera V., Luno J. Oxidative stress and inflammation, a link between chronic kidney disease and cardiovascular disease. Kidney Int. Suppl. 2008;74:S4–S9. doi: 10.1038/ki.2008.516. [DOI] [PubMed] [Google Scholar]
- 42.Blowey D.L., Alon U.S. Dialysis principles for primary health-care providers. Clin. Pediatr. (Phila) 2005;44:19–27. doi: 10.1177/000992280504400102. [DOI] [PubMed] [Google Scholar]
- 43.Divino Filho J.C. Preservation of renal function: HDF versus PD. The paramount role of peritoneal dialysis. Blood Purif. 2004;22(Suppl. 2):34–35. doi: 10.1159/000081872. [DOI] [PubMed] [Google Scholar]
- 44.Roob J.M. Comparison of clinical outcomes in peritoneal dialysis and hemodialysis. Wien. Klin. Wochenschr. 2005;117(Suppl. 6):60–68. doi: 10.1007/s00508-005-0494-9. [DOI] [PubMed] [Google Scholar]
- 45.Fenton S.S., Schaubel D.E., Desmeules M., Morrison H.I., Mao Y., Copleston P., Jeffery J.R., Kjellstrand C.M. Hemodialysis versus peritoneal dialysis: a comparison of adjusted mortality rates. Am. J. Kidney Dis. 1997;30:334–342. doi: 10.1016/s0272-6386(97)90276-6. [DOI] [PubMed] [Google Scholar]
- 46.Oreopoulos D.G., Ossareh S., Thodis E. Peritoneal dialysis: past, present, and future. Iran. J. Kidney Dis. 2008;2:171–182. [PubMed] [Google Scholar]
- 47.Ting G.O., Kjellstrand C., Freitas T., Carrie B.J., Zarghamee S. Long-term study of high-comorbidity ESRD patients converted from conventional to short daily hemodialysis. Am. J. Kidney Dis. 2003;42:1020–1035. doi: 10.1016/j.ajkd.2003.07.020. [DOI] [PubMed] [Google Scholar]
- 48.Williams A.W., Chebrolu S.B., Ing T.S., Ting G., Blagg C.R., Twardowski Z.J., Woredekal Y., Delano B., Gandhi V.C., Kjellstrand C.M. Early clinical, quality-of-life, and biochemical changes of “daily hemodialysis” (6 dialyses per week) Am. J. Kidney Dis. 2004;43:90–102. doi: 10.1053/j.ajkd.2003.09.017. [DOI] [PubMed] [Google Scholar]
- 49.Floridi A., Antolini F., Galli F., Fagugli R.M., Floridi E., Buoncristiani U. Daily haemodialysis improves indices of protein glycation. Nephrol. Dial. Transplant. 2002;17:871–878. doi: 10.1093/ndt/17.5.871. [DOI] [PubMed] [Google Scholar]
- 50.Rocco M.V. More frequent hemodialysis: back to the future? Adv. Chronic Kidney Dis. 2007;14:e1–e9. doi: 10.1053/j.ackd.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 51.Lornoy W., Becaus I., Billiouw J.M., Sierens L., Van Malderen P., D’Haenens P. On-line haemodiafiltration. Remarkable removal of beta2-microglobulin. Long-term clinical observations. Nephrol. Dial. Transplant. 2000;15(Suppl. 1):49–54. doi: 10.1093/oxfordjournals.ndt.a027964. [DOI] [PubMed] [Google Scholar]
- 52.Lin C.L., Yang C.W., Chiang C.C., Chang C.T., Huang C.C. Long-term on-line hemodiafiltration reduces predialysis beta-2-microglobulin levels in chronic hemodialysis patients. Blood Purif. 2001;19:301–307. doi: 10.1159/000046958. [DOI] [PubMed] [Google Scholar]
- 53.Rabindranath K.S., Strippoli G.F., Daly C., Roderick P.J., Wallace S., MacLeod A.M. Haemodiafiltration, haemofiltration and haemodialysis for end-stage kidney disease. Cochrane Database Syst. Rev. 2006:CD006258. doi: 10.1002/14651858.CD006258. [DOI] [PubMed] [Google Scholar]
- 54.Canaud B., Bragg-Gresham J.L., Marshall M.R., Desmeules S., Gillespie B.W., Depner T., Klassen P., Port F.K. Mortality risk for patients receiving hemodiafiltration versus hemodialysis: European results from the DOPPS. Kidney Int. 2006;69:2087–2093. doi: 10.1038/sj.ki.5000447. [DOI] [PubMed] [Google Scholar]
- 55.Thomas G., Jaber B.L. Convective therapies for removal of middle molecular weight uremic toxins in end-stage renal disease: a review of the evidence. Semin. Dial. 2009;22:610–614. doi: 10.1111/j.1525-139X.2009.00665.x. [DOI] [PubMed] [Google Scholar]
- 56.Penne E.L., van der Weerd N.C., Blankestijn P.J., van den Dorpel M.A., Grooteman M.P., Nube M.J., Ter Wee P.M., Levesque R., Bots M.L. Role of residual kidney function and convective volume on change in beta2-microglobulin levels in hemodiafiltration patients. Clin. J. Am. Soc. Nephrol. 2010;5:80–86. doi: 10.2215/CJN.03340509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Canaud B., Morena M., Leray-Moragues H., Chalabi L., Cristol J.P. Overview of clinical studies in hemodiafiltration: what do we need now? Hemodial. Int. 2006;10(Suppl. 1):S5–S12. doi: 10.1111/j.1542-4758.2006.01183.x. [DOI] [PubMed] [Google Scholar]
- 58.Locatelli F., Manzoni C., Cavalli A., Di Filippo S. Can convective therapies improve dialysis outcomes? Curr. Opin. Nephrol. Hypertens. 2009;18:476–480. doi: 10.1097/MNH.0b013e3283318e8b. [DOI] [PubMed] [Google Scholar]
- 59.Filiopoulos V., Hadjiyannakos D., Metaxaki P., Sideris V., Takouli L., Anogiati A., Vlassopoulos D. Inflammation and oxidative stress in patients on hemodiafiltration. Am. J. Nephrol. 2008;28:949–957. doi: 10.1159/000142724. [DOI] [PubMed] [Google Scholar]
- 60.Gonzalez-Diez B., Cavia M., Torres G., Abaigar P., Muniz P. Effect of a hemodiafiltration session with on-line regeneration of the ultrafiltrate on oxidative stress. Comparative study with conventional hemodialysis with polysulfone. Blood Purif. 2008;26:505–510. doi: 10.1159/000163848. [DOI] [PubMed] [Google Scholar]
- 61.Vaslaki L., Major L., Berta K., Karatson A., Misz M., Pethoe F., Ladanyi E., Fodor B., Stein G., Pischetsrieder M., Zima T., Wojke R., Gauly A., Passlick-Deetjen J. On-line haemodiafiltration versus haemodialysis: stable haematocrit with less erythropoietin and improvement of other relevant blood parameters. Blood Purif. 2006;24:163–173. doi: 10.1159/000090117. [DOI] [PubMed] [Google Scholar]
- 62.Calo L.A., Naso A., Carraro G., Wratten M.L., Pagnin E., Bertipaglia L., Rebeschini M., Davis P.A., Piccoli A., Cascone C. Effect of haemodiafiltration with online regeneration of ultrafiltrate on oxidative stress in dialysis patients. Nephrol. Dial. Transplant. 2007;22:1413–1419. doi: 10.1093/ndt/gfl783. [DOI] [PubMed] [Google Scholar]
- 63.Calo L.A., Naso A., Davis P.A. Hemodiafiltration and oxidative stress in dialysis patients. Blood Purif. 2009;28:59–60. doi: 10.1159/000210039. [DOI] [PubMed] [Google Scholar]
- 64.Kobras K., Schupp N., Nehrlich K., Adelhardt M., Bahner U., Vienken J., Heidland A., Sebekova K., Stopper H. Relation between different treatment modalities and genomic damage of end-stage renal failure patients. Kidney Blood Press. Res. 2006;29:10–17. doi: 10.1159/000092482. [DOI] [PubMed] [Google Scholar]
- 65.Kopple J.D. National Kidney Foundation K/DOQI clinical practice guidelines for nutrition in chronic renal failure. Am. J. Kidney Dis. 2001;37:S66–S70. doi: 10.1053/ajkd.2001.20748. [DOI] [PubMed] [Google Scholar]
- 66.Charra B., Calemard E., Cuche M., Laurent G. Control of hypertension and prolonged survival on maintenance hemodialysis. Nephron. 1983;33:96–99. doi: 10.1159/000182920. [DOI] [PubMed] [Google Scholar]
- 67.Charra B., Chazot C., Jean G., Hurot J.M., Vanel T., Terrat J.C., VoVan C. Long 3 × 8 hr dialysis: a three-decade summary. J. Nephrol. 2003;16(Suppl. 7):S64–S69. [PubMed] [Google Scholar]
- 68.Kliger A.S. More intensive hemodialysis. Clin. J. Am. Soc. Nephrol. 2009;4(Suppl. 1):S121–S124. doi: 10.2215/CJN.02920509. [DOI] [PubMed] [Google Scholar]
- 69.McFarlane P.A. More of the same: improving outcomes through intensive hemodialysis. Semin. Dial. 2009;22:598–602. doi: 10.1111/j.1525-139X.2009.00666.x. [DOI] [PubMed] [Google Scholar]
- 70.Ok E., Duman S., Asci G., Yilmaz M., Sertoz O.O., Toz H., Adam S.M., Basci A., Ozkahya M. Eight-hour nocturnal in-center hemodialysis provides survival benefit over four-hour conventional hemodialysis. J. Am. Soc. Nephrol. 2008;19:71A. [Google Scholar]
- 71.Kjellstrand C.M., Buoncristiani U., Ting G., Traeger J., Piccoli G.B., Sibai-Galland R., Young B.A., Blagg C.R. Short daily haemodialysis: survival in 415 patients treated for 1006 patient-years. Nephrol. Dial. Transplant. 2008;23:3283–3289. doi: 10.1093/ndt/gfn210. [DOI] [PubMed] [Google Scholar]
- 72.Punal J., Lema L.V., Sanhez-Guisande D., Ruano-Ravina A. Clinical effectiveness and quality of life of conventional haemodialysis versus short daily haemodialysis: a systematic review. Nephrol. Dial. Transplant. 2008;23:2634–2646. doi: 10.1093/ndt/gfn010. [DOI] [PubMed] [Google Scholar]
- 73.Koshikawa S., Akizawa T., Saito A., Kurokawa K. Clinical effect of short daily in-center hemodialysis. Nephron Clin. Pract. 2003;95:c23–c30. doi: 10.1159/000073015. [DOI] [PubMed] [Google Scholar]
- 74.Traeger J., Galland R., Delawari E., Arkouche W., Hadden R. Six years’ experience with short daily hemodialysis: do the early improvements persist in the mid and long term? Hemodial. Int. 2004;8:151–158. doi: 10.1111/j.1492-7535.2004.01089.x. [DOI] [PubMed] [Google Scholar]
- 75.Buoncristiani U., Fagugli R.M., Pinciaroli M.R., Kulurianu H., Ceravolo G., Bova C. Reversal of left-ventricular hypertrophy in uremic patients by treatment with daily hemodialysis (DHD) Contrib. Nephrol. 1996;119:152–156. doi: 10.1159/000425466. [DOI] [PubMed] [Google Scholar]
- 76.Galland R., Traeger J., Arkouche W., Delawari E., Fouque D. Short daily hemodialysis and nutritional status. Am. J. Kidney Dis. 2001;37:S95–S98. doi: 10.1053/ajkd.2001.20758. [DOI] [PubMed] [Google Scholar]
- 77.Reynolds J.T., Homel P., Cantey L., Evans E., Harding P., Gotch F., Wuerth D., Finkelstein S., Levin N., Kliger A., Simon D.B., Finkelstein F.O. A one-year trial of in-center daily hemodialysis with an emphasis on quality of life. Blood Purif. 2004;22:320–328. doi: 10.1159/000079186. [DOI] [PubMed] [Google Scholar]
- 78.Buoncristiani U. Fifteen years of clinical experience with daily haemodialysis. Nephrol. Dial. Transplant. 1998;13(Suppl. 6):148–151. doi: 10.1093/ndt/13.suppl_6.148. [DOI] [PubMed] [Google Scholar]
- 79.Fagugli R.M., Buoncristiani U., Ciao G. Anemia and blood pressure correction obtained by daily hemodialysis induce a reduction of left ventricular hypertrophy in dialysed patients. Int. J. Artif. Organs. 1998;21:429–431. [PubMed] [Google Scholar]
- 80.Nesrallah G.E., Suri R.S., Zoller R., Garg A.X., Moist L.M., Lindsay R.M. The International Quotidian Dialysis Registry: annual report 2006. Hemodial. Int. 2006;10:219–224. doi: 10.1111/j.1542-4758.2006.00103.x. [DOI] [PubMed] [Google Scholar]
- 81.Ayus J.C., Mizani M.R., Achinger S.G., Thadhani R., Go A.S., Lee S. Effects of short daily versus conventional hemodialysis on left ventricular hypertrophy and inflammatory markers: a prospective, controlled study. J. Am. Soc. Nephrol. 2005;16:2778–2788. doi: 10.1681/ASN.2005040392. [DOI] [PubMed] [Google Scholar]
- 82.Galland R., Traeger J. Short daily hemodialysis and nutritional status in patients with chronic renal failure. Semin. Dial. 2004;17:104–108. doi: 10.1111/j.0894-0959.2004.17205.x. [DOI] [PubMed] [Google Scholar]
- 83.Friedman A.N., Bostom A.G., Levey A.S., Rosenberg I.H., Selhub J., Pierratos A. Plasma total homocysteine levels among patients undergoing nocturnal versus standard hemodialysis. J. Am. Soc. Nephrol. 2002;13:265–268. doi: 10.1681/ASN.V131265. [DOI] [PubMed] [Google Scholar]
- 84.Fagugli R.M., Vanholder R., De Smet R., Selvi A., Antolini F., Lameire N., Floridi A., Buoncristiani U. Advanced glycation end products: specific fluorescence changes of pentosidine-like compounds during short daily hemodialysis. Int. J. Artif. Organs. 2001;24:256–262. [PubMed] [Google Scholar]
- 85.Yuen D., Richardson R.M., Fenton S.S., McGrath-Chong M.E., Chan C.T. Quotidian nocturnal hemodialysis improves cytokine profile and enhances erythropoietin responsiveness. ASAIO J. 2005;51:236–241. doi: 10.1097/01.mat.0000160578.43422.60. [DOI] [PubMed] [Google Scholar]
- 86.Hamlett L., Haragsim L. Quotidian hemodialysis and inflammation associated with chronic kidney disease. Adv. Chronic Kidney Dis. 2007;14:e35–e42. doi: 10.1053/j.ackd.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 87.Fragedaki E., Nebel M., Schupp N., Sebekova K., Volkel W., Klassen A., Pischetsrieder M., Frischmann M., Niwa T., Vienken J., Heidland A., Stopper H. Genomic damage and circulating AGE levels in patients undergoing daily versus standard haemodialysis. Nephrol. Dial. Transplant. 2005;20:1936–1943. doi: 10.1093/ndt/gfh898. [DOI] [PubMed] [Google Scholar]
- 88.Makoff R. Vitamin replacement therapy in renal failure patients. Miner. Electrolyte Metab. 1999;25:349–351. doi: 10.1159/000057473. [DOI] [PubMed] [Google Scholar]
- 89.Waxman S., Corcino J.J., Herbert V. Drugs, toxins and dietary amino acids affecting vitamin B12 or folic acid absorption or utilization. Am. J. Med. 1970;48:599–608. doi: 10.1016/0002-9343(70)90010-0. [DOI] [PubMed] [Google Scholar]
- 90.Muller C., Eisenbrand G., Gradinger M., Rath T., Albert F.W., Vienken J., Singh R., Farmer P.B., Stockis J.P., Janzowski C. Effects of hemodialysis, dialyser type and iron infusion on oxidative stress in uremic patients. Free Radic. Res. 2004;38:1093–1100. doi: 10.1080/10715760400011452. [DOI] [PubMed] [Google Scholar]
- 91.Tarng D.C., Huang T.P., Liu T.Y., Chen H.W., Sung Y.J., Wei Y.H. Effect of vitamin E-bonded membrane on the 8-hydroxy 2’-deoxyguanosine level in leukocyte DNA of hemodialysis patients. Kidney Int. 2000;58:790–799. doi: 10.1046/j.1523-1755.2000.00228.x. [DOI] [PubMed] [Google Scholar]
- 92.Cruz D.N., de Cal M., Ronco C. Oxidative stress and anemia in chronic hemodialysis: the promise of bioreactive membranes. Contrib. Nephrol. 2008;161:89–98. doi: 10.1159/000130413. [DOI] [PubMed] [Google Scholar]
- 93.Takouli L., Hadjiyannakos D., Metaxaki P., Sideris V., Filiopoulos V., Anogiati A., Vlassopoulos D. Vitamin E-coated cellulose acetate dialysis membrane: long-term effect on inflammation and oxidative stress. Ren. Fail. 2010;32:287–293. doi: 10.3109/08860221003615795. [DOI] [PubMed] [Google Scholar]
- 94.Matsumura M., Sasaki H., Sekizuka K., Sano H., Ogawa K., Shimizu C., Yoshida H., Kobayashi S., Koremoto M., Aritomi M., Ueki K. Improved management of intradialytic hypotension (IDH) using vitamin E-bonded polysulfone membrane dialyzer. Int. J. Artif. Organs. 2010;33:147–153. doi: 10.1177/039139881003300303. [DOI] [PubMed] [Google Scholar]
- 95.Schleicher E.D., Bierhaus A., Haring H.U., Nawroth P.P., Lehmann R. Chemistry and pathobiology of advanced glycation end products. In: D’Angelo A., Favaro S., Gambaro G., editors. Advanced Glycation End Products in Nephrology. Contrib. Nephrol. Vol. 131, pp. 1-9. Kager; Basel, Switzerland: 2001. [DOI] [PubMed] [Google Scholar]
- 96.Busch M., Franke S., Ruster C., Wolf G. Advanced glycation end-products and the kidney. Eur. J. Clin. Invest. 2010;40:742–755. doi: 10.1111/j.1365-2362.2010.02317.x. [DOI] [PubMed] [Google Scholar]
- 97.Brownlee M. Advanced protein glycosylation in diabetes and aging. Annu. Rev. Med. 1995;46:223–234. doi: 10.1146/annurev.med.46.1.223. [DOI] [PubMed] [Google Scholar]
- 98.Miyata T., Wada Y., Cai Z., Iida Y., Horie K., Yasuda Y., Maeda K., Kurokawa K., van Ypersele de Strihou C. Implication of an increased oxidative stress in the formation of advanced glycation end products in patients with end-stage renal failure. Kidney Int. 1997;51:1170–1181. doi: 10.1038/ki.1997.160. [DOI] [PubMed] [Google Scholar]
- 99.Miyata T., van Ypersele de Strihou C., Kurokawa K., Baynes J.W. Alterations in nonenzymatic biochemistry in uremia: origin and significance of “carbonyl stress” in long-term uremic complications. Kidney Int. 1999;55:389–399. doi: 10.1046/j.1523-1755.1999.00302.x. [DOI] [PubMed] [Google Scholar]
- 100.Agalou S., Ahmed N., Thornalley P.J., Dawnay A. Advanced glycation end product free adducts are cleared by dialysis. Ann. N. Y. Acad. Sci. 2005;1043:734–739. doi: 10.1196/annals.1333.085. [DOI] [PubMed] [Google Scholar]
- 101.Kalousova M., Zima T., Tesar V., Stipek S., Sulkova S. Advanced glycation end products in clinical nephrology. Kidney Blood Press. Res. 2004;27:18–28. doi: 10.1159/000075533. [DOI] [PubMed] [Google Scholar]
- 102.Miyata T. New aspects in the pathogenesis of dialysis-related amyloidosis: pathophysiology of advanced glycation end products in renal failure. Nippon Jinzo Gakkai Shi. 1996;38:191–197. [PubMed] [Google Scholar]
- 103.Miyata T., Kawai R., Taketomi S., Sprague S.M. Possible involvement of advanced glycation end-products in bone resorption. Nephrol. Dial. Transplant. 1996;11(Suppl. 5):54–57. doi: 10.1093/ndt/11.supp5.54. [DOI] [PubMed] [Google Scholar]
- 104.Miyata T., Kurokawa K., van Ypersele de Strihou C. Relevance of oxidative and carbonyl stress to long-term uremic complications. Kidney Int. Suppl. 2000;76:S120–S125. doi: 10.1046/j.1523-1755.2000.07615.x. [DOI] [PubMed] [Google Scholar]
- 105.Wendt T., Tanji N., Guo J., Hudson B.I., Bierhaus A., Ramasamy R., Arnold B., Nawroth P.P., Yan S.F., D’Agati V., Schmidt A.M. Glucose, glycation, and RAGE: implications for amplification of cellular dysfunction in diabetic nephropathy. J. Am. Soc. Nephrol. 2003;14:1383–1395. doi: 10.1097/01.asn.0000065100.17349.ca. [DOI] [PubMed] [Google Scholar]
- 106.Wautier M.P., Chappey O., Corda S., Stern D.M., Schmidt A.M., Wautier J.L. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am. J. Physiol. Endocrinol. Metab. 2001;280:E685–E694. doi: 10.1152/ajpendo.2001.280.5.E685. [DOI] [PubMed] [Google Scholar]
- 107.Schmidt A.M., Yan S.D., Yan S.F., Stern D.M. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J. Clin. Invest. 2001;108:949–955. doi: 10.1172/JCI14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hammes H.P., Du X., Edelstein D., Taguchi T., Matsumura T., Ju Q., Lin J., Bierhaus A., Nawroth P., Hannak D., Neumaier M., Bergfeld R., Giardino I., Brownlee M. Benfotiamine blocks three major pathways of hyperglycemic damage and prevents experimental diabetic retinopathy. Nat. Med. 2003;9:294–299. doi: 10.1038/nm834. [DOI] [PubMed] [Google Scholar]
- 109.Balakumar P., Rohilla A., Krishan P., Solairaj P., Thangathirupathi A. The multifaceted therapeutic potential of benfotiamine. Pharmacol. Res. 2010;61:482–488. doi: 10.1016/j.phrs.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 110.Peyroux J., Sternberg M. Advanced glycation endproducts (AGEs): Pharmacological inhibition in diabetes. Pathol. Biol. 2006;54:405–419. doi: 10.1016/j.patbio.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 111.Head K.A. Peripheral neuropathy: pathogenic mechanisms and alternative therapies. Altern. Med. Rev. 2006;11:294–329. [PubMed] [Google Scholar]
- 112.Frank T., Bitsch R., Maiwald J., Stein G. High thiamine diphosphate concentrations in erythrocytes can be achieved in dialysis patients by oral administration of benfontiamine. Eur. J. Clin. Pharmacol. 2000;56:251–257. doi: 10.1007/s002280000131. [DOI] [PubMed] [Google Scholar]
- 113.Lange K., Lonergan E.T., Semar M., Sterzel R.B. Transketolase inhibition as a mechanism in uremic neuropathy. Trans. Assoc. Am. Physicians. 1971;84:172–181. [PubMed] [Google Scholar]
- 114.Ueda K., Takada D., Mii A., Tsuzuku Y., Saito S.K., Kaneko T., Utsumi K., Iino Y., Katayama Y. Severe thiamine deficiency resulted in Wernicke’s encephalopathy in a chronic dialysis patient. Clin. Exp. Nephrol. 2006;10:290–293. doi: 10.1007/s10157-006-0440-9. [DOI] [PubMed] [Google Scholar]
- 115.Hung S.C., Hung S.H., Tarng D.C., Yang W.C., Chen T.W., Huang T.P. Thiamine deficiency and unexplained encephalopathy in hemodialysis and peritoneal dialysis patients. Am. J. Kidney Dis. 2001;38:941–947. doi: 10.1053/ajkd.2001.28578. [DOI] [PubMed] [Google Scholar]
- 116.Blumberg A., Hanck A., Sander G. Vitamin nutrition in patients on continuous ambulatory peritoneal dialysis (CAPD) Clin. Nephrol. 1983;20:244–250. [PubMed] [Google Scholar]
- 117.Eckert M., Schejbal P. Therapy of neuropathies with a vitamin B combination. Symptomatic treatment of painful diseases of the peripheral nervous system with a combination preparation of thiamine, pyridoxine and cyanocobalamin. Fortschr. Med. 1992;110:544–548. [PubMed] [Google Scholar]
- 118.Frank T., Bitsch R., Maiwald J., Stein G. Alteration of thiamine pharmacokinetics by end-stage renal disease (ESRD) Int. J. Clin. Pharmacol. Ther. 1999;37:449–455. [PubMed] [Google Scholar]
- 119.Schupp N., Dette E.M., Schmid U., Bahner U., Winkler M., Heidland A., Stopper H. Benfotiamine reduces genomic damage in peripheral lymphocytes of hemodialysis patients. Naunyn Schmiedebergs Arch. Pharmacol. 2008;378:283–291. doi: 10.1007/s00210-008-0310-y. [DOI] [PubMed] [Google Scholar]
- 120.Schmid U., Stopper H., Heidland A., Schupp N. Benfotiamine exhibits direct antioxidative capacity and prevents induction of DNA damage in vitro. Diabetes Metab. Res. Rev. 2008;24:371–377. doi: 10.1002/dmrr.860. [DOI] [PubMed] [Google Scholar]
- 121.Wu S., Ren J. Benfotiamine alleviates diabetes-induced cerebral oxidative damage independent of advanced glycation end-product, tissue factor and TNF-alpha. Neurosci. Lett. 2006;394:158–162. doi: 10.1016/j.neulet.2005.10.022. [DOI] [PubMed] [Google Scholar]
- 122.Ferechide D., Radulescu D. Hyperhomocysteinemia in renal diseases. J. Med. Life. 2009;2:53–59. [PMC free article] [PubMed] [Google Scholar]
- 123.Miyaki K. Genetic polymorphisms in homocysteine metabolism and response to folate intake: a comprehensive strategy to elucidate useful genetic information. J. Epidemiol. 2010;20:266–270. doi: 10.2188/jea.JE20100042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yi P., Melnyk S., Pogribna M., Pogribny I.P., Hine R.J., James S.J. Increase in plasma homocysteine associated with parallel increases in plasma S-adenosylhomocysteine and lymphocyte DNA hypomethylation. J. Biol. Chem. 2000;275:29318–29323. doi: 10.1074/jbc.M002725200. [DOI] [PubMed] [Google Scholar]
- 125.Dodge J.E., Okano M., Dick F., Tsujimoto N., Chen T., Wang S., Ueda Y., Dyson N., Li E. Inactivation of Dnmt3b in mouse embryonic fibroblasts results in DNA hypomethylation, chromosomal instability, and spontaneous immortalization. J. Biol. Chem. 2005;280:17986–17991. doi: 10.1074/jbc.M413246200. [DOI] [PubMed] [Google Scholar]
- 126.Fenech M. Micronucleus frequency in human lymphocytes is related to plasma vitamin B12 and homocysteine. Mutat. Res. 1999;428:299–304. doi: 10.1016/s1383-5742(99)00056-3. [DOI] [PubMed] [Google Scholar]
- 127.Vianna A.C., Mocelin A.J., Matsuo T., Morais-Filho D., Largura A., Delfino V.A., Soares A.E., Matni A.M. Uremic hyperhomocysteinemia: a randomized trial of folate treatment for the prevention of cardiovascular events. Hemodial. Int. 2007;11:210–216. doi: 10.1111/j.1542-4758.2007.00171.x. [DOI] [PubMed] [Google Scholar]
- 128.Chiu Y.W., Chang J.M., Hwang S.J., Tsai J.C., Chen H.C. Pharmacological dose of vitamin B12 is as effective as low-dose folinic acid in correcting hyperhomocysteinemia of hemodialysis patients. Ren. Fail. 2009;31:278–283. doi: 10.1080/08860220902780010. [DOI] [PubMed] [Google Scholar]
- 129.Cianciolo G., La Manna G., Coli L., Donati G., D’Addio F., Persici E., Comai G., Wratten M., Dormi A., Mantovani V., Grossi G., Stefoni S. 5-methyltetrahydrofolate administration is associated with prolonged survival and reduced inflammation in ESRD patients. Am. J. Nephrol. 2008;28:941–948. doi: 10.1159/000142363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ossareh S., Shayan-Moghaddam H., Salimi A., Asgari M., Farrokhi F. Different doses of oral folic acid for homocysteine-lowering therapy in patients on hemodialysis: a randomized controlled trial. Iran. J. Kidney Dis. 2009;3:227–233. [PubMed] [Google Scholar]
- 131.Heinz J., Kropf S., Domrose U., Westphal S., Borucki K., Luley C., Neumann K.H., Dierkes J. B vitamins and the risk of total mortality and cardiovascular disease in end-stage renal disease: results of a randomized controlled trial. Circulation. 2010;121:1432–1438. doi: 10.1161/CIRCULATIONAHA.109.904672. [DOI] [PubMed] [Google Scholar]
- 132.Jamison R.L., Hartigan P., Kaufman J.S., Goldfarb D.S., Warren S.R., Guarino P.D., Gaziano J.M. Effect of homocysteine lowering on mortality and vascular disease in advanced chronic kidney disease and end-stage renal disease: a randomized controlled trial. JAMA. 2007;298:1163–1170. doi: 10.1001/jama.298.10.1163. [DOI] [PubMed] [Google Scholar]
- 133.Brady C.B., Gaziano J.M., Cxypoliski R.A., Guarino P.D., Kaufman J.S., Warren S.R., Hartigan P., Goldfarb D.S., Jamison R.L. Homocysteine lowering and cognition in CKD: the Veterans Affairs homocysteine study. Am. J. Kidney Dis. 2009;54:440–449. doi: 10.1053/j.ajkd.2009.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Stopper H., Treutlein A.T., Bahner U., Schupp N., Schmid U., Brink A., Perna A., Heidland A. Reduction of the genomic damage level in haemodialysis patients by folic acid and vitamin B12 supplementation. Nephrol. Dial. Transplant. 2008;23:3272–3279. doi: 10.1093/ndt/gfn254. [DOI] [PubMed] [Google Scholar]
- 135.Schupp N., Schmid U., Rutkowski P., Lakner U., Kanase N., Heidland A., Stopper H. Angiotensin II-induced genomic damage in renal cells can be prevented by angiotensin II type 1 receptor blockage or radical scavenging. Am. J. Physiol. 2007;292:F1427–F1434. doi: 10.1152/ajprenal.00458.2006. [DOI] [PubMed] [Google Scholar]
- 136.Schmid U., Stopper H., Schweda F., Queisser N., Schupp N. Angiotensin II induces DNA damage in the kidney. Cancer Res. 2008;68:9239–9246. doi: 10.1158/0008-5472.CAN-08-1310. [DOI] [PubMed] [Google Scholar]
- 137.Luo L., Jiang L., Geng C., Cao J., Zhong L. Hydroquinone-induced genotoxicity and oxidative DNA damage in HepG2 cells. Chem. Biol. Interact. 2008;173:1–8. doi: 10.1016/j.cbi.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 138.Kim J., Kim N.H., Sohn E., Kim C.S., Kim J.S. Methylglyoxal induces cellular damage by increasing argpyrimidine accumulation and oxidative DNA damage in human lens epithelial cells. Biochem. Biophys. Res. Commun. 2010;391:346–351. doi: 10.1016/j.bbrc.2009.11.061. [DOI] [PubMed] [Google Scholar]
- 139.Rutkowski P. Are so-called uremic toxins always toxic? J. Ren. Nutr. 2008;18:7–11. doi: 10.1053/j.jrn.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 140.Erkekoglu P., Baydar T. Evaluation of the protective effect of ascorbic acid on nitrite- and nitrosamine-induced cytotoxicity and genotoxicity in human hepatoma line. Toxicol. Mech. Methods. 2010;20:45–52. doi: 10.3109/15376510903583711. [DOI] [PubMed] [Google Scholar]
- 141.Rothfuss A., O’Donovan M., De Boeck M., Brault D., Czich A., Custer L., Hamada S., Plappert-Helbig U., Hayashi M., Howe J., et al. Collaborative study on fifteen compounds in the rat-liver Comet assay integrated into 2- and 4-week repeat-dose studies. Mutat. Res. 2010;702:40–69. doi: 10.1016/j.mrgentox.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 142.Majer B.J., Hofer E., Cavin C., Lhoste E., Uhl M., Glatt H.R., Meinl W., Knasmuller S. Coffee diterpenes prevent the genotoxic effects of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) and N-nitrosodimethylamine in a human derived liver cell line (HepG2) Food Chem. Toxicol. 2005;43:433–441. doi: 10.1016/j.fct.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 143.Milligan J.R., Aguilera J.A., Paglinawan R.A., Ward J.F. Mechanism of DNA damage by thiocyanate radicals. Int. J. Radiat. Biol. 2000;76:1305–1314. doi: 10.1080/09553000050151574. [DOI] [PubMed] [Google Scholar]
- 144.Babbar N., Casero R.A., Jr. Tumor necrosis factor-alpha increases reactive oxygen species by inducing spermine oxidase in human lung epithelial cells: a potential mechanism for inflammation-induced carcinogenesis. Cancer Res. 2006;66:11125–11130. doi: 10.1158/0008-5472.CAN-06-3174. [DOI] [PubMed] [Google Scholar]