

Abstract
Murine MC3T3-E1 and MC-4 cells were stably transfected with −371/+70 bp of the murine cyclooxygenase-2 (COX-2) promoter fused to a luciferase reporter (Pluc371) or with Pluc371 carrying site-directed mutations. Mutations were made in (1) the cAMP response element (CRE) at −57/−52 bp, (2) the activating protein-1 (AP-1)–binding site at −69/−63 bp, (3) the nuclear factor of activated T-cells (NFAT)–binding site at −77/−73 bp, and (4) both the AP-1 and NFAT sites, which comprise a composite consensus sequence for NFAT/AP-1. Single mutation of CRE, AP-1, or NFAT sites decreased parathyroid hormone (PTH)–stimulated COX-2 promoter activity 40% to 60%, whereas joint mutation of NFAT and AP-1 abrogated the induction. On electrophoretic mobility shift analysis, PTH stimulated binding of phosphorylated CREB to an oligonucleotide spanning the CRE and binding of NFATc1, c-Fos, and c-Jun to an oligonucleotide spanning the NFAT/AP-1 composite site. Mutation of the NFAT site was less effective than mutation of the AP-1 site in competing binding to the composite element, suggesting that cooperative interactions of NFATc1 and AP-1 are more dependent on NFAT than on AP-1. Both PTH and forskolin, an activator of adenylyl cyclase, stimulated NFATc1 nuclear translocation. PTH- and forskolin-stimulated COX-2 promoter activity was inhibited 56% to 80% by calcium chelation or calcineurin inhibitors and 60% to 98% by protein kinase A (PKA) inhibitors. These results indicate an important role for the calcium-calcineurin-NFAT signaling pathway in the PTH induction of COX-2 and suggest that cross-talk between the cAMP/PKA pathway and the calcium-calcineurin-NFAT pathway may play a role in other functions of PTH in osteoblasts. © 2010 American Society for Bone and Mineral Research
Keywords: prostaglandin, MC3T3-E1, CREB, AP-1, protein kinase A
Introduction
Prostaglandins (PGs), especially PGE2, are abundantly produced by osteoblasts and can stimulate both formation and resorption of bone.(1) Cyclooxygenase (COX) catalyzes the committed step in the conversion of arachidonic acid, released from membrane phospholipids, to PGs. Although there are two kinds of COX, COX-1 and COX-2, the inducible enzyme, COX-2, is responsible for most PGs produced by osteoblasts.(1–3) Multiple factors can induce the expression of COX-2 in osteoblasts, including hormones, cytokines, and mechanical loading.(1)
Parathyroid hormone (PTH) is a major physiologic regulator of bone remodeling and calcium homeostasis. Although PTH stimulates both bone resorption and formation, PTH injected intermittently increases bone formation more than resorption. Recombinant human PTH(1-34) is the only anabolic therapy currently approved in the United States for treatment of osteoporosis.(4–6) PTH is a potent inducer of COX-2 in osteoblasts via the cyclic adenosine monophosphate (cAMP) protein kinase A (PKA) pathway.(7–9) Because PTH and PGE2 can have similar effects on bone remodeling, it is possible that some of the local effects of PTH may be mediated via the induction of COX-2 expression and PGE2 production.
Nuclear factor of activated T-cells (NFAT) is a family of transcription factors initially identified as regulating cytokine and early immune-response gene expression in T cells(10) and now known to be important in regulating gene expression and cell behavior in many nonimmune cells, including cardiomyocytes,(11) endothelial cells,(12,13) and glomerular mesangial cells.(14) There are five known NFAT members. NFAT5 is regulated by osmotic pressure. NFATc1 through NFATc4 are chiefly activated by the calcium-calcineurin pathway. Inactive, phosphorylated NFATs (c1–4) are located in the cytoplasm. When intracellular calcium ion concentration increases, calcineurin can be activated and dephosphorylate NFAT proteins, which then are translocated into the nucleus and become functional.(15) The activity of NFAT proteins may be modulated by interaction with transcription partners. NFAT proteins and AP-1 heterodimers (Fos/Jun) can bind cooperatively to a composite DNA site [GGA(N)9TCA] to activate gene expression.(16) NFATc1 plays an essential role in osteoclast differentiation,(17,18) and NFATc1 and NFATc2 are important in osteoblast differentiation.(19,20)
NFAT regulates COX-2 expression in several cell types. NFAT and AP-1 proteins were essential for increased COX-2 expression in T-lymphocytes in response to phorbol 12-myristate 13-acetate (PMA) and calcium ionophore.(21) NFAT also was shown to mediate the stimulation of COX-2 expression by vascular endothelial growth factor (VEGF) in endothelial cells(13) and by endothelin-1 in glomerular mesangial cells.(14)
In this study we show that PTH induces COX-2 transcription in murine osteoblastic cells via both CRE- and NFAT/AP-1–binding sites. This induction involves crosstalk between the cAMP-PKA pathway, which is thought to mediate the anabolic effects of PTH in bone, and the calcium-calcineurin-NFAT signaling pathway. Hence it is possible that some of the anabolic actions of PTH may involve the NFAT pathway.
Materials and Methods
Materials
The promoterless luciferase vector, the DNA constructs consisting of −371/+70 bp of the murine COX-2 gene (Pluc371), as well as 5′ deletions of the −371/+70 bp region, fused to a luciferase reporter gene in pXp-2 vector, have been described previously.(22–24) H-89, KT5720, and GF 109203X were purchased from Biomol (Plymouth Meeting, PA, USA). The NFAT-selective inhibitor VIVIT peptide was purchased from Calbiochem (San Diego, CA, USA). The PKA agonist 6-Bnz-cAMP and the Epac agonist 8-pCPT-2'-O-Me-cAMP were purchased from Biolog Life Science Institute (San Diego, CA, USA). The retroviral vector pMSCV-GFP and the pMSCV–constitutively active NFATc1 (caNFATc1)–GFP plasmid constructs were kind gifts of Dr Neil A Clipstone (Loyola University). Synthetic bovine PTH (1-34), forskolin, phorbol myristate acetate (PMA), and other chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Cell culture
MC3T3-E1 cells, immortalized from neonatal murine calvariae, were the kind gift of Dr Yoshiyuki Hakeda (Meikai University School of Dentistry, Sakado, Saitama, Japan). The MC-4 cell line, subcloned from MC3T3-E1 cells by Dr Franceschi at the University of Michigan,(25) was purchased from the American Type Culture Collection (CRL-2593, ATCC, Manassas, VA, USA). MC3T3-E1 cells were grown in phenol red–free Dulbecco's modified Eagle's medium (DMEM, Sigma-Aldrich), and MC-4 cells were grown in α modified Eagle's medium (α-MEM, (Invitrogen, Carlsbad, CA, USA). Both media contained 10% heat-inactivated fetal calf serum (FCS, Gibco, BRL, Gaithersburg, MD, USA), penicillin (100 U/mL), and streptomycin (50 µg/mL). Cells were plated in 6 well dishes at 5000/cm2 and grown until confluent in a humidified atmosphere of 5% CO2 at 37°C. MC3T3-E1 cells were changed to serum-free medium with 1% BSA 24 hours before treatment. Treatments were pulsed into MC-4 cultures without changing the medium to avoid the COX-2 induction effects of fresh serum. Inhibitors were pulsed 1 hour before treatment with PTH or forskolin.
Stable transfection
Stable transfections of MC3T3-E1 or MC-4 cells were performed as described previously.(26) After selection, colonies (>200) were pooled to minimize effects secondary to variable integration sites. Cells were grown in culture medium containing 200 µg/mL of G418 (Invitrogen). To maintain uniform cell phenotype, all constructs to be studied were transfected at the same time. Passage number after transfection was restricted to less than 10.
Luciferase activity
Luciferase activity was measured in soluble cell extracts prepared with a kit from Promega (Madison, WI, USA) using an automatic injection luminometer (Berthold Lumat, Wallac, Inc., Oak Ridge, TN, USA) For each experiment, 3 wells of a 6 well dish of cells were analyzed per treatment group. Luciferase activity was measured as relative light units per second (RLU/s) and normalized to total protein measured with a bicinchoninic acid (BCA) protein assay kit (Thermo Scientific, Rockford, IL, USA). Using these normalized values, fold induction of luciferase activity was calculated as the ratio of each sample to the mean activity for the appropriate control group.
Site-directed mutation
The template sequence for all mutations was the murine −371/+70 bp COX-2 DNA construct. The AP-1 sequence (5'-AGAGTCA-3') at −69/−63 bp was changed to 5'-AGAGTtg-3' (muAP-1) as described previously.(26) The CRE (5'-CGTCA-3') at −56/−52 bp was changed to 5'-atTCA-3' (muCRE) as described previously.(26,27) The NFAT sequence (5'-GGAAA-3') at −77/−73 bp was changed to 5'-ttAAA-3' (muNFAT) using the QuickChange Site-Directed Mutagenesis Kit from Stratagene (La Jolla, CA, USA) following the manufacturer's directions. After introduction of deletions or mutations, the COX-2 5'-flanking region was sequenced (Automated DNA Sequence Facility, University of Connecticut Health Center, Farmington, CT, USA). Mutated oligonucleotides were used as unlabeled competitors on electrophoretic mobility gel-shift assay (EMSA) to confirm that binding to the mutated sequence did not occur.
Electrophoretic mobility gel-shift assay (EMSA)
Cells were washed with PBS and collected by centrifugation, and nuclear extracts were obtained using a kit (BioVision Research Prodicts, Mountain View, CA, USA). Single-stranded oligonucleotides (Integrated DNA Technologies, Coralville, IA, USA) were annealed with complementary oligonucleotides, and the resulting double-stranded DNAs were end labeled with γ32P-ATP (PerkinElmer, Waltham, MA, USA) using T4 kinase (Invitrogen). The 6 µg of nuclear extract was incubated in 20 µL binding-reaction mixture [10 mM Tris HCl, pH 7.5, 1 mM DTT, 1 mM EDTA, 5% glycerol, and 2 µg poly dI-dC (Amersham Biosciences, Piscataway, NJ, USA)] with 50,000 cpm of purified labeled probe. Nondenaturing acrylamide gel (5%) electrophoresis was performed for 2 hours. Competitors (50 to 300 M excess) or supershifting antibodies (4 µg) were added to the binding mixture 30 minutes before addition of the probe, and incubation was continued for 30 minutes at 4°C. Antibodies to NFATc1-c4, c-Fos or c-Jun, and phosphorylated CREB were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Normal mouse and rabbit IgGs were purchased from Millipore (Billerica, MA, USA). Dried gels were exposed to X-ray film or phosphor image plate.
Western blot analysis
Nuclear and cytosolic extracts were obtained with a fractionation kit from BioVision. Protein concentrations were measured by BCA assay (Thermo Scientific). Equal amounts (25 or 40 µg) of protein were used for 10% SDS-PAGE and transferred to nitrocellulose membrane (Bio-Rad Lab, Helcules, CA, USA). Membranes were washed with Tris-buffered saline (TBS, pH 7.6), blocked with 5% wt/vol nonfat dry milk in 1 × TBS containing 0.05% Tween 20 (TBST) overnight at 4°C, and incubated with primary antibody (Santa Cruz) at 1:200 dilution. After washing with TBST, membranes were incubated with horseradish peroxidase–conjugated secondary antibody at 1:1,000 dilution. The signal was detected with LumiGLO chemiluminescent reagent (Cell Signaling Technology, Danvers, MA, USA).
Real-time (quantitative) PCR analysis
Gene expression studies were done using TaqMan fluorogenic probes. Total RNA was extracted with Trizol (Invitrogen) following the manufacturer's instructions. Then 5 µg of total RNA was converted to cDNA using the high-capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA, USA) according to the manufacturer's directions. Real-time PCR for COX-2 expression was performed in 96 well plates using the Assays-on-Demand gene expression system (Applied Biosystems) with TaqMan fluorogenic probes and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as the endogenous control. Each sample was amplified in duplicate. The PCR reaction mixture (20 mL/well including 2 × TaqMan Universal PCR Master Mix, 20 × Assays-on-Demand Gene Expression assay mix, and 100 ng of cDNA) was run in the ABI Prism 7300 Sequence machine universal thermal cycling parameters. The standard-curve method was used for data analysis.
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was performed using the EZ-CHIP Kit from Millipore following the manufacturer's instructions. MC-4 cells were grown to confluency, treated for 1 hour with PTH (10 nM) or vehicle, cross-linked with formaldehyde, lysed, sonicated with six sets of 20-second pulses, and soluble chromatin harvested. Supernatant was precleared with protein G agarose and purified by centrifugation. Immunoprecipitation was performed by incubating the samples overnight at 4°C with 2 µg of mouse NFATc1 antibody (Santa Cruz) or 1 µg anti-RNA polymerase antibody (positive control) or 1 µg of mouse IgG antibody (negative control), followed by incubation with protein G agarose for 1 hour at 4°C to collect antibody-antigen-DNA complexes. Protein-DNA complexes were eluted from the agarose and reverse cross-linked to free the DNA, which was then purified using spin columns. The DNA solution also was used as the template for SYBR Green quantitative polymerase chain reaction (PCR). Specific primers encompassing the NFAT binding sites on the COX-2 promoter (from nucleotide −135 to −4 bp) were designed using the NCBI/Primer-BLAST program (http://www.ncbi.nlm.nih.gov/BLAST). Experimentally validated murine primer pairs for actin (ID 6671509a1) were found in PrimerBank (http://pga.mgh.harvard.edu/primerbank). Primers were made by Integrated DNA Technologies. The PCR reaction mixture for SYBR Green assays contained 2× SYBR Green PCR Master Mix (Applied Biosystems) forward and reverse primer mix at final concentration for each primer of 200 nM and 2 µL of DNA solution. The reaction was run in triplicate for each sample under a common PCR thermal profile: 50°C for 2 minutes, 95°C for 10 minutes, 95°C for 15 seconds, 60°C for 30 seconds, and 72°C for 30 seconds. PCR products were analyzed by gel electrophoresis (37 cycles) and by real-time PCR (40 cycles). For real-time PCR, we started with equal amounts of chromatin for each sample, and the Ct values for each sample were calculated without normalization. The ChIP assay was done in duplicate.
Statistical analysis
Means of groups were compared by one-way analysis of variance (ANOVA) using SigmaStat for Windows, Version 2.03 (San Rafael, CA, USA). Significance of differences was determined by post hoc testing using Bonferonni t test or the Tukey test.
Results
Site-directed mutation of CRE, AP-1, and NFAT elements
Studies were done in both preosteoblastic MC3T3-E1 cells and MC-4 cells. MC-4 cells were subcloned from MC3T3-E1 cells because of concerns that MC3T3-E1 cells had become a mixed population of phenotypes after years of being carried by various laboratories and were shown to have high differentiation and mineralization potential.(25) All studies were done in stably transfected cells grown for 6 to 7 days after plating. PTH has been shown to rapidly induce transcription and expression of COX-2 in MC3T3-E1 cells.(8,9) In both MC3T3-E1 and MC-4 cells stably transfected with Pluc371, PTH-stimulated luciferase activity peaked at 2 to 3 hours and returned to baseline by 8 hours (1A, B). We used the 3 hour time point for all further studies of luciferase activity.
Fig. 1.

Induction of murine COX-2 promoter activity by PTH in MC3T3-E1 and MC-4 cells is dependent on the CRE, AP-1, and NFAT sites. Cells stably transfected with wild-type (WT) Pluc371 or Pluc371 with 2 bp mutations in the CRE, AP-1, and NFAT sites were treated with vehicle or PTH (10 nM). Unless otherwise specified, cells were treated for 3 hours. Luciferase activity was calculated as relative light units (RLU)/s and normalized to total protein. Using the normalized values, fold induction of luciferase activity was calculated as the ratio of each sample activity to the mean activity for the appropriate control group. Time course for the PTH induction of COX-2 promoter luciferase reporter activity in (A) MC3T3-E1 cells and in (B) MC-4 cells carrying Pluc371. (C) Location in the murine COX-2 5′-flanking region of the mutated cis-acting elements. (D) Comparison of nucleotide sequences in the −80/−60 bp COX-2 5'-flanking regions of the human, rat and mouse genes. (E) Effects of mutating the CRE, AP-1, or both CRE and AP-1 sites in MC3T3-E1 cells on basal luciferase activity and (F) PTH-stimulated fold increase in luciferase activity. (G) Effects of mutating the AP-1, NFAT, or both AP-1 and NFAT sites in MC3T3-E1 cells on basal luciferase activity, and (H) PTH-stimulated fold increase in luciferase activity. (I) Effects of mutating the CRE, AP-1, and NFAT sites in MC-4 cells on basal luciferase activity, and (J) PTH-stimulated fold increase in luciferase activity. Bars or symbols are means ± SEM for n = 3 wells of cells. aSignificant effect of PTH, p < .01. bSignificantly different from WT, p < .01. cp < .05. dSignificantly different from all other mutations, p < .01. eSignificantly different from the single NFAT mutation, p < .05.
We made mutations in the cAMP response element (CRE) that is important for the regulation of COX-2 expression by multiple agents,(27) an AP-1-binding site that partially mediates effects of PMA and serum on COX-2,(2) and an NFAT consensus sequence(21) (see 1C). The sequence 5′-GGAAAGACAGAGTCA-3 spanning the NFAT and AP-1 sites is identical to an NFAT/AP-1 composite consensus sequence GGA(N)9TCA(12) and is conserved among the human, rat, and mouse genomic sequences(21) (see 1D). NFAT and AP-1 (Fos/Jun) proteins have been shown to interact cooperatively to mediate transcription.(12,28,29) We compared mutated constructs only in cells transfected simultaneously with those constructs.
Mutations of the CRE, the AP-1 site, and both sites were compared in MC3T3-E1 cells. A representative experiment is shown in 1E, F. PTH (10 nM) significantly induced luciferase activity 7.2-fold in one experiment (see 1F) and 3.3- and 5.3-fold in two other experiments (data not shown). AP-1 and CRE mutations significantly decreased the PTH-stimulated fold activity on average 60% and 50%, respectively. There was no additional reduction in the response to PTH with the combination of AP-1 and CRE mutations in any of the three experiments. Although the basal activity was variable among experiments, the joint AP-1 and CRE mutation consistently reduced basal luciferase activity (on average 76%) more than the individual mutations (40% to 46%).
Mutations of the AP-1 site, the NFAT site, and both sites also were compared in MC3T3-E1 cells. A representative experiment is shown in 1G, H. PTH significantly induced luciferase activity 5.6-fold (see 1H) in one experiment and 3.8- and 8.9-fold in the other two experiments (data not shown). Single mutation of the AP-1 site significantly reduced the PTH stimulated fold induction in two of the three experiments by 39%. Single mutation of the NFAT site significantly reduced the fold induction of PTH in all three experiments by 18% to 40%. In contrast, the PTH-stimulated fold increase was decreased 82%, 94%, and 96% by the joint AP-1 and NFAT mutations. The mutations produced no consistent differences in basal activity.
Mutations of the CRE, the AP-1 site, the NFAT site, and both the AP-1 and NFAT sites were compared in MC-4 cells. A representative experiment is shown in 1I, J. In this experiment, PTH induced a 10-fold increase in luciferase activity that was reduced 61%, 54%, 64%, and 93% by the CRE, AP-1, NFAT, and joint AP-1 and NFAT mutations, respectively. In a second experiment, PTH induced a 5.8-fold increase in luciferase activity that was significantly reduced 50%, 48%, 38%, and 98%, respectively, by the CRE, AP-1, NFAT, and joint AP-1 and NFAT mutations, respectively (data not shown). In a third experiment, PTH induced a 7-fold increase in luciferase activity that was significantly reduced 48%, 50%, and 94% by the AP-1, NFAT, and combined mutations, respectively (data not shown).
In the six independent experiments in MC3T3-E1 and MC-4 cells, there was no statistically significant induction of luciferase activity by PTH in any group with joint AP-1 and NFAT mutations, suggesting that the composite NFAT/AP-1 element plays a critical role in the PTH induction of COX-2 promoter activity. As noted earlier, basal activity was quite variable even in repeated assays on cells carrying the same construct, suggesting that despite pooling of multiple clones for the transfection, some phenotypic differences occurred during the selection process or during passage. The joint NFAT/AP-1 mutation increased basal activity significantly in three experiments, reduced basal activity significantly in two experiments, and had no effect on basal activity in the remaining experiment. Hence the abrogation of PTH-stimulated luciferase activity by the combined AP-1 and NFAT mutations was independent of effects on basal activity.
Binding of nuclear proteins to CRE, AP-1, and NFAT elements
We performed EMSA on MC-4 cells treated with vehicle or PTH (10 nM) for 1 hour. There was constitutive binding of a 32P-labeled COX-2 oligonucleotide (−63/−39 bp) spanning the CRE site on EMSA (2A, band a in lanes 2, 3, 8, and 9), with increased binding seen on some gels after PTH treatment. This binding was competed by unlabeled oligonucleotide (lanes 4 and 5) but not by unlabeled oligonucleotide with mutation of the CRE (MUT; lanes 6 and 7). Antibody to phosphorylated CREB (pCREB) supershifted a band (lane 11, band b) in PTH-treated nuclear extracts but not in vehicle-treated samples (lane 10). No supershift was observed in extracts incubated with nonspecific IgG (lanes 12 and 13).
Fig. 2.

PTH induces binding of nuclear proteins to the CRE, NFAT, and AP-1 elements. EMSA and ChIP analyses were performed on MC-4 cells treated with PTH (10 nM) for 1 hour. (A) Binding of nuclear proteins extracted from MC-4 cells to a 32P-labeled COX-2 oligonucleotide (−63/−39 bp) spanning the CRE (−57/−52 bp). Binding (arrow a, lanes 2, 3, 8, and 9) was competed by unlabeled wild-type (WT) oligonucleotide (lanes 4 and 5) but not by oligonucleotide with 2 bp mutation of the CRE (lanes 6 and 7) and supershifed by antibody to phoshorylated CREB (pCREB) (arrow b, lane 11) but not by IgG (lane 13). Lane 1: Free probe. (B) Binding of nuclear proteins extracted from MC-4 cells to a 32P-labeled COX-2 oligonucleotide (−85/−57 bp) spanning the composite NFAT/AP-1 element (−77/−63 bp). PTH-induced binding (arrow a, lanes 2 and 3) was competed by unlabeled WT oligonucleotide (lanes 4 and 5) but not by oligonucleotide with mutation of the AP-1 and NFAT sites (lanes 6 and 7). Supershift was seen with antibody to NFATc1 (arrow b, lane 11) but not with antibodies to NFATc2–4 or IgG (lanes 13, 15, 17, and 19). Supershift also was seen with antibodies to c-Fos (arrow c, lane 21) and c-Jun (lane 23). Lane 1: Free probe. (C) ChIP assay for binding of NFATc1 to the native COX-2 promoter. Soluble chromatin extracts from MC-4 cells treated with vehicle or PTH were incubated with anti-NFATc1 antibody, nonimmune IgG, or no antibody (input). Immunoprecipitated DNA was analyzed by PCR using primers spanning −135/−4 bp of the COX-2 promoter region. PCR products were run on 2% agarose gel and stained with ethidium bromide.
PTH induced binding of a 32P-labeled oligonucleotide (−85 to −57 bp) that spanned the NFAT and AP-1 composite site (see 2B, lane 3, band a). This binding was competed by unlabeled oligonucleotide (lanes 4 and 5) but not by unlabeled oligonucleotide with mutations in both AP-1 and NFAT sites (lanes 6 and 7). Antibody to NFATc1 supershifted a band in PTH-treated extracts (lane 11, band b). In contrast, antibodies to NFATc2 (lanes 12 and 13), NFATc3 (lanes 14 and 15), and NFATc4 (lanes 16 and 17) did not alter the position of this band. Antibodies to c-Fos and c-Jun also gave rise to retarded bands (lanes 21 and 23, respectively). No supershifted band was seen with IgG (lane 19). Similar results were seen with EMSA in MC3T3-E1 cells (data not shown).
To demonstrate binding of NFATc1 to native chromatin in MC-4 cells, we performed ChIP using primers spanning −135/−4 bp of the COX-2 promoter region. PCR products run on agarose gel and stained with ethidium bromide are shown in 2C. Products also were analyzed by real-time PCR. The difference in Ct values between control and PTH-treated samples was 4.8 in one experiment and 3.9 in a second experiment, indicating an increase in PTH-stimulated NFATc1 binding of about 20-fold. We cannot rule out binding to several other potential NFAT elements in the −135/−4 bp promoter sequence, which spans two additional GGAAA sequences at −90/−86 and −111/−107 bp. However, mutation of either of these sites in combination with mutation of the AP-1 site studied in stably transfected MC-4 cells resulted in no greater decrease in PTH-stimulated luciferase activity than the AP-1 mutation alone (data not shown).
To study the cooperative binding of NFAT and AP-1 proteins to the composite NFAT/AP-1 element, we compared the ability of oligonucleotides carrying the single AP-1 mutation (muAP-1) or NFAT mutation (muNFAT) to compete the binding of nuclear proteins to the NFAT/AP-1 element, as described previously(29) (Fig. 3). Unlabeled oligonucleotide with muAP-1 (lanes 16 to 18) was almost as effective as the wild-type oligonucleotide (lanes 4 to 6) at competing for the formation of the protein-DNA complexes and much more effective than the oligonucleotide with muNFAT (lanes 22 to 24). These observations suggest that interactions at the composite NFAT/AP-1 element are more dependent on NFAT than on AP-1.
Fig. 3.

Mutating the NFAT element has a greater effect on nuclear protein binding to the composite NFAT/AP-1 sequence than mutating the AP-1 element. EMSA was performed on nuclear extracts from MC-4 cells treated with PTH (10 nM) for 1 hour using a 32P-labeled COX-2 oligonucleotide (−85/−57 bp) spanning the composite NFAT/AP-1 element (−77/−63 bp). Unlabeled wild-type oligonucleotide (WT, lanes 4 to 6) or unlabeled oligonucleotide with mutations in both AP-1 and NFAT (lanes 10 to 12), mutations in AP-1 only (muAP-1, lanes 16 to 18) or NFAT only (muNFAT, lanes 22 to 24) were used at 50, 100, and 300 M excess. Lanes 1, 7, 13, and 19: Free probe.
Involvement of the cAMP-PKA pathway
PTH exerts its effect on bone metabolism by binding to the G protein–coupled PTH/PTH-related peptide receptor.(30,31) PTH binding to this receptor stimulates adenylate cyclase through Gαs protein, thereby increasing cAMP and activating PKA. Although PTH is thought to mediate most of its biologic effects in osteoblastic cells via cAMP-PKA signaling, PTH also can activate the protein kinase C (PKC) pathway.(32,33) H-89, a PKA inhibitor, inhibits PTH induction of COX-2 transcription and COX-2 mRNA and protein expression in MC3T3-E1 cells.(8,9) However, H-89 also strongly inhibits the induction of COX-2 by PMA, a PKC agonist.(34–36) To rule out involvement of the PKC pathway, we examined effects of a specific PKC inhibitor GF 109203X on the induction of luciferase activity in MC-4 cells in response to PMA (1 µM) and forskolin (1 µM), an adenylate cyclase agonist. As reported previously,(34–36) GF 109203X inhibited the PMA induction (2.6-fold) of luciferase activity by 80% (data not shown), with no effect on the forskolin induction (7.1-fold) of luciferase activity (data not shown). GF 109203X had no effect on the PTH induction of luciferase activity (4A). Consistent with earlier studies, H-89 abrogated the PTH induction of luciferase activity (4B). A more specific PKA inhibitor, KT 5720,(37) significantly inhibited the forskolin induction (7.9-fold) of luciferase activity by 64% (data not shown) and the PTH induction of luciferase by 60% (see 4B).
Fig. 4.
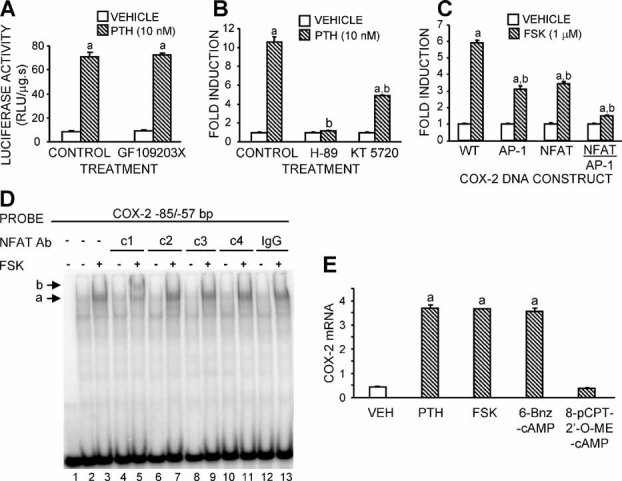
PTH induction of COX-2 promoter activity depends on the cAMP-PKA pathway. (A, B) Effects of the specific PKC inhibitor GF109203X (0.6 µM) and the PKA inhibitors H-89 (30 µM) and KT 5720 (10 µM) on the luciferase activity or fold induction in response to PTH. MC-4 cells stably transfected with Pluc371 were treated with vehicle, PTH, or forskolin (FSK). Inhibitors or vehicle (Control) were added 1 hour before PTH. (C) Effects of mutating the AP-1 site, the NFAT site, and both on the response to forskolin. MC-4 cells stably transfected with wild-type (WT) Pluc371 or Pluc371 with mutations in the AP-1 and NFAT sites were treated for 3 hours with vehicle or forskolin (FSK). Luciferase activity was calculated as relative light units (RLU)/s and normalized to total protein. Using the normalized values, fold induction of luciferase activity was calculated as the ratio of each sample to the mean activity for the appropriate control group. Bars are means ± SEM for n = 3 replicates. aSignificant effect of PTH or forskolin, p < .01. bSignificant effect of inhibitor or mutation, p < .01. (D) EMSA of forskolin-induced nuclear binding to a 32P-labeled COX-2 oligonucleotide (−85/−57 bp) spanning the composite NFAT/AP-1 element (−77/−63 bp). MC-4 cells were treated with forskolin (FSK, 1 µM) for 1 hour. Forskolin-induced binding (arrow a, lane 3) was supershifted by antibody to NFATc1 (arrow b, lane 5) but not by antibodies to NFATc2–4 (lanes 7, 9, and 11) or IgG (lane 13). Lane 1: Free probe. (E) Effects of specific PKA and Epac agonists on COX-2 mRNA expression, measured by real-time RT-PCR. MC-4 cells were treated with vehicle (VEH), PTH (10 nM), forskolin (FSK, 1 µM), 6-Bnz-cAMP (1 mM), and 8-pCPT-2′-O-Me-cAMP (1 mM) for 1 hour. Bars are means ± SEM for n = 3 replicates. aSignificant effect of agonist, p < .01.
Similar to the effects seen with PTH, the forskolin induction of luciferase activity in stably transfected MC-4 cells was inhibited 50% to 60% by mutation of the AP-1 or NFAT site and abrogated by joint mutation of these two sites (see 4C). On EMSA, forskolin treatment induced binding (band a in lane 3) that was supershifted by antibody to NFATc1 (band b in lane 5) but not by antibodies to other members of the NFAT family (see 4D).
Exchange protein directly activated by cAMP (Epac) is another target of cAMP that mediates effects of cAMP independent of the PKA pathway. Epac is expressed in MC-4 cells and was shown to mediate effects of PTH on cell proliferation.(38) We compared treatment with high doses (1 mM) of the specific PKA agonist 6-Bnz-cAMP and the specific Epac agonist 8-pCPT-2′-O-Me-cAMP on COX-2 mRNA expression in MC-4 cells.(39) 6-Bnz-cAMP induced COX-2 mRNA more than 8-fold, similar to induction by PTH or forskolin, whereas 8-pCPT-2′-O-Me-cAMP had no effect on COX-2 message (see 4E).
Regulation of calcium-calcineurin-NFAT signaling by the cAMP-PKA pathway
Increased concentration of intracellular calcium activates calcineurin, and calcineurin activates NFATc1 protein in the cytoplasm by dephosphorylation.(15) Dephosphorylated NFAT1c translocates to the nucleus. Consequently, cellular localization of NFAT1c is an indication of its activation status. We examined by Western blot the distribution of NFAT proteins in the cytoplasm and nucleus in MC-4 cells treated with vehicle or PTH. Nuclear concentration of NFATc1 was increased by PTH (5A). Forskolin also stimulated nuclear translocation of NFATc1, but PMA did not (5B).
Fig. 5.

NFATc1 nuclear translocation is induced by PTH and forskolin but not by PMA in MC-4 cells. MC-4 cells were treated for 30 to 60 minutes. Factionated nuclear and cytoplasmic proteins were extracted as described in “Materials and Methods.” (A) NFATc1 nuclear translocation induced by treatment with PTH (10 nM). (B) NFATc1 nuclear translocation induced by 1 hour of treatment with vehicle (VEH), forskolin (FSK, 1 µM), or PMA (1 µM). β-actin and histone H1 are shown to assess loading on gel.
PTH-stimulated COX-2 mRNA expression in MC-4 cells was inhibited inhibited 88% by the calcium chelator BAPTA-AM and 58% and 65%, respectively, by the calcineurin inhibitors FK506 and cyclosporin A (CsA) (6A). PTH-stimulated Pluc371 activity in MC-4 cells was inhibited 99% by BAPTA-AM and 56% to 60% by the calcineurin inhibitors FK506, CsA, and VIVIT in the experiment shown in 6B. In a second experiment, PTH-stimulated Pluc371 activity was inhibited 81% by BAPTA-AM and 76% to 81% by FK506 and CsA (p < .01; data not shown). Forskolin-stimulated Pluc371 activity was inhibited 76% by BAPTA-AM and 64% to 65% by FK506 and CsA (see 6C). These data indicate that PTH can stimulate COX-2 mRNA and promoter activity via crosstalk between the cAMP-PKA and calcineurin-NFAT signaling pathways.
Fig. 6.

PTH induction of COX-2 expression and promoter activity depends on calcium-calcineurin signaling. MC-4 cells stably transfected with Pluc371 were treated with vehicle, PTH (10 nM), or forskolin (FSK, 1 µM) for 1 hour (mRNA) or 3 hours (luciferase activity). BAPTA-AM (BAPTA, 15 µM), FK506 (10 µg/mL), cyclosporin A (CsA, 10 µg/mL), or VIVIT (1 µM) were added 1 hour before PTH or forskolin. Vehicle (CON) for the inhibitors, DMSO, also was added 1 hour before PTH or forskolin. Luciferase activity was normalized to total protein and used to calculate the fold induction as the ratio of treatment to control. (A) Effects of the calcium chelator BAPTA-AM and the calcineurin inhibitors FK506 and CsA on PTH-induced COX-2 mRNA expression measured by real-time RT-PCR. (B) Effect of BAPTA-AM and the calcineurin inhibitors FK506, CsA, and VIVIT on the PTH induction of luciferase activity. (C) Effect of BAPTA-AM, FK506, and CsA on the forskolin induction of luciferase activity. Bars are means ± SEM for n = 3 wells of cells. aSignificant effect of agonist, p < .01. bSignificant effect of inhibitor, p < .01.
Enhancement of COX-2 mRNA expression by NFATc1 overexpression
We stably transfected MC-4 cells with a constitutively active mutant form of NFATc1 (caNFATc1) or its control vector (pMSCV-GFP).(40) Nuclear expression of NFATc1 protein was increased in cells stably transfected with caNFATc1 compared with control cells (7A). Basal and PTH-induced COX-2 mRNA expression was increased 3.3- and 1.6-fold, respectively, in the caNFATc1 expressing cells compared with control cells (7B).
Fig. 7.

NFATc1 overexpression enhances basal COX-2 expression and induction by PTH. MC-4 cells stably transfected with pMSCV-GFP (vector) or pMSCV-caNFATc1-GFP plasmid constructs were treated with vehicle or PTH (10 nM) for 1 hour. (A) Nuclear protein extraction and Western blot were performed as described in “Materials and Methods.” (B) COX-2 mRNA levels measured by real-time RT-PCR. Bars are means ± SEM for n = 3 wells of cells. aSignificant effect of PTH, p < .01. bSignificant effect of caNFATc1 overexpression, p < .01.
Discussion
Our results are consistent with earlier studies that reported the PTH induction of COX-2 expression and promoter activity to be mediated via the cAMP-PKA pathway.(8,9) As expected, the CRE, which is involved in the induction of COX-2 promoter activity by multiple factors,(26,27,41) was found to play a significant role in the PTH induction of COX-2 promoter activity. PTH increased binding of phosphorylated CREB-1 to the CRE on EMSA, and a 2-bp mutation in the CRE reduced PTH-stimulated COX-2 promoter-luciferase activity 50% to 60%.
Transcriptional regulation of COX-2 by most factors appears to involve multiple cis-acting sites.(1,42) Along with the CRE, the AP-1-binding element at −69/−63 bp has been shown to mediate induction of COX-2 by serum and PMA.(26) PTH induced c-Fos and c-Jun binding to the oligonucleotide spanning the AP-1-binding site on EMSA, and a 2-bp mutation of this AP-1 element reduced PTH-stimulated luciferase activity 40% to 60%. A role for AP-1 factors in mediating PTH effects has been found in several other systems. For example, PTH can induce collagenase expression(43,44) and c-Fos expression(45) via AP-1 factors.
The reduction in PTH stimulated COX-2 promoter activity by a 2 bp mutation of the NFAT element alone ranged from 18% to 64%. The joint mutation of the AP-1 and NFAT elements, which form a consensus NFAT/AP-1 composite binding sequence, consistently abrogated the PTH induction of COX-2 promoter activity. EMSA showed that PTH stimulated NFATc1 but not NFATc2–4 binding to a sequence spanning the composite site and that NFAT played a dominant role in the cooperative binding of NFAT and AP-1 proteins to the composite site. Binding of NFAT and AP-1 proteins to a composite NFAT/AP-1 site is reported to be important in the regulation of expression of cytokine and surface receptor genes in immunocytes(16,46) and in the induction of COX-2 by PMA and calcium ionophore in human T-lymphocytes(21) and by HIV-Tat in glial cells.(47) Our data indicate that this composite NFAT/AP-1 element is also important for COX-2 induction by PTH in osteoblasts and that constitutively active NFATc1 can enhance the basal and PTH-stimulated expression of COX-2 in osteoblast cultures.
Although regulation of NFAT activity has been shown to be involved in mediating COX-2 transcription by a variety of agents,(13,14,21,47,48) this is the first study to report regulation by an agent thought to act largely via the cAMP-PKA pathway. Previous studies on the relationship between the cAMP-PKA pathway and the calcium-calcineurin-NFAT pathway have reported differing results. PKA had inhibitory effects on NFAT activity in T-lymphocytes and mouse lymphoma EL-4 cells(49–51) but activated NFAT proteins in pancreatic islet β cells.(52,53) In this study, the adenylate cyclase agonist forskolin mimicked the effects of PTH, stimulating binding of nuclear proteins to the NFAT/AP-1 composite element and activating NFATc1 nuclear translocation. Inhibitors of PKA inhibited the stimulation of COX-2 promoter activity by PTH and forskolin similarly. Calcium chelation as well as calcineurin inhibitors significantly blunted effects of both PTH and forskolin on COX-2 promoter activity. PTH has been shown to activate ERK phosphorylation via Epac in MC-4 cells.(38) Although we cannot rule out a role for Epac, our data strongly suggest that PTH activates the calcium-calcineurin-NFAT pathway via cAMP-PKA signaling.
One possible mechanism for the association of cAMP-PKA and calcium-calcineurin activity in osteoblasts might be the family of scaffold proteins called A kinase-anchoring proteins (AKAPs). In a variety of cells, including T-lymphocyte, adipocytes, cardiomyocytes, and myocytes, PKA can form a signal-transduction complex with AKAPs located in different subcellular compartments through interactions with lipids or other proteins.(54) Our preliminary studies show expression of AKAP79 and AKAP18 proteins in MC-4 cells (data not shown). Some studies report that calcineurin can be a part of the AKAP complex and that PKA activated by cAMP can sensitize calcium channels on the cell compartment membrane, increasing the concentration of calcium ions in the cytosol, which, in turn, activates calcineurin and NFAT proteins.(55–57)
Although NFAT proteins were identified originally as controlling cytokine and early immune-response gene expression in T cells,(10) they are now found to regulate gene expression in a variety of cells.(11–14) NFATc1 plays a pivotal role in osteoclast differentiation.(17,18) NFAT proteins are also likely to be important in osteoblast differentiation and function. Although one study showed that overexpression of NFATc1 in vitro inhibited osteoblast differentiation,(58) other studies have shown that NFATc1 deficiency suppressed osteoblast differentiation in vitro(19); NFATc1 or NFATc2 deficiency caused a defect in bone formation in vivo(19,20); and NFATc1 overexpression in vivo resulted in increased osteoblast proliferation and high bone mass.(20)
PTH stimulates bone formation and resorption by acting on cells of the osteoblastic lineage to increase both their differentiation into mature matrix-producing cells and their essential support for the differentiation of osteoclasts. The results of our study show that PTH can activate the calcium-calcineurin-NFAT signaling pathway and that this activation is important for the PTH induction of COX-2 expression in osteoblasts. These results suggest that crosstalk between the cAMP/PKA pathway and the calcium-calcineurin-NFAT pathway may play a role in other functions of PTH that are important for the regulation of bone turnover.
Acknowledgments
This work was funded by NIH Grant DK48361 (CP). We are grateful for the careful reading and thoughtful comments of the reviewers, whose efforts greatly improved the quality of this manuscript.
Disclosures
All the authors state that they do not have any conflicts of interest.
References
- 1.Pilbeam C, Choudhary S, Blackwell K, Raisz LG. Prostaglandins and bone metabolism. In: Bilezikian JPRL, Martin TJ, editors. Principles of Bone Biology. 3rd ed. San Dirgo: Elsevier/Academic Press; 2008. pp. 1235–1271. [Google Scholar]
- 2.Smith WL, Garavito RM, DeWitt DL. Prostaglandin endoperoxide H synthases (cyclooxygenases)–1 and –2. J Biol Chem. 1996;271:33157–33160. doi: 10.1074/jbc.271.52.33157. [DOI] [PubMed] [Google Scholar]
- 3.Herschman HR, Reddy ST, Xie W. Function and regulation of prostaglandin synthase-2. Adv Exp Med Biol. 1997;407:61–66. doi: 10.1007/978-1-4899-1813-0_9. [DOI] [PubMed] [Google Scholar]
- 4.Rodan GA, Martin TJ. Therapeutic approaches to bone diseases. Science. 2000;289:1508–1514. doi: 10.1126/science.289.5484.1508. [DOI] [PubMed] [Google Scholar]
- 5.Potts JT, Gardella TJ. Progress, paradox, and potential: parathyroid hormone research over five decades. Ann NY Acad Sci. 2007;1117:196–208. doi: 10.1196/annals.1402.088. [DOI] [PubMed] [Google Scholar]
- 6.Girotra M, Rubin MR, Bilezikian JP. The use of parathyroid hormone in the treatment of osteoporosis. Rev Endocr Metab Disord. 2006;7:113–121. doi: 10.1007/s11154-006-9007-z. [DOI] [PubMed] [Google Scholar]
- 7.Kawaguchi H, Raisz LG, Voznesensky OS, Alander CB, Hakeda Y, Pilbeam CC. Regulation of the two prostaglandin G/H synthases by parathyroid hormone, interleukin-1, cortisol, and prostaglandin E2 in cultured neonatal mouse calvariae. Endocrinology. 1994;135:1157–1164. doi: 10.1210/endo.135.3.8070358. [DOI] [PubMed] [Google Scholar]
- 8.Tetradis S, Pilbeam CC, Liu Y, Kream BE. Parathyroid hormone induces prostaglandin G/H synthase-2 expression by a cyclic adenosine 3',5'-monophosphate-mediated pathway in the murine osteoblastic cell line MC3T3-E1. Endocrinology. 1996;137:5435–5440. doi: 10.1210/endo.137.12.8940368. [DOI] [PubMed] [Google Scholar]
- 9.Tetradis S, Pilbeam CC, Liu Y, Herschman HR, Kream BE. Parathyroid hormone increases prostaglandin G/H synthase-2 transcription by a cyclic adenosine 3',5'-monophosphate-mediated pathway in murine osteoblastic MC3T3-E1 cells. Endocrinology. 1997;138:3594–3600. doi: 10.1210/endo.138.9.5391. [DOI] [PubMed] [Google Scholar]
- 10.Shaw JP, Utz PJ, Durand DB, Toole JJ, Emmel EA, Crabtree GR. Identification of a putative regulator of early T-cell activation genes. Science. 1988;241:202–205. [PubMed] [Google Scholar]
- 11.Morel E, Marcantoni A, Gastineau M, Birkedal R, Rochais F, Garnier A, Lompre AM, Vandecasteele G, Lezoualc'h F. cAMP-binding protein Epac induces cardiomyocyte hypertrophy. Circ Res. 2005;97:1296–1304. doi: 10.1161/01.RES.0000194325.31359.86. [DOI] [PubMed] [Google Scholar]
- 12.Cockerill GW, Bert AG, Ryan GR, Gamble JR, Vadas MA, Cockerill PN. Regulation of granulocyte-macrophage colony-stimulating factor and E-selectin expression in endothelial cells by cyclosporin A and the T-cell transcription factor NFAT. Blood. 1995;86:2689–2698. [PubMed] [Google Scholar]
- 13.Hernandez GL, Volpert OV, Iniguez MA, Lorenzo E, Martinez-Martinez S, Grau R, Fresno M, Redondo JM. Selective inhibition of vascular endothelial growth factor-mediated angiogenesis by cyclosporin A: roles of the nuclear factor of activated T cells and cyclooxygenase 2. J Exp Med. 2001;193:607–620. doi: 10.1084/jem.193.5.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sugimoto T, Haneda M, Sawano H, Isshiki K, Maeda S, Koya D, Inoki K, Yasuda H, Kashiwagi A, Kikkawa R. Endothelin-1 induces cyclooxygenase-2 expression via nuclear factor of activated T-cell transcription factor in glomerular mesangial cells. J Am Soc Nephrol. 2001;12:1359–1368. doi: 10.1681/ASN.V1271359. [DOI] [PubMed] [Google Scholar]
- 15.Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003;17:2205–2232. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- 16.Macian F, Lopez-Rodriguez C, Rao A. Partners in transcription: NFAT and AP-1. Oncogene. 2001;20:2476–2489. doi: 10.1038/sj.onc.1204386. [DOI] [PubMed] [Google Scholar]
- 17.Hirotani H, Tuohy NA, Woo JT, Stern PH, Clipstone NA. The calcineurin/nuclear factor of activated T cells signaling pathway regulates osteoclastogenesis in RAW264.7 cells. J Biol Chem. 2004;279:13984–13992. doi: 10.1074/jbc.M213067200. [DOI] [PubMed] [Google Scholar]
- 18.Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, Saiura A, Isobe M, Yokochi T, Inoue J, Wagner EF, Mak TW, Kodama T, Taniguchi T. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3:889–901. doi: 10.1016/s1534-5807(02)00369-6. [DOI] [PubMed] [Google Scholar]
- 19.Koga T, Matsui Y, Asagiri M, Kodama T, de Crombrugghe B, Nakashima K, Takayanagi H. NFAT and Osterix cooperatively regulate bone formation. Nat Med. 2005;11:880–885. doi: 10.1038/nm1270. [DOI] [PubMed] [Google Scholar]
- 20.Winslow MM, Pan M, Starbuck M, Gallo EM, Deng L, Karsenty G, Crabtree GR. Calcineurin/NFAT signaling in osteoblasts regulates bone mass. Dev Cell. 2006;10:771–782. doi: 10.1016/j.devcel.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 21.Iniguez MA, Martinez-Martinez S, Punzon C, Redondo JM, Fresno M. An essential role of the nuclear factor of activated T cells in the regulation of the expression of the cyclooxygenase-2 gene in human T lymphocytes. J Biol Chem. 2000;275:23627–23635. doi: 10.1074/jbc.M001381200. [DOI] [PubMed] [Google Scholar]
- 22.Kujubu DA, Fletcher BS, Varnum BC, Lim RW, Herschman HR. TIS10, a phorbol ester tumor promoter-inducible mRNA from Swiss 3T3 cells, encodes a novel prostaglandin synthase/cyclooxygenase homologue. J Biol Chem. 1991;266:12866–12872. [PubMed] [Google Scholar]
- 23.Fletcher BS, Kujubu DA, Perrin DM, Herschman HR. Structure of the mitogen-inducible TIS10 gene and demonstration that the TIS10-encoded protein is a functional prostaglandin G/H synthase. J Biol Chem. 1992;267:4338–4344. [PubMed] [Google Scholar]
- 24.Bazan NG, Fletcher BS, Herschman HR, Mukherjee PK. Platelet-activating factor and retinoic acid synergistically activate the inducible prostaglandin synthase gene. Proc Natl Acad Sci U S A. 1994;91:5252–5256. doi: 10.1073/pnas.91.12.5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang D, Christensen K, Chawla K, Xiao G, Krebsbach PH, Franceschi RT. Isolation and characterization of MC3T3-E1 preosteoblast subclones with distinct in vitro and in vivo differentiation/mineralization potential. J Bone Miner Res. 1999;14:893–903. doi: 10.1359/jbmr.1999.14.6.893. [DOI] [PubMed] [Google Scholar]
- 26.Okada Y, Voznesensky O, Herschman H, Harrison J, Pilbeam C. Identification of multiple cis-acting elements mediating the induction of prostaglandin G/H synthase-2 by phorbol ester in murine osteoblastic cells. J Cell Biochem. 2000;78:197–209. [PubMed] [Google Scholar]
- 27.Xie W, Herschman HR. Transcriptional regulation of prostaglandin synthase 2 gene expression by platelet-derived growth factor and serum. J Biol Chem. 1996;271:31742–31748. doi: 10.1074/jbc.271.49.31742. [DOI] [PubMed] [Google Scholar]
- 28.Jain J, McCaffrey PG, Miner Z, Kerppola TK, Lambert JN, Verdine GL, Curran T, Rao A. The T-cell transcription factor NFATp is a substrate for calcineurin and interacts with Fos and Jun. Nature. 1993;365:352–355. doi: 10.1038/365352a0. [DOI] [PubMed] [Google Scholar]
- 29.Rooney JW, Sun YL, Glimcher LH, Hoey T. Novel NFAT sites that mediate activation of the interleukin-2 promoter in response to T-cell receptor stimulation. Mol Cell Biol. 1995;15:6299–6310. doi: 10.1128/mcb.15.11.6299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lanske B, Divieti P, Kovacs CS, Pirro A, Landis WJ, Krane SM, Bringhurst FR, Kronenberg HM. The parathyroid hormone (PTH)/PTH-related peptide receptor mediates actions of both ligands in murine bone. Endocrinology. 1998;139:5194–5204. doi: 10.1210/endo.139.12.6361. [DOI] [PubMed] [Google Scholar]
- 31.Juppner H, Abou-Samra AB, Freeman M, Kong XF, Schipani E, Richards J, Kolakowski LF, Jr, Hock J, Potts JT, Jr, Kronenberg HM, et al. A G protein-linked receptor for parathyroid hormone and parathyroid hormone-related peptide. Science. 1991;254:1024–1026. doi: 10.1126/science.1658941. [DOI] [PubMed] [Google Scholar]
- 32.Fujimori A, Cheng SL, Avioli LV, Civitelli R. Structure-function relationship of parathyroid hormone: activation of phospholipase-C, protein kinase-A and -C in osteosarcoma cells. Endocrinology. 1992;130:29–36. doi: 10.1210/endo.130.1.1727705. [DOI] [PubMed] [Google Scholar]
- 33.Swarthout JT, D'Alonzo RC, Selvamurugan N, Partridge NC. Parathyroid hormone-dependent signaling pathways regulating genes in bone cells. Gene. 2002;282:1–17. doi: 10.1016/s0378-1119(01)00798-3. [DOI] [PubMed] [Google Scholar]
- 34.Wadhwa S, Choudhary S, Voznesensky M, Epstein M, Raisz L, Pilbeam C. Fluid flow induces COX-2 expression in MC3T3-E1 osteoblasts via a PKA signaling pathway. Biochem Biophys Res Commun. 2002;297:46–51. doi: 10.1016/s0006-291x(02)02124-1. [DOI] [PubMed] [Google Scholar]
- 35.Mehrotra M, Saegusa M, Wadhwa S, Voznesensky O, Peterson D, Pilbeam C. Fluid flow induces Rankl expression in primary murine calvarial osteoblasts. J Cell Biochem. 2006;98:1271–1283. doi: 10.1002/jcb.20864. [DOI] [PubMed] [Google Scholar]
- 36.Choudhary S, Kumar A, Kale RK, Raisz LG, Pilbeam CC. Extracellular calcium induces COX-2 in osteoblasts via a PKA pathway. Biochem Biophys Res Commun. 2004;322:395–402. doi: 10.1016/j.bbrc.2004.07.129. [DOI] [PubMed] [Google Scholar]
- 37.Kase H, Iwahashi K, Nakanishi S, Matsuda Y, Yamada K, Takahashi M, Murakata C, Sato A, Kaneko M. K-252 compounds, novel and potent inhibitors of protein kinase C and cyclic nucleotide-dependent protein kinases. Biochem Biophys Res Commun. 1987;142:436–440. doi: 10.1016/0006-291x(87)90293-2. [DOI] [PubMed] [Google Scholar]
- 38.Fujita T, Meguro T, Fukuyama R, Nakamuta H, Koida M. New signaling pathway for parathyroid hormone and cyclic AMP action on extracellular-regulated kinase and cell proliferation in bone cells. Checkpoint of modulation by cyclic AMP. J Biol Chem. 2002;277:22191–22200. doi: 10.1074/jbc.M110364200. [DOI] [PubMed] [Google Scholar]
- 39.Christensen AE, Selheim F, de Rooij J, Dremier S, Schwede F, Dao KK, Martinez A, Maenhaut C, Bos JL, Genieser HG, Doskeland SO. cAMP analogue mapping of Epac1 and cAMP kinase: discriminating analogues demonstrate that Epac and cAMP kinase act synergistically to promote PC-12 cell neurite extension. J Biol Chem. 2003;278:35394–35402. doi: 10.1074/jbc.M302179200. [DOI] [PubMed] [Google Scholar]
- 40.Porter CM, Clipstone NA. Sustained NFAT signaling promotes a Th1-like pattern of gene expression in primary murine CD4+ T cells. J Immunol. 2002;168:4936–4945. doi: 10.4049/jimmunol.168.10.4936. [DOI] [PubMed] [Google Scholar]
- 41.Wadleigh DJ, Herschman HR. Transcriptional regulation of the cyclooxygenase-2 gene by diverse ligands in murine osteoblasts. Biochem Biophys Res Commun. 1999;264:865–870. doi: 10.1006/bbrc.1999.1606. [DOI] [PubMed] [Google Scholar]
- 42.Kang YJ, Mbonye UR, DeLong CJ, Wada M, Smith WL. Regulation of intracellular cyclooxygenase levels by gene transcription and protein degradation. Prog Lipid Res. 2007;46:108–125. doi: 10.1016/j.plipres.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Porte D, Tuckermann J, Becker M, Baumann B, Teurich S, Higgins T, Owen MJ, Schorpp-Kistner M, Angel P. Both AP-1 and Cbfa1-like factors are required for the induction of interstitial collagenase by parathyroid hormone. Oncogene. 1999;18:667–678. doi: 10.1038/sj.onc.1202333. [DOI] [PubMed] [Google Scholar]
- 44.Selvamurugan N, Chou WY, Pearman AT, Pulumati MR, Partridge NC. Parathyroid hormone regulates the rat collagenase-3 promoter in osteoblastic cells through the cooperative interaction of the activator protein-1 site and the runt domain binding sequence. J Biol Chem. 1998;273:10647–10657. doi: 10.1074/jbc.273.17.10647. [DOI] [PubMed] [Google Scholar]
- 45.McCauley LK, Koh AJ, Beecher CA, Rosol TJ. Proto-oncogene c-fos is transcriptionally regulated by parathyroid hormone (PTH) and PTH-related protein in a cyclic adenosine monophosphate-dependent manner in osteoblastic cells. Endocrinology. 1997;138:5427–5433. doi: 10.1210/endo.138.12.5587. [DOI] [PubMed] [Google Scholar]
- 46.Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol. 1997;15:707–747. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- 47.Blanco A, Alvarez S, Fresno M, Munoz-Fernandez MA. Extracellular HIV-Tat induces cyclooxygenase-2 in glial cells through activation of nuclear factor of activated T cells. J Immunol. 2008;180:530–540. doi: 10.4049/jimmunol.180.1.530. [DOI] [PubMed] [Google Scholar]
- 48.Flockhart RJ, Diffey BL, Farr PM, Lloyd J, Reynolds NJ. NFAT regulates induction of COX-2 and apoptosis of keratinocytes in response to ultraviolet radiation exposure. FASEB J. 2008 doi: 10.1096/fj.08-113076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Negulescu PA, Shastri N, Cahalan MD. Intracellular calcium dependence of gene expression in single T-lymphocytes. Proc Natl Acad Sci USA. 1994;91:2873–2877. doi: 10.1073/pnas.91.7.2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lacour M, Arrighi JF, Muller KM, Carlberg C, Saurat JH, Hauser C. cAMP up-regulates IL-4 and IL-5 production from activated CD4+ T cells while decreasing IL-2 release and NF-AT induction. Int Immunol. 1994;6:1333–1343. doi: 10.1093/intimm/6.9.1333. [DOI] [PubMed] [Google Scholar]
- 51.Tsuruta L, Lee HJ, Masuda ES, Koyano-Nakagawa N, Arai N, Arai K, Yokota T. Cyclic AMP inhibits expression of the IL-2 gene through the nuclear factor of activated T cells (NF-AT) site, and transfection of NF-AT cDNAs abrogates the sensitivity of EL-4 cells to cyclic AMP. J Immunol. 1995;154:5255–5264. [PubMed] [Google Scholar]
- 52.Lawrence MC, Bhatt HS, Easom RA. NFAT regulates insulin gene promoter activity in response to synergistic pathways induced by glucose and glucagon-like peptide-1. Diabetes. 2002;51:691–698. doi: 10.2337/diabetes.51.3.691. [DOI] [PubMed] [Google Scholar]
- 53.Lester LB, Faux MC, Nauert JB, Scott JD. Targeted protein kinase A and PP-2B regulate insulin secretion through reversible phosphorylation. Endocrinology. 2001;142:1218–1227. doi: 10.1210/endo.142.3.8023. [DOI] [PubMed] [Google Scholar]
- 54.McConnachie G, Langeberg LK, Scott JD. AKAP signaling complexes: getting to the heart of the matter. Trends Mol Med. 2006;12:317–323. doi: 10.1016/j.molmed.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 55.Pare GC, Bauman AL, McHenry M, Michel JJ, Dodge-Kafka KL, Kapiloff MS. The mAKAP complex participates in the induction of cardiac myocyte hypertrophy by adrenergic receptor signaling. J Cell Sci. 2005;118:5637–5646. doi: 10.1242/jcs.02675. [DOI] [PubMed] [Google Scholar]
- 56.Crabtree GR, Olson EN. NFAT signaling: choreographing the social lives of cells. Cell. 2002;109(Suppl):S67–79. doi: 10.1016/s0092-8674(02)00699-2. [DOI] [PubMed] [Google Scholar]
- 57.Dodge-Kafka KL, Kapiloff MS. The mAKAP signaling complex: integration of cAMP, calcium, and MAP kinase signaling pathways. Eur J Cell Biol. 2006;85:593–602. doi: 10.1016/j.ejcb.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 58.Yeo H, Beck LH, McDonald JM, Zayzafoon M. Cyclosporin A elicits dose-dependent biphasic effects on osteoblast differentiation and bone formation. Bone. 2007;40:1502–1516. doi: 10.1016/j.bone.2007.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]