

Abstract
To function optimally as vaccines, dendritic cells (DCs) must actively migrate to lymphoid organs and maintain a viable, mature state for sufficient time to effectively present their Ag to cognate T cells. Unfortunately, mature DCs rapidly lose viability and function after injection, and only a minority leaves the vaccine site and migrates to lymph nodes. We show that all of these functions can be enhanced in DCs by removal of IL-1R–associated kinase M (IRAK-M). We found that IRAK-M is induced in DCs by TLR ligation and that its absence from these cells leads to increased activation of the p38-MAPK and NF-κB pathways, which, in turn, improves DC migration to lymph nodes, increases their longevity, and augments their secretion of Th1-skewing cytokines and chemokines. These biological effects have immunological consequences. IRAK-M−/− DCs increase the proliferation and activation of Ag-specific T cells, and a single vaccination with Ag-pulsed, LPS-matured IRAK-M−/− DCs eliminates established tumors and prolongs the survival of EG7 or B16.f10 tumor-bearing mice, without discernible induction of autoimmune disease. Thus, manipulation of IRAK-M levels can increase the potency of DC vaccines by enhancing their Ag-presenting function, migration, and longevity.
Dendritic cells (DCs) are the most potent APCs known (1–3), and they are being increasingly exploited as vaccines for cancer (4–9). In one particularly successful trial, vaccination with idiotype-pulsed DCs yielded progression-free survival in 70% of treated B cell lymphoma patients (8). Unfortunately, most other clinical studies have been less successful, with objective tumor responses seen in only a minority of cases, underscoring the need for improvement (4, 9–12).
DC vaccines must fulfill three major requirements for induction of an optimal T cell response: migration to lymphoid tissues to present the immunizing Ag, acquisition and maintenance of a mature stimulatory phenotype, and longevity. Migration to lymph nodes requires acquisition of a migratory phenotype, including expression of the chemokine receptor, CCR7, which directs mature DCs to the T cell areas of lymphoid organs in response to the homeostatic chemokines CCL19 and CCL21 (13–16). However, clinical studies of DC vaccines have shown that <5% of DCs reach the lymph node, even if injected in close proximity (17); the remaining cells die in situ, effectively reducing the vaccine dose by >1 log. Although direct infusion of DCs into lymphatic vessels may overcome migratory deficiencies and improve antitumor immunity (4, 7, 18), this is technically challenging. After migration, DCs must retain their mature immunostimulatory phenotype and persist, so that they can continue to stimulate adequate numbers of T cells for sufficient time to eliminate infection or tumor. Most DC vaccines are matured ex vivo using combinations of cytokines and TLR ligands, but these maturation signals become attenuated following injection. Consequently, DCs succumb rapidly to endogenous inhibitors, and their immunostimulatory functions remain short-lived (19–21).
IL-1R–associated kinase M (IRAK-M) inhibits cytokine secretion in monocytes and macrophages (22). Its loss leads to hyperactivation of the innate immune system, and altered levels of IRAK-M have been associated with conditions such as osteoporosis, cirrhosis, and sepsis (23–25). We aimed to determine whether IRAK-M is also an inhibitor of DC functions and, if so, whether its absence in tumor Ag-expressing DC vaccines would result in enhanced activation of tumor Ag-specific immunity and improved tumor clearance. We show that IRAK-M is expressed in murine DCs and that abrogation of this single molecular target enhances activity through the NF-κB and p38-MAPK pathways after TLR ligation, and thereby it promotes DC migration to lymph nodes, maintains their maturity, and prolongs their survival. As a consequence, Ag-pulsed IRAK-M−/− DCs increase proliferation of Ag-specific CD4+ and CD8+ T cells in vivo and enhance antitumor activity.
Materials and Methods
Mice
C57BL/6, BALB/c, and B6(Cg)-Tyrc-2J/J mice were purchased from The Jackson Laboratory (Bar Harbor, ME). IRAK-M−/− mice were obtained from the laboratory of Richard A. Flavell (Yale University School of Medicine, New Haven, CT) and were described previously (22). Mice were maintained in a pathogen-free mouse facility at Baylor College of Medicine, according to institutional guidelines. This study was approved by the Institutional Animal Care and Use Committee of Baylor College of Medicine.
Peptides, chemicals, proteins, and cell lines
H2-Kb–restricted OT-I (SIINFEKL) and I-Ad–restricted OT-II (ISQAVHAAHAEINEAGR) (20) peptides were synthesized and purified by HPLC to >95% purity by Genemed Synthesis (San Antonio, TX). TRP2 (SVYDFFVWL) and gp100 (EGSRNQDWL) peptides were from ProImmune (Bradenton, FL). Peptides were dissolved in DMSO before final dilution in endotoxin-free PBS (Sigma-Aldrich, St. Louis, MO). Ammonium pyrrolidine dithiocarbamate was from EMD Biosciences (San Diego, CA). U0126 was from Cell Signaling Technology (Danvers, MA). SB203580 was from InvivoGen (San Diego, CA). Escherichia coli LPS and OVA protein were from Sigma-Aldrich. Recombinant human CCL-19 and CCL-21 were from PeproTech (Rocky Hill, NJ). Recombinant mouse CD40L was from R&D Systems (Minneapolis, MN). The EL4 thymoma cell line (H2-b) was obtained from American Type Culture Collection (Manassas, VA). The EG.7 thymoma cell line (H2-b) was kindly provided by D. Spencer (Baylor College of Medicine). The B16.f10 melanoma cell line (H2-b) was obtained from American Type Culture Collection. FACS Abs CD3, CD4, CD11c, CD86, CD80, MHC-II, CD40, IL-6, TNF-α, IFN-γ, and Annexin V were from BD Pharmingen (San Jose, CA); FACS Abs CCR7, CD8, GITRL, and OX40-L were from eBioscience (San Diego, CA).
Cell culture and flow-cytometric analysis
Mouse bone marrow-derived DCs (BMDCs) were obtained, as described (20), with some modifications. Bone marrow was flushed from hind limbs, passed through nylon mesh filters, and depleted of RBCs by incubation at room temperature in RBC Lysing Buffer (Sigma-Aldrich). Cells were maintained in HyClone RPMI 1640 (Logan, UT), supplemented with 10% FBS (Summit Biotechnology, Fort Collins, CO), nonessential amino acids, HEPES buffer, glutamax, β-ME, IL-4 (20 ng/ml), and GM-CSF (20 ng/ml; PeproTech) at 37°C, 5% CO2. After 48 h in culture, nonadherent cells were removed, and fresh media and cytokines were added. On day 5–6 of culture, >80% of cells expressed DC markers, as determined by FACS analysis (CD11c+, H2-kb+, CD8−, CD14−). To avoid possible activation by FBS, DCs were incubated overnight in Cellgenix DC media (Freiburg, Germany) prior to ELISA, Western blot, or FACS. Intracellular cytokine staining was performed using the BD Cytofix/Cytoperm Fixation/Permeabilization Solution Kit with GolgiPlug (brefeldin A) (BD Biosciences, San Jose, CA) added during the last 4–6 h of incubation. Flow cytometry was performed with a FACSCalibur (BD Biosciences) and analyzed using FCS Express software (De Novo Software, Los Angeles, CA).
Migration assays
Chemotaxis in response to chemokines was determined by measuring the number of cells migrating through a 5.0-μm-pore polycarbonate membrane insert (Corning, Corning, NY). For each experiment, 2–3 × 106 DCs were labeled with 100 μCi [51Cr] as sodium chromate (Amersham Biosciences, Piscataway, NJ) for 1 h at 37°C in 1 ml PBS. Cells were washed three times with PBS and counted. The upper chamber contained 5 × 105 DCs stimulated as indicated and suspended in 100 μl serum-free Aim V media (Invitrogen, Carlsbad, CA). The bottom chamber contained 600 μl serum-free Aim V media with or without chemokines, as indicated. Chemokines were used at 100 ng/ml. For some assays, cells were preincubated with chemical inhibitors for 1 h (25 μM) before addition of LPS. Cells migrated for 3 h at 37°C in a 5% CO2 incubator, and cells accumulating in the bottom chamber were counted with a γ counter. For migration and phenotyping assays, chemotaxis was performed as above, but without [51Cr] labeling, followed by staining and flow-cytometric analysis using BD TruCount tubes.
Alloreactions/T cell proliferation
Spleens were harvested from BALB/c mice and disrupted to obtain a single-cell suspension. T cells were purified using MACS CD8a (Ly-2) or CD4 (L3T4) MicroBeads (Miltenyi Biotec, Auburn, CA). IRAK-M−/− or wild-type (WT) DCs, stimulated as indicated, were washed and plated in doubling dilutions, starting at 2 × 104 cells/well in a round-bottom 96-well microtiter plate in triplicate. A total of 1 × 105 purified T cells were added to each well, for a total volume of 200 μl complete RPMI 1640. In most experiments, 100 μl media was removed and stored at −20°C for cytokine ELISA prior to radioactive labeling. Proliferation was measured after 3 or 6 d by adding 1 μCi [3H]thymidine per well for the last 16–18 h of culture.
Cytokine ELISA
Cytokine levels were measured using the supernatants of cell cultures at the indicated time points by ELISA analysis (IL-6, IL-10, IFN-γ, MCP-1; BD Biosciences; CCL-19, IL-12, R&D Systems), according to the manufacturer's instructions.
Western blot analysis
To avoid possible activation by FBS, DCs were first incubated overnight in Cellgenix DC media. DCs were stimulated for the indicated times with 50 ng/ml LPS, then harvested, washed with PBS, and lysed with cell-lysis buffer (20 mM NaPO3, 150 mM NaCl, 5 mM EDTA, 1× Triton X-100 + Halt Protease inhibitor mixture; Pierce, Rockford, IL). Cellular debris was removed by centrifugation, and lysates were stored at −80°C until further use. Where indicated, nuclear fractionation was performed using the Abcam protocol (cytoplasmic buffer: 10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 0.05% IGEPAL; nuclear buffer: 5 mM HEPES, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, 26% glycerol [v/v]). Protein levels were determined by standard Bio-Rad protein assay (Bio-Rad, Hercules, CA). An equal amount of lysate (20–50 μg/lane) was subjected to SDS-PAGE and then blotted onto Immobilon-P Transfer membranes (Millipore, Bedford, MA) and blocked in 2.5% nonfat milk-TBS-0.025% Tween-20 buffer. Blots were then incubated in primary Ab (actin from Cell Signaling Technology; CCR7 from Abcam, Cambridge, MA), followed by appropriate secondary IgG-conjugated HRP (Santa Cruz Biotechnology, Santa Cruz, CA). Blots were developed using ECL Plus Reagents (Amersham Biosciences) and printed on Biomax light film (Kodak, Rochester, NY).
DC immunization and tumor models
DCs were obtained from IRAK-M−/− or C57BL/6 WT control mice, as described. DCs were pulsed with OVA protein for 8 h and cultured for an additional 12–16 h with LPS. DCs were washed in PBS and injected in the rear footpad of naive C57BL/6 mice at 1 × 106 cells/mouse. Mice were sacrificed on indicated days; inguinal lymph nodes and spleens were removed for intracellular staining and ELISPOT, respectively. Inguinal lymph nodes were dissociated and plated in complete RPMI 1640 containing OT-I and OT-II peptides (10 μg/ml each) for an overnight stimulation at 37°C, followed by intracellular staining. For ELISPOT (20), spleens were dissociated and purified using MACS CD4 (L3T4) or CD8 (Ly-2) MicroBeads (Miltenyi Biotec). Cells were plated in triplicate with OT-II or OT-I peptide (10 μg/ml) or anti-CD3/CD28 for positive control. Plates were incubated overnight, and IFN-γ secretion was assessed with Abs from Mabtech (Nacka, Sweden). For the tumor models, C57BL/6 mice were injected s.c. with 5 × 105 E.G7-OVA or 2.5 × 105 B16.f10 tumor cells, as indicated. Five days later, mice were vaccinated with 1 × 106 nonpulsed or Ag-pulsed (OVA for E.G7, TRP2 + gp100 peptides for B16.f10), LPS-matured IRAK-M−/− or WT DCs, as indicated. Tumor volumes were measured two or three times per week with a digital caliper.
Statistical analyses
Statistical analyses were performed using SPSS 16.0 software (SPSS, Chicago, IL) with a 95% confidence limit, defined as p < 0.05.
Results
IRAK-M−/− DCs can be generated and mature normally
To discover whether DC function and longevity are enhanced in the absence of IRAK-M, we first determined whether IRAK-M was expressed in WT DCs and whether we could still differentiate and mature DCs from IRAK−/− mice in its absence. Fig. 1A shows expression of low levels of IRAK-M in WT DCs cultured in GM-CSF and IL-4 prior to maturation induction, with a further increase in protein levels for up to 2 h after addition of the TLR4 ligand LPS. No IRAK-M was detected in DCs from IRAK-M−/− mice (data not shown).
FIGURE 1.

Removal of IRAK-M does not negatively affect DC maturation. A, BMDCs were taken from naive WT C57BL/6 mice, cultured with or without LPS (100 ng/ml), and subjected to Western blot for IRAK-M. Actin is shown for loading control. B, DCs taken from IRAK-M−/− or WT animals were cultured in CellGenix DC media for 6 h with or without LPS (50 ng/ml) and then analyzed for expression of maturation markers by flow cytometry. Plots are gated on CD11c+ cells and normalized to isotype control. Data are representative of at least three experiments C, Unstimulated or LPS-stimulated DCs (C57BL/6) were cultured with freshly isolated allogeneic splenocytes (BALB/c) for 6 d at the indicated ratios, with [3H]thymidine added during the last 18 h of culture. Results shown are representative of three experiments performed in triplicate. *p < 0.05; **p < 0.01; ***p < 0.001. D, Unstimulated or LPS-stimulated DCs (C57BL/6) were cultured with freshly isolated MACS-purified BALB/c T cells at the indicated ratios for 6 d. Media was removed and examined by ELISA for IFN-γ. Data are shown as the average of four experiments performed in triplicate.
Despite the lack of IRAK-M, we observed no significant difference in the yield of immature DCs from the bone marrow of IRAK-M−/− mice compared with WT mice, and IRAK-M−/− DCs matured normally in response to LPS, as demonstrated by their expression of CD80, CD86, and MHC class II (Fig. 1B). Indeed, expression of several maturation markers was enhanced in CD11c-gated LPS-matured IRAK-M−/− DCs compared with WT DCs (MHCII+: mean fluorescence intensity [MFI], 1323.6 ± 71.3 versus 756.6 ± 65.6; CD86+: MFI, 734.6 ± 163.2 versus 585.1 ± 149.0; and CD80+: MFI, 246.0 ± 44.6 versus 176.4 ± 34.9). There was no significant difference in expression of OX40L, GITRL, or CD40. Thus, the absence of IRAK-M does not adversely affect DC generation or maturation.
IRAK-M−/− DCs enhance activation of effector T cells
Despite the modest phenotypic differences identified, LPS-matured and unstimulated IRAK-M−/− DCs stimulated the proliferation of allogeneic splenocytes more potently than did LPS-matured or immature WT DCs, as measured by [3H]thymidine uptake at day 3 (data not shown) and day 6 (Fig. 1C). Additionally, as shown in Fig. 1D, allogeneic CD4+ and CD8+ splenocytes cultured with IRAK-M−/− DCs produced significantly greater amounts of IFN-γ than after culture with WT DCs (14.9 ± 3.8 ng/ml versus 7.0 ± 0.9 ng/ml at 1:5 DC/T cell ratio for CD8+ T cells, p < 0.01; 13.6 ± 1.1 ng/ml versus 5.9 ± 0.8 ng/ml at 1:20 DC/T cell ratio for CD4+ T cells, p < 0.05). Together, these data demonstrate the superior ability of IRAK-M−/− DCs to activate and induce T cell proliferation.
Expression and activity of p38 and NF-κB
TLR signaling in DCs regulates multiple cellular responses, including migration (through MAPK pathway activation) and survival (through the NF-κB pathway) (26–28). To evaluate the effect of IRAK-M removal on these pathways, we compared the kinetics of NF-κB and MAPK activity after TLR activation in IRAK-M−/− and WT DCs. p38 activity was measured in IRAK-M−/− and WT DCs stimulated with LPS after overnight incubation in CellGenix DC medium to avoid serum-induced maturation. As shown by Western blot (Fig. 2A), IRAK-M−/− DCs had increased phosphorylation of the Th1-polarizing p38-MAPK, but not of the Th2-polarizing ERK1/2 (p42/44) (29, 30), prior to and throughout LPS stimulation. To evaluate NF-κB activity, we studied phosphorylation of RelA (p65) and the NF-κB inhibitor IκB-α, whose activity is inversely correlated to that of NF-κB. Increased RelA activity was seen in IRAK-M−/− DCs compared with WT cells, as indicated by higher levels of phospho-RelA in nuclear fractions relative to actin controls (Fig. 2C). In agreement, WT DCs showed increased IκB-α protein compared with IRAK-M−/− DCs (Fig. 2B).
FIGURE 2.

IRAK-M−/− DCs demonstrate increased activity through p38 and NF-κB pathways. IRAK-M−/− or WT DCs were cultured for the indicated amounts of time with LPS (50 ng/ml), lysed with buffer containing protease and phosphatase inhibitors, then subjected to Western blot. A, Blots were probed for p38 or ERK1/2 and then stripped and reprobed for the corresponding phosphoprotein. Actin is shown for loading control; densitometry is shown in the right panels. B, Blots were probed for IκB-α and then stripped and reprobed for actin. C, Nuclear/cytoplasmic fractionation was performed and then blots were probed for NF-κB phospho-p65 (RelA). Actin was used to control for loading, and tubulin was used to control for the separation itself. Results shown are representative of at least three independent replicates. Abs and buffers used are listed in Materials and Methods.
IRAK-M−/− DCs produce more Th1 cytokines than WT DCs
Because MAPK and NF-κB control cytokine secretion, migration, and longevity, we next evaluated those functions in IRAK-M−/− DCs. Following LPS stimulation, IRAK-M−/− DCs produced significantly increased levels of the proinflammatory cytokines IL-12p70, TNF-α, and IL-6 compared with their WT counterparts (peak values knockout [KO] versus WT IL-12p70: 3775.3 ± 1367.4 pg/ml versus 468.4 ± 222.4 pg/ml; TNF-α: 8755.0 ± 1471.1 pg/ml versus 4376.8 ± 768.3 pg/ml; IL-6: 3489.5 ± 364.6 pg/ml versus 2354.2 ± 468.1 pg/ml; Fig. 3A). IRAK-M−/− DCs also produced lower levels of the Th2 cytokine IL-10 (peak values KO versus WT IL-10: 364.0 ± 95.3 pg/ml versus 1435.5 ± 380.5 pg/ml; Fig. 3B), indicating overall Th1 skewing. In addition, LPS-stimulated IRAK-M−/− DCs produced significantly less of the Th2-skewing chemokine MCP-1/CCL-2 (31) than did LPS-stimulated WT DCs at all time points evaluated (p < 0.01; Fig. 3C) and significantly more of the Th1-skewing chemokine CCL19, an effect that persisted up to 72 h after LPS stimulation (p < 0.05; Fig. 3C). CCL19 induces DC maturation (32) and, thereby, it may contribute to the superior ability of IRAK-M−/− DCs to activate allogeneic T cells. To assess whether the enhanced IL-12p70 secretion by IRAK-M−/− DCs is specific only to TLR4 stimulation, we applied other TLR agonists, including peptidoglycan (TLR2) and CpG:oligodeoxynucleotides (TLR9) and then performed ELISA. As shown in Supplemental Fig. 1, enhanced IL-12p70 production was observed by IRAK-M−/− DCs compared with WT DCs under all conditions, although we noted that LPS stimulation led to the highest levels of IL-12p70 production. Together, these data show that removal of IRAK-M enhances DC maturation and favors polarization toward a Th1-promoting phenotype, indicating that IRAK-M is a negative regulator of DC maturation and cytokine secretion.
FIGURE 3.

IRAK-M−/− DCs demonstrate a Th1-skewed cytokine and chemokine profile compared with WT DCs. A and B, DCs were plated in 24-well plates at 2 × 106 cells/ml in CellGenix DC media. Cells were stimulated with LPS for the indicated times, and medium was removed and examined by ELISA for the proinflammatory cytokines IL-12p70, TNF-α, and IL-6, as well as the anti-inflammatory cytokine IL-10. Data shown are averages of at least four experiments performed in triplicate. C, DCs were plated in 12-well plates, and media were removed and examined by ELISA for secretion of the chemokines CCL-19 and MCP-1 (CCL-2). Data shown are representative of at least three experiments performed in triplicate. *p < 0.05; **p < 0.01; ***p < 0.001.
IRAK-M−/− DCs migrate more effectively than WT DCs in vitro and in vivo
To evaluate DC migration, we measured expression of the chemokine receptor CCR7 (required for migration to lymph nodes after pathogen encounter) and of CCR2, CCR5, and CCR6 (important for the migration of immature DCs to sites of injury and inflammation) (16, 33). Fig. 4A shows enhanced expression of CCR7 by CD11c-gated IRAK-M−/− DCs compared with their WT counterparts in the unstimulated condition, as well as after 6 and 24 h of LPS stimulation (CCR7+: MFI, 541.69 versus 397.79 at 6 h, 621.29 versus 364.69 at 24 h). Western blotting for CCR7 in IRAK-M−/− and WT DCs following LPS exposure confirmed these data. As anticipated, expression of inflammatory chemokine receptors, such as CCR2, CCR6, and CCR9, was low, and no significant difference was observed between IRAK-M−/− and WT DCs (data not shown).
FIGURE 4.

IRAK-M−/− DCs migrate more effectively in vitro and in vivo than WT DCs. A, IRAK-M−/− and WT DCs were cultured with or without LPS (50 ng/ml) for the indicated times and then analyzed by intracellular staining for expression of the chemokine receptor CCR7. Black line, IRAK-M−/−; gray line, WT; shaded graph, isotype control. Plots are gated on CD11c+ cells and normalized to isotype control. B, DCs were analyzed for migratory capacity toward CCL-19 or CCL-20 (100 ng/ml) using a standard Transwell-assay system. DCs were stimulated with LPS (50 ng/ml), labeled with [51Cr], loaded into the top chamber of a 5.0-μm-pore Transwell plate, and allowed to migrate for 3 h at 37°C. Where indicated, saturating concentrations of blocking Abs to CCR7 (a-CCR7) or CCR6 (α-CCR6) were added directly to the Transwell assay plate. C, DCs were preincubated (or not) with p38-specific inhibitor SB203580 or ERK1/2-specific inhibitor U0126 (25 μM each), followed by maturation with LPS and chemotaxis assay. Data are averages of at least three separate experiments. D, LPS-matured DCs were allowed to migrate in Transwell inserts for 3 h and then cells in the top chamber (nonmigrated) and bottom chamber (migrated) were stained separately and analyzed by FACS. Numbers represent MFI (top number, IRAK-M−/−; bottom number, WT). Migrated samples were acquired using BD TruCount tubes; height of peak represents absolute number of cells. E, A total of 1.5 × 106 LPS-stimulated DCs were labeled with CFSE (1.5 μM), washed, and injected into the left footpad of naive recipient C57BL/6 mice. Mice were euthanized after 48 h, and the target tissues were examined for CD11c+CFSE+ migratory DCs. Top panel, Representative plot of inguinal lymph nodes from mice receiving WT or IRAK-M−/− CFSE-labeled DCs. Bottom panels, Number of CD11c+CFSE+ migrated cells found in inguinal lymph nodes or spleen; average of three experiments with four or five mice/group. *p < 0.05; **p < 0.01; ***p < 0.001. ns, not significant.
We performed transwell migration assays to determine whether IRAK-M−/− DCs migrated more effectively toward CCR7 ligands. As shown in Fig. 4B, a significantly higher percentage of IRAK-M−/− DCs migrated in response to the CCR7 ligands CCL19 and CCL21 compared with WT DCs (15.36 ± 1.3% versus 8.58 ± 1.46% for CCL19; 19.66 ± 0.6% versus 9.12 ± 0.3% for CCL21). Further, Ab blockade of CCR7 significantly inhibited migration of IRAK-M−/− and WT DCs (KO: 15.36 ± 1.5% decreased to 5.22 ± 0.4%; WT: 8.58 ± 1.5% reduced to 3.94 ± 0.5%). Of note, IRAK-M−/− DCs consistently showed a higher level of basal migration in the absence of exogenous cytokines, suggesting an intrinsically more migratory phenotype. However, attempts to induce migration via inflammatory receptors, such as CCR6, CCR5, and CCR2, using their respective ligands CCL20 (MIP-3α), CCL5, (RANTES), and CCL2 (MCP-1), produced no specific migration (Fig. 4B, data not shown).
We next compared the phenotype of migrating and nonmigrating cells and performed quantitative FACS to determine the absolute numbers of migrating cells. In agreement with our data above, more IRAK-M−/− DCs migrated in response to CCR7 ligands compared with WT DCs (CD86+: 19,181 and 10,222, respectively; CD80+: 18,107 and 9,315, respectively; Fig. 4C). Further, migratory cells exhibited higher expression of classic maturation markers, whereas nonmigratory cells expressed lower levels of CD80 and CD86, indicating a less mature phenotype (Fig. 4C). To compare the in vivo migration of IRAK-M−/− and WT DCs, cells were stimulated with LPS (50 ng/ml) overnight, then harvested, stained with CFSE, and injected into the footpad of WT recipient mice, which were sacrificed 48 h later. Inguinal lymph nodes and spleens were removed and examined by quantitative FACS for the presence of CFSE+/CD11c+ migratory DCs. As shown in Fig. 4D, significantly larger numbers of IRAK-M−/− DCs migrated to inguinal lymph nodes and spleens of recipient mice. These results agree with our in vitro data and demonstrate that IRAK-M is a negative regulator of murine DC migration in vivo.
To confirm that enhanced DC migration resulted from the observed increase in p38 activity, we included a chemical inhibitor of p38 (SB203580) in our migration assays (Fig. 4E), which led to a significant reduction in migration of IRAK-M−/− and WT DCs (KO: 16.55 ± 0.8% decreased to 4.33 ± 0.3%; WT: 9.92 ± 0.4% decreased to 5.18 ± 0.3%). By contrast, preincubation with the ERK1/2- or p65-specific inhibitors U0126 or ammonium pyrrolidine dithiocarbamate, respectively, had no effect. Taken together, these results indicate that DCs lacking IRAK-M have enhanced p38-dependent migration in response to the CCR7 ligands CCL19 and CCL21.
IRAK-M−/− BMDCs are longer lived than WT BMDCs
To determine whether increased activity of the NF-κB pathway in IRAK-M−/− DCs affected the long-term viability and function of DCs, we analyzed their viability over 7 d of culture without growth factors in the presence or absence of LPS. Western immunoblotting showed increased expression of Bcl-2 in IRAK-M−/− DCs compared with WT DCs for as long as 5 d (Fig. 5A), in agreement with the sustained increase in activity through the NF-κB pathway shown in Fig. 2.
FIGURE 5.
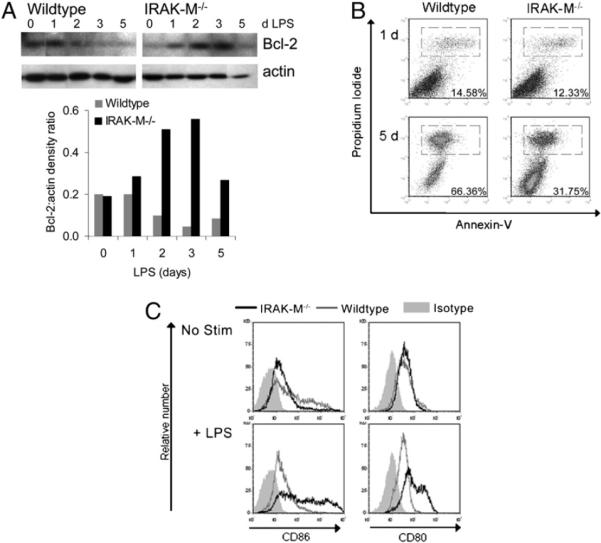
IRAK-M−/− DCs live longer than WT DCs. IRAK-M−/− and WT DCs were cultured in CellGenix media without growth factors plus 50 ng/ml LPS. A, At the times indicated, cells were harvested, lysed with buffer containing protease and phosphatase inhibitors, and subjected to Western blot. Blots were probed for Bcl-2, stripped, and reprobed for actin; densitometry is also shown (lower panel). Abs used are listed in Materials and Methods. B, At the times indicated, cells were harvested and stained for Annexin V and PI; numbers indicate the percentage of gated (dead) cells. Data are representative of four independent experiments. C, After 5 d of the indicated stimulus, cells were harvested, stained, and analyzed by quantitative flow cytometry. Plots are gated on viable cells and shown as nonnormalized to indicate the relative number of events.
We also enumerated viable DCs at increasing times after LPS stimulation using trypan blue, Annexin V, and propidium iodide (PI) staining. As shown in Fig. 5B, greater numbers of IRAK-M−/− DCs remained viable (PI−) after long-term growth factor deprivation and exposure to LPS (day 5: CD11c+/PI−, 53.38 ± 5.5% versus 31.94 ± 7.0%). In addition, IRAK-M−/− DCs maintained their mature phenotype for 6 d after deprivation of cytokines and exposure to LPS. Under the same conditions, WT DCs seemed to lose their maturation markers (Fig. 5C). As expected from the decreased DC death rate, quantitative flow cytometry showed that we recovered 3.2-fold more CD11c+/CD86+ and 2.4-fold more CD11c+/CD80+ IRAK-M−/− DCs than WT DCs on day 6 after addition of LPS. Similar results were obtained upon stimulation of DCs with additional TLR agonists, including peptidoglycan (TLR2) or CpG:oligodeoxynucleotides (TLR9) (Supplemental Fig. 1). Hence, WT DCs can become mature in response to short-term TLR stimulation (Fig. 1B), but their mature and viable state is not maintained long-term.
Vaccination with IRAK-M−/− BMDCs leads to an enhanced immune response
DC survival and maintenance of their mature immunostimulatory phenotype described above suggested that IRAK-M−/− DCs should induce superior immune responses compared with WT DCs. Therefore, we immunized WT mice in the footpad with a single dose of 1 × 106 IRAK-M−/− or WT DCs that had been pulsed with OVA protein (100 μg/ml, 6–8 h), followed by LPS maturation overnight. Recipient mice were euthanized on various days following immunization. As shown in Fig. 6A and Supplemental Fig. 2, inguinal lymph nodes removed from mice immunized with IRAK-M−/− DCs and restimulated overnight with OT-I and OT-II peptides (10 μg/ml each) all contained higher frequencies of OVA-specific CD4+ and CD8+ T cells, as measured in intracellular IFN-γ assays. Thus, 8.82 and 10.19% of total lymph node CD8+ and CD4+ T cells from IRAK-M−/− vaccinees secreted IFN-γ in response to OVA on day 7, compared with 4.8 and 6.19% of mice vaccinated with WT DCs. This advantage persisted for ≥2 wk following vaccination, because mice sacrificed at day 14 after DC vaccination showed the same activity (CD8+IFN-γ+: 10.19 ± 2.12% versus 4.21 ± 0.44%; CD4+IFN-γ+: 11.44 ± 1.67% versus 6.99 ± 0.71%). MACS-purified CD8+ and CD4+ splenocytes from IRAK-M−/− DC-vaccinated mice secreted increased IFN-γ by ELISPOT compared with WT recipients at all time points studied, as shown in Fig. 6B (day 7: 139.42 ± 41.56 spots versus 17.42 ± 4.86 spots per 1 ± 106 CD8+; 381.71 ± 18.00 spots versus 110.75 ± 24.69 spots per 1 ± 106 CD4+).
FIGURE 6.
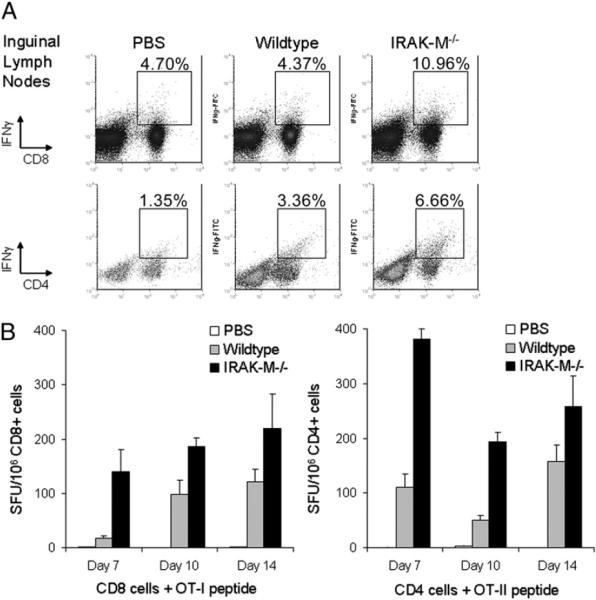
Vaccination with IRAK-M−/− DCs results in an enhanced immune response. Naive C57BL/6 mice were vaccinated in the left footpad with a single dose of 1 × 106 OVA-pulsed, LPS-matured WT or IRAK-M−/− DCs or PBS control. Mice were euthanized at indicated time points, and inguinal lymph nodes and spleens were removed. A, Inguinal lymph nodes from vaccinated mice were restimulated overnight with OT-I and OT-II peptides (10 μg/ml each), with GolgiPlug (brefeldin A) added during the last 6 h of culture. Cells were harvested, washed, and analyzed by intracellular staining for IFN-γ; representative day 14 lymph nodes are shown. B, Spleens from vaccinated mice were purified using CD8+ or CD4+ MACS beads and plated in triplicate with peptide (OT-I or OT-II; 10 μg/ml) or CD3/CD28 Ab; all samples responded to positive control. Plates were incubated overnight, and IFN-γ secretion was assessed by ELISPOT. Results are averages of two to four experiments, with four or five mice per group.
To determine whether IRAK-M−/− DCs enhanced antitumor immunity, naive WT C57BL/6 mice were inoculated on day 0 with the EG7 lymphoma (EL4 cells expressing OVA) at 5 × 105 cells/mouse. Immunization 5 d later with a single dose of 1 × 106 OVA-pulsed, LPS-matured IRAK-M−/− DCs significantly inhibited the growth of EG7 tumors (KO versus WT; p < 0.001), with complete elimination seen in nearly half the mice. By contrast, injection of OVA-pulsed, LPS-matured WT DCs had minimal impact on tumor growth, and no mice cleared their tumors (WT versus PBS, p = 0.082; Fig. 7A). Mice receiving IRAK-M−/− DC vaccine had a median survival > 50 d (Fig. 7B); most mice in the IRAK-M−/− treatment group were terminated at day 50 for histological analysis, but 5 mice were not, and these survived beyond 90 d. Animals receiving WT DC vaccine had a median survival of 40 d (range, 10–45 d), and those receiving PBS control injection had a median survival of 30 d (range, 10–35 d).
FIGURE 7.
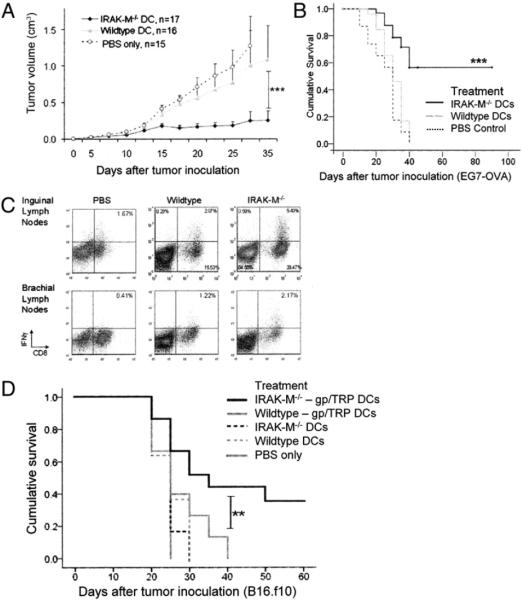
Vaccination with IRAK-M−/− DCs results in enhanced tumor clearance. A, Naive C57BL/6 mice were inoculated with 5 × 105 EG7-OVA cells at day 0, followed by a single vaccination of OVA-pulsed, LPS-matured WT or IRAK-M−/− DCs or PBS control on day 5. Tumor growth was monitored using a caliper. Results shown are averages of four independent experiments. B, Survival of tumor-bearing mice was monitored over time, with end points defined as death of an animal or the point at which the tumor burden became so large that the animal had to be euthanized. Results shown are compiled from four independent experiments. C, Inguinal and brachial lymph nodes were removed from tumor-bearing animals and re-stimulated overnight with OVA peptides (10 μg/ml each); BD GolgiPlug (brefeldin A) was added during the last 6 h of culture. Cells were harvested, washed, and analyzed by intracellular staining for IFN-γ; representative staining is shown. D, Naive C57BL/6 mice were inoculated with 2.5 × 105 B16.f10 cells at day 0, followed by a single vaccination of gp100-TRP2–pulsed, LPS-matured WT or IRAK-M−/− DCs; non-pulsed, LPS-matured WT or IRAK-M−/− DCs; or PBS control on day 5. Survival of tumor-bearing mice was monitored over time, with end points defined as death of an animal or the point at which the tumor burden became so large that the animal had to be euthanized. Results shown are compiled from three independent experiments. **p < 0.01; ***p < 0.001.
To characterize the T cell immune responses in tumor-bearing mice, tissues were removed and analyzed for OVA-specific T cells. OVA-specific cytotoxicity against tumor cell lines was demonstrated by splenic T cells removed from tumor-bearing mice (data not shown), whereas inguinal and brachial tumor-draining lymph nodes from mice immunized with IRAK-M−/− DCs contained a higher percentage of CD4+ and CD8+ IFN-γ+–secreting T cells than mice immunized with WT DCs (CD8+IFN-γ+ inguinal: 5.40 versus 2.07%; CD8+IFN-γ+ brachial: 1.17 versus 0.17%; Fig. 7C).
To apply this vaccination technique to more physiologically relevant Ags, we performed tumor studies using the melanoma line B16.f10. This cell line expresses multiple tumor-associated Ags, such as TRP2, gp100, and Melan-A/MART-1, which have also been studied in human clinical trials (4, 34). Naive WT C57BL/6 mice were inoculated on day 0 with 2.5 × 105 B16.f10 cells/mouse, followed by vaccination on day 5 with 1 × 106 IRAK-M−/− or WT DCs that had been pulsed with TRP2 and gp100 peptides and then matured with LPS. Fig. 7D shows the survival curve obtained from these studies; vaccination with IRAK-M−/− DCs was also able to significantly prolong survival of B16.f10 tumor-bearing mice in comparison with WT DCs (p = 0.009) or PBS control injection (p < 0.001).
To demonstrate the antigenic specificity of our vaccination, we also included treatment groups receiving nonpulsed LPS-matured WT and IRAK-M−/− DCs. As shown in Fig. 7D, these mice were not able to control their tumors and succumbed rapidly, indicating that Ag is necessary for the enhanced in vivo function of our inhibitor-silenced DC vaccine.
Discussion
IRAK-M, an inhibitor of MyD88-dependent TLR and IL-1 signaling, is expressed only in cells of myeloid origin, such as macrophages, monocytes, and osteoclasts (22, 35, 36). It functions by binding to the IRAK-1/IRAK-4/TRAF6 complex downstream of MyD88, thereby inhibiting its dissociation and subsequent signaling. We have now shown that IRAK-M is expressed in murine DCs, where it also acts as a negative regulator of TLR signaling. After TLR ligation, DCs from IRAK-M−/− mice demonstrated increased activation of the p38-MAPK and NF-κB pathways compared with WT DCs (Fig. 2), resulting in enhanced secretion of Th1 cytokines and chemokines, as well as superior Ag-presenting function (Fig. 3). IRAK-M−/− DCs expressed higher levels of CCR7 and migrated more effectively than their WT counterparts, in vitro and in vivo (Fig. 4), and showed increased longevity (Fig. 5). As a consequence, IRAK-M−/− DCs induced a stronger T cell immune response in vivo, eliminating established tumors and prolonging the survival of tumor-bearing mice (Figs. 6, 7).
The IRAK family consists of four members, the ubiquitously expressed IRAKs 1, 2, and 4 and the myeloid-lineage−restricted IRAK-M (36). IRAK-M−/− mice were found to exhibit unexpectedly enhanced immune responses following bacterial challenge, as well as delayed endotoxin tolerance, leading to the conclusion that unlike other IRAKs, IRAK-M functions as a TLR-signaling inhibitor (22). Like other negative regulators of immune responses, such as SOCS-1 and A20, IRAK-M likely serves to shut down the immune response following pathogen clearance and prevent overwhelming inflammatory responses; indeed, altered IRAK-M levels have been associated with sepsis, osteoporosis, and cirrhosis in murine models and in patients (23–25, 35). However, unlike A20 and SOCS-1, IRAK-M is highly myeloid lineage restricted and, likely for that reason, IRAK-M knockout in mice does not cause premature lethality. Most studies of IRAK-M have concentrated on the role of IRAK-M in innate immunity with a focus on sepsis, shock, and endotoxin tolerance, but relatively little is known about the role of IRAK-M in adaptive immunity.
Because decreased IRAK-M in macrophages and monocytes results in enhanced and prolonged activity through the p38 and NF-κB pathways (22, 24, 35) and because these signaling pathways influence migration, longevity, and cytokine/chemokine secretion, we reasoned that if IRAK-M is expressed in DCs, its removal would result in enhanced activity through these pathways. Indeed, we observed increased migration and prolonged lifespan of IRAK-M−/− DCs compared with WT DCs. The enhanced migration was dependent on p38 (but not ERK1/2) and likely correlated with increased expression of the CCR7 receptor (27, 37). An unexpected benefit of IRAK-M removal in DCs was Th1 skewing, a DC attribute that has proved essential for the induction of antitumor immune responses (38, 39). IRAK-M–modified DCs continued to secrete significant amounts of the pivotal Th1 cytokine IL-12 for 5 d compared with WT DCs, as well as greater amounts of IL-6, TNF-α, and the Th1-skewing chemokine CCL19.
An additional aspect of DC biology that is crucial to vaccine efficacy is viability. After capturing Ag and trafficking to the lymph nodes, DCs must remain in a mature and viable state for sufficient time topresent the Ag to cognate T cells. It has been suggested that the lifespan of mature DCs is on the order of days and varies by subset (40); however, IRAK-M−/− DCs survived significantly longer than their WT counterparts while retaining a functional, mature pheno-type. The apparent loss of maturation markers by WT DCs may be due to exhaustion and death following stimulation (41), leaving behind a less mature population. As observed in Fig. 1B, WT DCs are able to become phenotypically mature following TLR ligation; however, the majority of the population does not maintain a mature and viable state long-term as do the IRAK-M−/− DCs.
The enhanced viability observed in IRAK-M−/− DCs could be explained by the prolonged activity of the NF-κB pathway and expression of the antiapoptotic protein Bcl-2, which were previously shown to control the longevity of murine DCs (26, 28, 40, 42). During natural pathogen infections, continued production of TLR ligands maintains a flow of DCs from the infection site to the lymph node until the infection is contained, so that DC longevity may not be crucial. However, tumors are poorly immunogenic, do not produce TLR ligands, and create an immunosuppressive micro-environment; therefore, after vaccination, DC longevity becomes critical (43, 44).
The characteristics imparted to DCs by inhibition of IRAK-M signaling should translate into more potent DC vaccines, the current efficacy of which is limited by their ability to migrate to lymph nodes and maintain potency and viability in vivo. Less than 4% of injected mature DCs migrate to lymph nodes, and those that remain at the injection site lose viability and are cleared by infiltrating CD163+ macrophages within 48 h (17). In our study, IRAK-M−/− DCs were detected with lower frequency at the injection site, as measured by CFSE staining (data not shown), and at greater frequency in local and distant lymph nodes, consistent with migration from the injection site to lymphoid tissues. These migrated DCs induced higher frequencies of Ag-specific T cells over a longer time compared with WT DC vaccines.
Several strategies to surmount the deficits of DC vaccines have been evaluated in mouse models and in the clinic. The systemic infusion of adjuvants or cytokines can prolong in vivo DC stimulation, but it may have significant off-target effects, such as toxicity and activation of regulatory T cells (9, 38). To increase tumor targeting, TLR agonists have been injected directly into tumor sites, leading to tumor regression with minimal systemic side effects (45, 46); however, tumor sites may be multiple, inaccessible, or unidentified. To circumvent the requirement for migration, injections of Ag-pulsed DCs have been given directly into lymph nodes or lymphatic vessels of lymphoma and melanoma patients, yielding immunological responses in more than half of the cases but few complete remissions (4, 6). These complicated procedures might be avoided by a single injection of IRAK-M‣silenced DCs that could naturally migrate to lymph nodes and maintain their mature phenotype without requiring an in vivo adjuvant.
Genetic strategies have also been used, either to enhance maturation signals or to inhibit endogenous inhibitors in preclinical studies (5, 20, 21, 44, 47, 48). Genetic modification affects only the DCs presenting the tumor Ags of interest and renders the treatment highly specific. Modifications leading to enhancement of positive-regulatory pathways include inducible or constitutive activation of stimulatory pathways, like Akt1 or CD40 (5, 44); coexpression of receptor and ligand pairs, like receptor activator of NF-κB and RANKL (48); expression of T cell-attracting chemokines; and expression of Th1-skewing cytokines, such as IL-12, IL-18, and/or TNF-α (47). These approaches produced tumor regression in animal models, but enhanced DCs may remain susceptible to endogenous inhibitors.
Therefore, the alternative approach has been to inhibit negative regulators and, hence, improve and prolong the viability and function of mature DCs. Small interfering RNA (siRNA) targeting of A20, a negative regulator of NF-κB signaling, led to increased IL-6 production, inhibition of regulatory T cells, and enhanced Ag-specific antitumor activity (21). Silencing SOCS1 in DCs led to enhanced IL-12 production and regression of established murine tumors (19, 20). Although highly effective, both of these strategies required injection of multiple doses of vaccine and/or use of an in vivo adjuvant. Furthermore, these genetic modifications regulated one or two characteristics of DCs (cytokine secretion and/or longevity and/or migration) but did not affect all three (19‣21, 44, 47). Because IRAK-M regulates the NF-κB and p38 pathways, we reasoned that removal of IRAK-M would enhance cytokine secretion and increase DC longevity and migration, producing multiple benefits from a single modification. Indeed, a single dose of 1 × 106 Ag-loaded, mature IRAK-M−/− DCs was able to suppress tumor growth without additional boosting. To generate a clinically relevant vaccine product, IRAK-M could also be targeted in human DCs using an siRNA approach. Although we are not aware of any small molecule inhibitors of IRAK-M, systemic administration of such an inhibitor or viral-mediated delivery of an siRNA should cause little toxicity because of the tissue-specific expression of IRAK-M.
Absence of functional IRAK-M in DCs does not seem to be associated with adverse effects in our studies. No autoimmunity was observed in IRAK-M−/− DC-vaccinated mice that were followed for>6 mo; these mice remained active and alert, with no discernible changes in dietary or grooming habits. This is in contrast to mice that underwent systemic administration of cytokines or TLR ligands (49). Indeed, IRAK-M−/− mice are viable, fertile, and do not exhibit any gross abnormalities until later in life when osteoporosis may develop (22, 35). This relative well-being contrasts with the effects observed in other inhibitor KO mice. SOCS1−/− mice exhibit lymphocyte-dependent dysregulation of cytokine signaling, resulting in overproduction of IFN-γ and death by 3 wk of age (50). A20−/− mice develop severe innate immune cell-mediated inflammation and die within 6 wk of birth (51). These phenotypic differences may be due largely to tissue distribution; although SOCS1 and A20 are widely expressed, IRAK-M displays limited expression only in cells of myeloid origin. Therefore, removal or silencing of IRAK-M will affect a specific and limited lineage, whereas targeting ubiquitously expressed proteins could affect multiple tissues and cause widespread adverse effects.
In summary, we demonstrated that IRAK-M is an inhibitor of multiple DC activities and that removal of this molecule enhances DC survival, migration, and Ag presentation, with beneficial effects on antitumor immune responses. Given the importance of these three functions to the development of an effective DC vaccine, IRAK-M may be a useful target for inhibition when an enhanced and specific immune response is desired.
Supplementary Material
Acknowledgments
We thank R. Paylor for valuable help with statistics and M.C. Ngo for technical assistance.
This work was supported in part by National Institutes of Health-National Cancer Institute Grants PO1 CA94234-01A1, P50 CA126752, and T32 HL 092332-06 (to M.E.T.) and by the Leukemia and Lymphoma Society Specialized Center of Research Grant 7018-04.
Abbreviations used in this paper
- BMDC
bone marrow-derived dendritic cell
- DC
dendritic cell
- IRAK-M
IL-1R–associated kinase M
- KO
knockout
- MFI
mean fluorescence intensity
- ns
not significant
- PI
propidium iodide
- siRNA
small interfering RNA
- WT
wild-type
Footnotes
The online version of this article contains supplemental material.
Disclosures The authors have no financial conflicts of interest.
References
- 1.Steinman RM. Some interfaces of dendritic cell biology. APMIS. 2003;111:675–697. doi: 10.1034/j.1600-0463.2003.11107802.x. [DOI] [PubMed] [Google Scholar]
- 2.Trombetta ES, Mellman I. Cell biology of antigen processing in vitro and in vivo. Annu. Rev. Immunol. 2005;23:975–1028. doi: 10.1146/annurev.immunol.22.012703.104538. [DOI] [PubMed] [Google Scholar]
- 3.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 4.Grover A, Kim GJ, Lizée G, Tschoi M, Wang G, Wunderlich JR, Rosenberg SA, Hwang ST, Hwu P. Intralymphatic dendritic cell vaccination induces tumor antigen-specific, skin-homing T lymphocytes. Clin. Cancer Res. 2006;12:5801–5808. doi: 10.1158/1078-0432.CCR-05-2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lapteva N, Seethammagari MR, Hanks BA, Jiang J, Levitt JM, Slawin KM, Spencer DM. Enhanced activation of human dendritic cells by inducible CD40 and Toll-like receptor-4 ligation. Cancer Res. 2007;67:10528–10537. doi: 10.1158/0008-5472.CAN-07-0833. [DOI] [PubMed] [Google Scholar]
- 6.Maier T, Tun-Kyi A, Tassis A, Jungius KP, Burg G, Dummer R, Nestle FO. Vaccination of patients with cutaneous T-cell lymphoma using intranodal injection of autologous tumor-lysate-pulsed dendritic cells. Blood. 2003;102:2338–2344. doi: 10.1182/blood-2002-08-2455. [DOI] [PubMed] [Google Scholar]
- 7.Nestle FO, Alijagic S, Gilliet M, Sun Y, Grabbe S, Dummer R, Burg G, Schadendorf D. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat. Med. 1998;4:328–332. doi: 10.1038/nm0398-328. [DOI] [PubMed] [Google Scholar]
- 8.Timmerman JM, Czerwinski DK, Davis TA, Hsu FJ, Benike C, Hao ZM, Taidi B, Rajapaksa R, Caspar CB, Okada CY, et al. Idiotype-pulsed dendritic cell vaccination for B-cell lymphoma: clinical and immune responses in 35 patients. Blood. 2002;99:1517–1526. doi: 10.1182/blood.v99.5.1517. [DOI] [PubMed] [Google Scholar]
- 9.Wierecky J, Müller MR, Wirths S, Halder-Oehler E, Dörfel D, Schmidt SM, Häntschel M, Brugger W, Schröder S, Horger MS, et al. Immunologic and clinical responses after vaccinations with peptide-pulsed dendritic cells in metastatic renal cancer patients. Cancer Res. 2006;66:5910–5918. doi: 10.1158/0008-5472.CAN-05-3905. [DOI] [PubMed] [Google Scholar]
- 10.Schuler G, Schuler-Thurner B, Steinman RM. The use of dendritic cells in cancer immunotherapy. Curr. Opin. Immunol. 2003;15:138–147. doi: 10.1016/s0952-7915(03)00015-3. [DOI] [PubMed] [Google Scholar]
- 11.Osada T, Clay TM, Woo CY, Morse MA, Lyerly HK. Dendritic cell-based immunotherapy. Int. Rev. Immunol. 2006;25:377–413. doi: 10.1080/08830180600992456. [DOI] [PubMed] [Google Scholar]
- 12.Vieweg J, Jackson A. Modulation of antitumor responses by dendritic cells. Springer Semin. Immunopathol. 2005;26:329–341. doi: 10.1007/s00281-004-0175-1. [DOI] [PubMed] [Google Scholar]
- 13.Allan RS, Waithman J, Bedoui S, Jones CM, Villadangos JA, Zhan Y, Lew AM, Shortman K, Heath WR, Carbone FR. Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity. 2006;25:153–162. doi: 10.1016/j.immuni.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 14.Allenspach EJ, Lemos MP, Porrett PM, Turka LA, Laufer TM. Migratory and lymphoid-resident dendritic cells cooperate to efficiently prime naive CD4 T cells. Immunity. 2008;29:795–806. doi: 10.1016/j.immuni.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Vries IJ, Krooshoop DJ, Scharenborg NM, Lesterhuis WJ, Diepstra JH, Van Muijen GN, Strijk SP, Ruers TJ, Boerman OC, Oyen WJ, et al. Effective migration of antigen-pulsed dendritic cells to lymph nodes in melanoma patients is determined by their maturation state. Cancer Res. 2003;63:12–17. [PubMed] [Google Scholar]
- 16.Förster R, Schubel A, Breitfeld D, Kremmer E, Renner-Müller I, Wolf E, Lipp M. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 1999;99:23–33. doi: 10.1016/s0092-8674(00)80059-8. [DOI] [PubMed] [Google Scholar]
- 17.Verdijk P, Aarntzen EH, Lesterhuis WJ, Boullart AC, Kok E, van Rossum MM, Strijk S, Eijckeler F, Bonenkamp JJ, Jacobs JF, et al. Limited amounts of dendritic cells migrate into the T-cell area of lymph nodes but have high immune activating potential in melanoma patients. Clin. Cancer Res. 2009;15:2531–2540. doi: 10.1158/1078-0432.CCR-08-2729. [DOI] [PubMed] [Google Scholar]
- 18.Reichardt VL, Milazzo C, Brugger W, Einsele H, Kanz L, Brossart P. Idiotype vaccination of multiple myeloma patients using monocyte-derived dendritic cells. Haematologica. 2003;88:1139–1149. [PubMed] [Google Scholar]
- 19.Evel-Kabler K, Song XT, Aldrich M, Huang XF, Chen SY. SOCS1 restricts dendritic cells' ability to break self tolerance and induce anti-tumor immunity by regulating IL-12 production and signaling. J. Clin. Invest. 2006;116:90–100. doi: 10.1172/JCI26169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shen L, Evel-Kabler K, Strube R, Chen SY. Silencing of SOCS1 enhances antigen presentation by dendritic cells and antigen-specific anti-tumor immunity. Nat. Biotechnol. 2004;22:1546–1553. doi: 10.1038/nbt1035. [DOI] [PubMed] [Google Scholar]
- 21.Song XT, Evel-Kabler K, Shen L, Rollins L, Huang XF, Chen SY. A20 is an antigen presentation attenuator, and its inhibition overcomes regulatory T cell-mediated suppression. Nat. Med. 2008;14:258–265. doi: 10.1038/nm1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kobayashi K, Hernandez LD, Galán JE, Janeway CA, Jr., Medzhitov R, Flavell RA. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 2002;110:191–202. doi: 10.1016/s0092-8674(02)00827-9. [DOI] [PubMed] [Google Scholar]
- 23.Deng JC, Cheng G, Newstead MW, Zeng X, Kobayashi K, Flavell RA, Standiford TJ. Sepsis-induced suppression of lung innate immunity is mediated by IRAK-M. J. Clin. Invest. 2006;116:2532–2542. doi: 10.1172/JCI28054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tazi KA, Quioc JJ, Saada V, Bezeaud A, Lebrec D, Moreau R. Upregulation of TNF-alpha production signaling pathways in monocytes from patients with advanced cirrhosis: possible role of Akt and IRAK-M. J. Hepatol. 2006;45:280–289. doi: 10.1016/j.jhep.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 25.Wiersinga WJ, van't Veer C, van den Pangaart PS, Dondorp AM, Day NP, Peacock SJ, van der Poll T. Immunosuppression associated with interleukin-1R-associated-kinase-M upregulation predicts mortality in Gram-negative sepsis (melioidosis) Crit. Care Med. 2009;37:569–576. doi: 10.1097/CCM.0b013e318194b1bf. [DOI] [PubMed] [Google Scholar]
- 26.Hou WS, Van Parijs L. A Bcl-2-dependent molecular timer regulates the lifespan and immunogenicity of dendritic cells. Nat. Immunol. 2004;5:583–589. doi: 10.1038/ni1071. [DOI] [PubMed] [Google Scholar]
- 27.Jung ID, Lee JS, Kim YJ, Jeong YI, Lee CM, Lee MG, Ahn SC, Park YM. Sphingosine kinase inhibitor suppresses dendritic cell migration by regulating chemokine receptor expression and impairing p38 mitogen-activated protein kinase. Immunology. 2007;121:533–544. doi: 10.1111/j.1365-2567.2007.02601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ouaaz F, Arron J, Zheng Y, Choi Y, Beg AA. Dendritic cell development and survival require distinct NF-kappaB subunits. Immunity. 2002;16:257–270. doi: 10.1016/s1074-7613(02)00272-8. [DOI] [PubMed] [Google Scholar]
- 29.Randolph GJ, Sanchez-Schmitz G, Angeli V. Factors and signals that govern the migration of dendritic cells via lymphatics: recent advances. Springer Semin. Immunopathol. 2005;26:273–287. doi: 10.1007/s00281-004-0168-0. [DOI] [PubMed] [Google Scholar]
- 30.Agrawal S, Agrawal A, Doughty B, Gerwitz A, Blenis J, Van Dyke T, Pulendran B. Cutting edge: different Toll-like receptor agonists instruct dendritic cells to induce distinct Th responses via differential modulation of extracellular signal-regulated kinase-mitogen-activated protein kinase and c-Fos. J. Immunol. 2003;171:4984–4989. doi: 10.4049/jimmunol.171.10.4984. [DOI] [PubMed] [Google Scholar]
- 31.Daly C, Rollins BJ. Monocyte chemoattractant protein-1 (CCL2) in inflammatory disease and adaptive immunity: therapeutic opportunities and controversies. Microcirculation. 2003;10:247–257. doi: 10.1038/sj.mn.7800190. [DOI] [PubMed] [Google Scholar]
- 32.Marsland BJ, Bäattig P, Bauer M, Ruedl C, Lässing U, Beerli RR, Dietmeier K, Ivanova L, Pfister T, Vogt L, et al. CCL19 and CCL21 induce a potent proinflammatory differentiation program in licensed dendritic cells. Immunity. 2005;22:493–505. doi: 10.1016/j.immuni.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 33.Gunn MD. Chemokine mediated control of dendritic cell migration and function. Semin. Immunol. 2003;15:271–276. doi: 10.1016/j.smim.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 34.Butterfield LH, Comin-Anduix B, Vujanovic L, Lee Y, Dissette VB, Yang JQ, Vu HT, Seja E, Oseguera DK, Potter DM, et al. Adenovirus MART-1-engineered autologous dendritic cell vaccine for metastatic melanoma. J. Immunother. 2008;31:294–309. doi: 10.1097/CJI.0b013e31816a8910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H, Cuartas E, Cui W, Choi Y, Crawford TD, Ke HZ, Kobayashi KS, Flavell RA, Vignery A. IL-1 receptor-associated kinase M is a central regulator of osteoclast differentiation and activation. J. Exp. Med. 2005;201:1169–1177. doi: 10.1084/jem.20041444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wesche H, Gao X, Li X, Kirschning CJ, Stark GR, Cao Z. IRAK-M is a novel member of the Pelle/interleukin-1 receptor-associated kinase (IRAK) family. J. Biol. Chem. 1999;274:19403–19410. doi: 10.1074/jbc.274.27.19403. [DOI] [PubMed] [Google Scholar]
- 37.MartIn-Fontecha A, Sebastiani S, Höpken UE, Uguccioni M, Lipp M, Lanzavecchia A, Sallusto F. Regulation of dendritic cell migration to the draining lymph node: impact on T lymphocyte traffic and priming. J. Exp. Med. 2003;198:615–621. doi: 10.1084/jem.20030448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Giermasz AS, Urban JA, Nakamura Y, Watchmaker P, Cumberland RL, Gooding W, Kalinski P. Type-1 polarized dendritic cells primed for high IL-12 production show enhanced activity as cancer vaccines. Cancer Immunol. Immunother. 2009;58:1329–1336. doi: 10.1007/s00262-008-0648-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 40.Chen M, Huang L, Shabier Z, Wang J. Regulation of the lifespan in dendritic cell subsets. Mol. Immunol. 2007;44:2558–2565. doi: 10.1016/j.molimm.2006.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaka AS, Foster AE, Weiss HL, Rooney CM, Leen AM. Using dendritic cell maturation and IL-12 producing capacity as markers of function: a cautionary tale. J. Immunother. 2008;31:359–369. doi: 10.1097/CJI.0b013e318165f5d2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gautier EL, Huby T, Saint-Charles F, Ouzilleau B, Chapman MJ, Lesnik P. Enhanced dendritic cell survival attenuates lipopolysaccharide-induced immunosuppression and increases resistance to lethal endotoxic shock. J. Immunol. 2008;180:6941–6946. doi: 10.4049/jimmunol.180.10.6941. [DOI] [PubMed] [Google Scholar]
- 43.Elliott B, Scolyer RA, Suciu S, Lebecque S, Rimoldi D, Gugerli O, Musat E, Sharma RN, Lienard D, Keilholz U, et al. European Organization for Research and Treatment of Cancer Melanoma Group Long-term protective effect of mature DC-LAMP+ dendritic cell accumulation in sentinel lymph nodes containing micrometastatic melanoma. Clin. Cancer Res. 2007;13:3825–3830. doi: 10.1158/1078-0432.CCR-07-0358. [DOI] [PubMed] [Google Scholar]
- 44.Park D, Lapteva N, Seethammagari M, Slawin KM, Spencer DM. An essential role for Akt1 in dendritic cell function and tumor immuno-therapy. Nat. Biotechnol. 2006;24:1581–1590. doi: 10.1038/nbt1262. [DOI] [PubMed] [Google Scholar]
- 45.Heckelsmiller K, Beck S, Rall K, Sipos B, Schlamp A, Tuma E, Rothenfusser S, Endres S, Hartmann G. Combined dendritic cell-and CpG oligonucleotide-based immune therapy cures large murine tumors that resist chemotherapy. Eur. J. Immunol. 2002;32:3235–3245. doi: 10.1002/1521-4141(200211)32:11<3235::AID-IMMU3235>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 46.Broomfield SA, van der Most RG, Prosser AC, Mahendran S, Tovey MG, Smyth MJ, Robinson BW, Currie AJ. Locally administered TLR7 agonists drive systemic antitumor immune responses that are enhanced by anti-CD40 immunotherapy. J. Immunol. 2009;182:5217–5224. doi: 10.4049/jimmunol.0803826. [DOI] [PubMed] [Google Scholar]
- 47.Tatsumi T, Huang J, Gooding WE, Gambotto A, Robbins PD, Vujanovic NL, Alber SM, Watkins SC, Okada H, Storkus WJ. Intratumoral delivery of dendritic cells engineered to secrete both interleukin (IL)-12 and IL-18 effectively treats local and distant disease in association with broadly reactive Tc1-type immunity. Cancer Res. 2003;63:6378–6386. [PubMed] [Google Scholar]
- 48.Wiethe C, Dittmar K, Doan T, Lindenmaier W, Tindle R. Enhanced effector and memory CTL responses generated by incorporation of receptor activator of NF-kappa B (RANK)/RANK ligand costimulatory molecules into dendritic cell immunogens expressing a human tumor-specific antigen. J. Immunol. 2003;171:4121–4130. doi: 10.4049/jimmunol.171.8.4121. [DOI] [PubMed] [Google Scholar]
- 49.Nogai A, Siffrin V, Bonhagen K, Pfueller CF, Hohnstein T, Volkmer-Engert R, Brück W, Stadelmann C, Kamradt T. Lipopolysaccharide injection induces relapses of experimental autoimmune encephalomyelitis in nontransgenic mice via bystander activation of autoreactive CD4+ cells. J. Immunol. 2005;175:959–966. doi: 10.4049/jimmunol.175.2.959. [DOI] [PubMed] [Google Scholar]
- 50.Marine JC, Topham DJ, McKay C, Wang D, Parganas E, Stravopodis D, Yoshimura A, Ihle JN. SOCS1 deficiency causes a lymphocyte-dependent perinatal lethality. Cell. 1999;98:609–616. doi: 10.1016/s0092-8674(00)80048-3. [DOI] [PubMed] [Google Scholar]
- 51.Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, Ma A. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289:2350–2354. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.