

Abstract
The importance of canonical transforming growth factor β (TGF-β) and bone morphogenetic protein (BMP) signaling during cartilage and joint development is well established, but the necessity for noncanonical (SMAD-independent) signaling during these processes is largely unknown. TGF-β activated kinase 1 (TAK1) is a MAP3K activated by TGF-β, BMP, and other mitogen-activated protein kinase (MAPK) signaling components. We set out to define the potential role for noncanonical, TAK1-mediated signaling in cartilage and joint development via deletion of Tak1 in chondrocytes (Col2Cre;Tak1f/f) and the developing limb mesenchyme (Prx1Cre;Tak1f/f). Deletion of Tak1 in chondrocytes resulted in novel embryonic developmental cartilage defects including decreased chondrocyte proliferation, reduced proliferating chondrocyte survival, delayed onset of hypertrophy, reduced Mmp13 expression, and a failure to maintain interzone cells of the elbow joint, which were not observed previously in another Col2Cre;Tak1f/f model. Deletion of Tak1 in limb mesenchyme resulted in widespread joint fusions likely owing to the differentiation of interzone cells to the chondrocyte lineage. The Prx1Cre;Tak1f/f model also allowed us to identify novel columnar chondrocyte organization and terminal maturation defects owing to the interplay between chondrocytes and the surrounding mesenchyme. Furthermore, both our in vivo models and in vitro cell culture studies demonstrate that loss of Tak1 results in impaired activation of the downstream MAPK target p38, as well as diminished activation of the BMP/SMAD signaling pathway. Taken together, these data demonstrate that TAK1 is a critical regulator of both MAPK and BMP signaling and is necessary for proper cartilage and joint development. © 2010 American Society for Bone and Mineral Research.
Keywords: TAK1, MAPK, BMP, cartilage, joint
Introduction
Limb development is a complicated process culminating in the formation of a highly functional limb skeleton composed of cartilage, bone, connective tissues, and mobile joints. The limb skeleton originally derives from mesenchymal cells of the lateral-plate mesoderm that migrate, condense, proliferate, differentiate, and undergo terminal maturation. As condensed mesenchymal cells undergo the process of chondrogenesis, several layers of cells at prospective joint regions adopt a nonchondrogenic fate, forming the mesenchymal interzone. Interzone cells ultimately give rise to the synovium, joint capsule, and articular cartilage tissues of adult synovial joints.(1) Genetic studies have implicated several signaling factors in the proper formation and maintenance of joints, including Wnt9a, Wnt4, Erg, Noggin, Gdf5, Bmpr1a, and Bmpr1b.(1–8)
Mesenchymal cells outside interzone regions complete the process of chondrogenesis primarily through the activities of Sox family transcription factors—Sox9, Sox5, and Sox6.(9,10) After initial formation of the individual cartilage rudiments, coupled processes of chondrocyte proliferation and maturation are responsible for longitudinal growth. Chondrocyte proliferation serves as the initial driving force of longitudinal growth, but ultimately proliferating cells near the center of cartilage elements withdraw from the cell cycle and undergo chondrocyte maturation or hypertrophic differentiation. Progression of chondrocyte maturation can be followed via examination of key markers of maturation: Prehypertrophic and early hypertrophic chondrocytes express Indian hedgehog (Ihh) and Runx2, whereas hypertophic chondrocytes generating a mineralizing matrix are characterized by type X collagen (Col10a1) expression, and finally, the most terminally hypertrophic chondrocytes express matrix metalloproteinase 13 (Mmp13), an enzyme that controls degradation and removal of the cartilage matrix.(11) Completion of chondrocyte maturation is necessary for proper vascular invasion, establishment of the marrow cavity, and normal growth plate development.
The transforming growth factor β (TGF-β) superfamily represents a group of molecules critical for normal skeletal growth and development. Recent mouse genetic studies have provided important insights into the contributions of each of these molecules to the processes of chondrogenesis, chondrocyte maturation, and joint morphogenesis. Mouse models lacking the TGF-β type II receptor in chondrocytes (Col2Cre;Tgfbr2f/f) exhibit defects in the postnatal regulation of chondrocyte maturation primarily within the axial skeleton.(12) Deletion of Tgfbr2 in early limb mesenchyme (Prx1Cre;Tgfbr2f/f) resulted in delayed cartilage formation, reduced chondrocyte proliferation, and malformation of joints within the digits.(13) Bone morphogenetic protein (BMP) signaling molecules are also known to be important regulators of chondrogenesis, chondrocyte differentiation, joint formation, proliferation, and apoptosis.(4,14–20). Mice lacking BMP receptors 1a and 1b within cartilage (Col2Cre;Bmpr1af/f, Bmpr1b+/−, and Col2Cre;Bmpr1a f/f;Bmpr1b−/−) or lacking canonical BMP targets Smads 1 and 5 within cartilage (Col2Cre;Smad1f/f;Smad5f/f) have reduced chondrocyte proliferation, delayed onset of chondrocyte maturation, reduced proliferating chondrocyte survival, and delayed progression of terminal maturation.(16,17) Conditional deletion of the BMP ligands, BMP-2 and BMP-4, using the Prx1Cre transgene (Prx1Cre;Bmp2f/f;Bmp4f/f) resulted in shorter and thinner limbs, delayed differentiation and marrow cavity formation, disorganized clearance of hypertrophic chondrocytes, joint fusion of the stylopod and zeugopod elements with little effect on the autopod, and incomplete bone formation.(14) Conventional deletion of another BMP family member, Gdf5, also demonstrated effects on the limb skeleton, resulting in decreased limb length and various joint fusions/abnormalities.(4,8,15)
TGF-β superfamily signaling is initiated when ligand binds to the type II serine/threonine kinase receptor, leading to activation of the type I receptor and induction of downstream signaling mechanisms.(21) Canonical TGF-β and BMP signaling occurs via activation of receptor Smads (R-Smads) 2/3 and 1/5/8, respectively. While the TGF-β and BMP canonical pathways play critical and differential roles during chondrogenesis and chondrocyte maturation, in vitro studies have demonstrated recently a role for noncanonical signaling via the activation of MAPKs. Noncanonical signaling by the TGF-β and BMP pathways is thought to activate MAPK signaling via induction of transforming growth factor β activated kinase 1 (TAK1).(21) On activation, TAK1 directly phosphorylates MKK 3/6, leading to the subsequent phosphorylation and activation of p38.(22,23)
The importance of TAK1 activation by TGF-β and BMP in cartilage remains unclear. In vitro data suggest that noncanonical signaling via MAPKs can regulate chondrogenesis, hypertrophic differentiation, and proliferation via TGF-β-, Gdf5-, and BMP-mediated activation of p38.(24–29) Recent studies aimed directly at MAPK pathway components have suggested that both overexpression and inhibition of the MAPK factors can generate similar in vitro chondrocyte phenotypes, leading investigators to question whether MAPK signaling supports a single event or if it is the balance of its activity that is important.(24,27) Use of these various gain- and loss-of-function mouse models has similarly demonstrated that activation or inhibition of the MAPK pathway results in analogous cartilage and bone phenotypes that manifest primarily during postnatal development. Loss-of-function mouse models, dominant-negative (dn) P38 and Atf2- deficient mice, have demonstrated that disruption of MAPK signaling results in postnatal chondrodysplasia, decreased chondrocyte proliferation, and decreased chondrocyte survival.(30–32) Meanwhile, transgenic mice overexpressing constitutively active MKK6 in chondrocytes are dwarfed, have decreased chondrocyte proliferation, delayed onset of hypertrophic differentiation during embryonic development, and a shortened zone of hypertrophic chondrocytes.(33) Recently, Shim and colleagues(34) showed the importance of TAK1 in postnatal chondrocytes when they analyzed a group of Col2Cre;Tak1f/f mice. Their analyses indicated that loss of Tak1 in chondrocytes resulted in postnatal skeletal defects, including decreased chondrocyte proliferation, increased apoptosis, elbow dislocations, and deficient BMP and MAPK signaling. Despite the clear postnatal skeletal phenotypes presented by the disruption of MAPK signaling components, including TAK1, the roles of these pathways have not been thoroughly examined during embryonic skeletal development. Therefore, we set out to determine whether TAK1 represents an important upstream component of the MAPK and TGF-β/BMP signaling pathways during embryonic cartilage and joint development by deleting floxed Tak1 alleles in cartilage using a different Col2Cre transgenic line and in early limb mesenchyme using the Prx1Cre transgene.
Materials and Methods
Tak1f/f mice were generated as described previously.(35) Tak1f/f mice were crossed with Col2Cre transgenic mice(36) to delete Tak1 floxed alleles within the developing cartilage. The Col2Cre;Tak1f/f mice are referred to as Tak1Col2 mutants. Tak1 deletion within the early limb mesenchyme was performed by crossing Tak1f/f mice with the Prx1Cre transgenic mouse.(37) Prx1Cre;Tak1f/f mice are referred to as Tak1Prx mutants.
Skeletal preparation, histology, in situ hybridization, and immunohistochemistry
Whole embryos for alcian blue/alizarin red skeletal staining were prepared as described previously.(38) Embryos harvested for histology were dissected in PBS, fixed in 10% neutral buffered formalin (NBF), decalcified in 14% EDTA, processed in paraffin, and embedded prior to sectioning at 6 µm. Hematoxylin and eosin (H&E), safranin O, and fast green or alcian blue, hematoxylin, and orange G stains were performed on various sections to identify tissue/cell morphology of the limbs. In situ hybridization was performed on longitudinal forelimb sections using 35S radiolabeled probes as described previously.(39) Many of our antisense probes have been described previously.(3,38–40) Riboprobes not previously described were generated from the following cDNA clones: Wnt9a (30435371, Open Biosystems, Huntsville, AL, USA), Erg (40131153, Open Biosystems), Gdf5,(15) and Agc1 (5345931, Open Biosystems). Immunohistochemistry was performed for p-Smad 1/5/8 (1:100 dilution; Cell Signaling, Danvers, MA, USA) and type II collagen (Thermo Scientific, Freemont, CA, USA) on paraffin sections using standard protocols.
Chondrocyte proliferation and apoptosis
For cell proliferation assays, pregnant female mice were administered BrdU via i.p. injection 2 hours prior to killing. Embryos were dissected, fixed, decalcified, and processed as described earlier. BrdU immunohistochemistry was performed on three limb sections for three separate embryos of each genotype using the Invitrogen BrdU Kit (Invitrogen, Carlsbad, CA, USA). Proliferation rate is reported as the percentage of BrdU-positive nuclei in the proximal humerus relative to wild-type littermates. Changes in chondrocyte apoptosis were determined using the In Situ Cell Death Kit (Roche, Indianapolis, IN, USA) according to manufacturer's protocol and reported as absolute number of terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate–biotin nick end labeling (TUNEL)+ apoptotic cells in the proximal humerus. Cells were counted and averaged for three separate wild-type and mutant sections from three independent litters.
Western blot analysis of limb cartilage
Dissected cartilage for Western blotting was lysed using RIPA buffer (150 mM NaCl, 1.0% Triton-X, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris. pH 8.0) containing minicomplete protease inhibitor (Roche) and PhosSTOP phosphatase inhibitor tablets (Roche). Whole-cell lysate was quantified, normalized, and run on 10% Bis-Tris gels (Invitrogen). Blots were incubated with primary antibodies including total TAK1 (Cell Signaling) and β-actin (Sigma). Blots then were washed in Tris-buffered saline with Tween-20 (TBST), incubated in secondary antibody, and visualized using chemiluminescence (Pierce, Rockford, IL, USA).
Cell culture and signaling
Primary sternal chondrocytes were isolated from 2- to 3-day-old Tak1f/f mice as described previously(41–43) or from E14.5 cartilage elements of the limb. All cells were seeded as follows: 1 × 106 cells per well in a six-well plate and maintained in DMEM containing 10% FBS and 1% penicillin/streptomycin. In vitro deletion was performed using Ad5-CMV-GFP (control) or Ad5-CMV-Cre viruses (Baylor Vector Development, Houston, TX, USA). Cells were infected for 48 hours and allowed to recover overnight before treatment. For alkaline phosphatase staining, cells were fixed in 4% paraformaldehyde, washed, and stained using NTB/BCIP (Thermo Scientific).
Rat chondrosarcoma (RCS) cells were cultured in DMEM containing 10% FBS and 1% penicillin/streptomycin.(44) For Western blotting, cells were serum-starved overnight, pretreated with the cis-enol resorcylic acid lactone–selective TAK1 inhibitor (TI-2), LLZ1640-2 (Bioaustralis, Smithfield, NSW, Australia),(45) the inactive homologue (TI-4), LLZ1640-4, or DMSO (Sigma) and then treated for 60 minutes with 100 ng/mL of BMP2 (Peprotech, Rocky Hill, NJ, USA). Cells were lysed using NP-40 lysis buffer (150 mM NaCl, 1% NP-40, 50 mM Tris-HCl, pH 8.0) containing mini-complete protease inhibitor (Roche), PhosSTOP phosphatase inhibitor tablets (Roche), 1 mM sodium orthovanadate, 10 mM NaF, and 0.5 mM okadaic acid. Lysates were run on 10% Bis-Tris gels (Invitrogen), and antibodies used included TAK1, pSmad 1/5/8, pP38, tP38, atubulin (Cell Signaling), and total Smad 1/5/8 (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Gene expression studies used RNA extracted with Trizol reagent (Invitrogen) according to manufacturer's protocol. Real-time RT-PCR was performed on a RotorGene RT Machine (Qiagen, Valencia, CA, USA) using 1 µL of cDNA and PerfeCTa SYBR Green reagent (Quanta BioSciences, Gaithersburg, MD, USA). For reporter assays, cells were transfected with 12×-SBE luciferase reporter(46) using Fugene-HD (Roche) according to the manufacturer's protocol. Cells were serum-starved overnight and pretreated with TAK1 inhibitor for 30 minutes prior to treatment with 100 ng/mL of BMP2. Luciferase activity was measured 24 hours later using the Dual-Promoter Luciferase Assay Kit (Promega) and normalized to Renilla luciferase. Triplicates of each treatment group were analyzed, and statistical significance was determined by t test.
Results
Loss of Tak1 within cartilage results in neonatal lethality
Tak1 is expressed throughout the cartilage and surrounding tissues during endochondral bone development and during in vitro chondrocyte maturation (Supplemental Fig. 1A, B). Interestingly, cartilage deletion of Tak1 floxed alleles using a Col2Cre transgenic line resulted in neonatal lethality immediately following birth. X-ray and micro–computed tomographic (µCT) analyses of newborn pups on postnatal day 0 (P0) demonstrated that Col2Cre;Tak1f/f (Tak1Col2) mutant mice exhibit decreased skeletal growth and have a significantly smaller ribcage compared with littermate controls (Supplemental Fig. 2A). Alcian blue and alizarin red skeletal staining for cartilaginous components of the airway revealed an undermineralized hyoid and a fusion of the hyoid to the larynx in Tak1Col2 mutant pups (Supplemental Fig. 2B). These data and the visual observation of pups at birth indicated that Tak1Col2 mutants do not survive owing to respiratory distress.
Tak1Col2 mutants display defects in cartilage proliferation, maturation, and apoptosis
Examination of limbs isolated from alcian blue– and alizarin red–stained embryos at 14.5 days of gestation (E14.5) clearly demonstrated a decrease in the size of both forelimb (Fig. 1A, A1 and A2) and hindlimb (Fig. 1A, A3 and A4) elements of Tak1Col2 mutants compared with wild-type littermate controls. Western blot analysis performed on protein extracted from E14.5 humeri cartilages verified deletion of Tak1 in chondrocytes of the developing skeleton, although low levels of TAK1 protein remained detectable likely owing to contaminating perichondrium and surrounding soft tissues inadequately removed during isolation procedures (Fig. 1B). H&E staining of humerus sections at E14.5 revealed a shorter limb in Tak1Col2 mutants and a much smaller hypertrophic region (Fig. 1C, C1 and C2). Molecular analyses of chondrocyte maturation via in situ hybridization demonstrated a delay in the onset of hypertrophy, as evidenced by the decreased domain of expression for the prehypertrophic marker Ihh (Fig. 1C, C3 and C4), the hypertrophic marker Col10a1 (Fig. 1C, C5 and C6), and the complete absence of the terminal hypertrophic marker Mmp13 (Fig. 1C, C7 and C8). H&E staining of E15.5 humerus sections demonstrated that the primary center of ossification had begun to form in both wild-type and Tak1Col2 mutant limbs (Fig. 1C, C9 and C10), but uniform development of the marrow cavity in the Tak1Col2 mutant was incomplete owing to a continuous delay in chondrocyte maturation (Fig. 1C, C9 abd C10, red dashed contours). Molecular analysis of the maturational markers Ihh, Col10a1, and Mmp13 demonstrated that the spatial expression domains of Ihh (Fig. 1C, C11 and C12) and Col10a1 (Fig. 1C, C13 and C14) are less well separated in Tak1Col2 mutants compared with littermate controls, whereas Mmp13 expression (Fig. 1C, C15 and C16) is markedly reduced in Tak1Col2 mutant limbs at E15.5. To determine if the delayed marrow development was due to impaired vascularization, we performed in situ hybridization to assess angiopoietin 1 (Angpt1) expression within the developing marrow cavity of wild-type and Tak1Col2 mutant limbs. However, the results were not remarkable (data not shown), suggesting that vascularization of the primary center of ossification occurred normally, and the primary defect is in chondrocyte maturation. Interestingly, analyses of E18.5 histologic sections and in situ hybridization for the chondrocyte maturation markers demonstrated a relatively normal appearance of Ihh and Col10a1 expression domains in Tak1Col2 mutants at E18.5 (Fig. 1C, C17 through C22) as well as the distinct presence of Mmp13 expression at the chondroosseous junction, although Mmp13 expression remained slightly reduced (Fig. 1C, C23 amd C24), and the overall growth of the elements still was delayed.
Fig. 1.
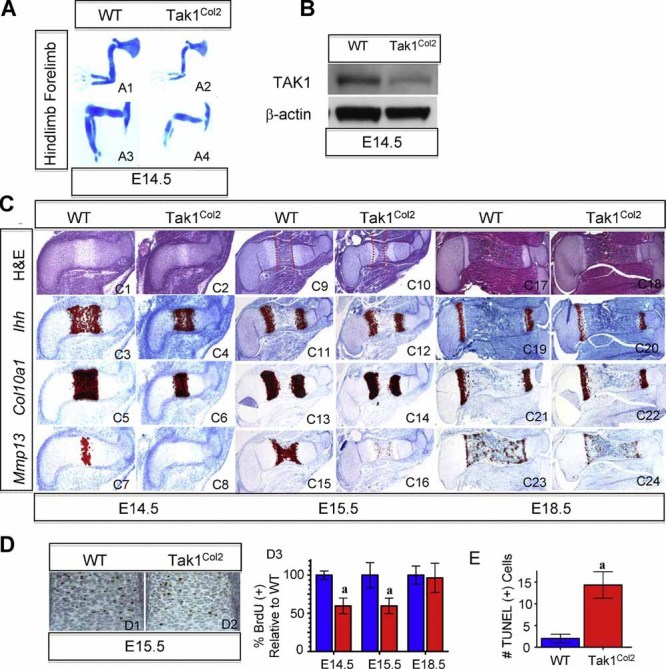
TAK1 regulates chondrocyte proliferation, maturation, and immature chondrocyte survival. (A) Alcian blue skeletal staining of forelimbs (A1, A2) and hindlimbs (A3, A4) from wild-type and Tak1Col2 embryos at E14.5. (B) Lysates from cartilage of E14.5 humerus immunoblotted using anti-TAK1 antibody. (C) Hematoxylin and eosin staining of E14.5 (C1, C2), E15.5 (C9, C10), and E18.5 (C17, C18) humerus sections. (C9, C10) Red dashed lines indicate the chondroosseous junction. In situ hybridization for Ihh, Col10a1, and Mmp13 at E14.5 (C3–C8), E15.5 (C11–C16), and E18.5 (C19–C24). (D) Representative IHC for BrdU of E15.5 wild-type (D1) and Tak1Col2 (D2) embryos. (D3) Quantification of BrdU of cells in the proximal humerus of wild-type (blue) and Tak1Col2 (red) embryos at E14.5, E15.5, and E18.5. (D2) Quantification of TUNEL+ cells in the proximal humerus of wild-type and Tak1Col2 embryos at E18.5 (ap < .05 versus wild type).
To further address cartilage growth discrepancies between Tak1Col2 mutants and littermate controls, we performed BrdU and TUNEL labeling experiments on humeral sections of E14.5, E15.5, and E18.5 embryos. A representative section of BrdU immunohistochemistry (IHC) in the proliferative cells of the proximal humerus demonstrated that loss of Tak1 leads to decreased chondrocyte proliferation at E15.5 (Fig. 1D, D1 and D2). Quantification of BrdU incorporation at E14.5 and E15.5 revealed that the loss of Tak1 results in a significant decrease (40%) in chondrocyte proliferation, but by E18.5, chondrocyte proliferation is restored to levels near that of wild-type littermate controls (Fig. 1D, D3). TUNEL labeling studies further demonstrated that Tak1Col2 mutants exhibit significant elevations in resting/proliferating zone chondrocyte apoptosis of the developing humerus at E14.5 and E18.5 (Fig. 1E; and data not shown). Collectively, these data demonstrate that loss of Tak1 in cartilage resulted in early delays in chondrocyte maturation, reductions in chondrocyte proliferation, and increased levels of immature chondrocyte apoptosis.
Tak1Prx mice exhibit delays in both the onset and progression of chondrocyte maturation
Owing to the neonatal lethality and interesting cartilage phenotypes of the Tak1Col2 mice, we generated Prx1Cre;Tak1f/f mice (Tak1Prx) to determine whether TAK1 controls endochondral ossification in a broader context during both embryonic and postnatal development. While the Prx1Cre transgene targets both the cartilage and surrounding tissues derived from the early limb mesenchyme,(37) it remains restricted primarily to the appendicular skeleton and thus would avoid any potential respiratory complications responsible for the neonatal death of Tak1Col2 mice. Many of these animals survive to the weaning stage (3 to 4 weeks), although additional skeletal anomalies affect their long-term feeding and survival (Supplemental Fig. 2C).
We first analyzed the limb skeleton phenotypes of Tak1Prx mutant mice at E14.5 and E18.5. Prx1Cre mediated deletion of Tak1 was verified by immunoblotting for total TAK1 protein using lysates obtained from isolated limb cartilage elements of E14.5 embryos (Fig. 2A). H&E staining (Fig. 2B, B1 and B2), as well as in situ hybridization, on humeral sections from E14.5 wild-type and Tak1Prx mutants supports a role for TAK1 in promoting the onset of chondrocyte hypertrophy because the expression domains of Ihh (Fig. 2B, B3 and B4), Col10a1 (Fig. 2B, B5 and B6), and Mmp13 (Fig. 2B, B7 and B8) were again both markedly reduced in size and level of expression. Interestingly, histologic examination of Tak1Prx embryos at E18.5 indicated that in addition to the early delays in the onset of chondrocyte hypertrophy observed at E14.5 (Fig. 2B), loss of Tak1 in surrounding mesenchymal tissues delayed the progression of chondrocyte maturation toward the terminal hypertrophic state, which was not observed in the Tak1Col2 embryos at this stage. H&E staining clearly demonstrated that E18.5 Tak1Prx mutants have thinner cartilage elements and surrounding perichondrium, a disorganized and shortened columnar zone of chondrocytes (brackets), and an increased size of the hypertrophic zone (double-headed arrows) (Fig. 2C, C1 through C4). In situ hybridization for chondrocyte maturation markers was performed on the E18.5 humeral sections of Tak1Prx and wild-type embryos. Tak1Prx mutants exhibited reduced Ihh expression (Fig. 2C, C5 and C6), expansion of the Col10a1 expression domain (Fig. 2C, C7 and C8) corresponding to the increased size of the hypertrophic region, and an expanded domain of Mmp13 expression within the terminally hypertrophic chondrocyes at the chondroosseus junction (Fig. 2C, C9 and C10). Assessment of BrdU incorporation at E14.5 and E18.5 in Tak1Prx embryos reveals that in addition to the decreased proliferation seen at E14.5 in Tak1Col2 embryos, loss of Tak1 in limb mesenchyme results in a decrease in proliferation that persists at E18.5 (Fig. 2D). Similar to Tak1Col2 mutants, Tak1Prx mice exhibited increased apoptosis in proliferating chondrocytes without any apparent change in apoptosis at the ends of the growth plate (Fig. 2E). Taken together, these data argue that TAK1 not only regulates chondrocyte proliferation, immature chondrocyte survival, and the onset of chondrocyte hypertrophy in cartilage but also controls the organization of columnar chondrocytes and the progression of chondrocyte terminal maturation via signaling within adjacent mesenchymal derived tissues.
Fig. 2.
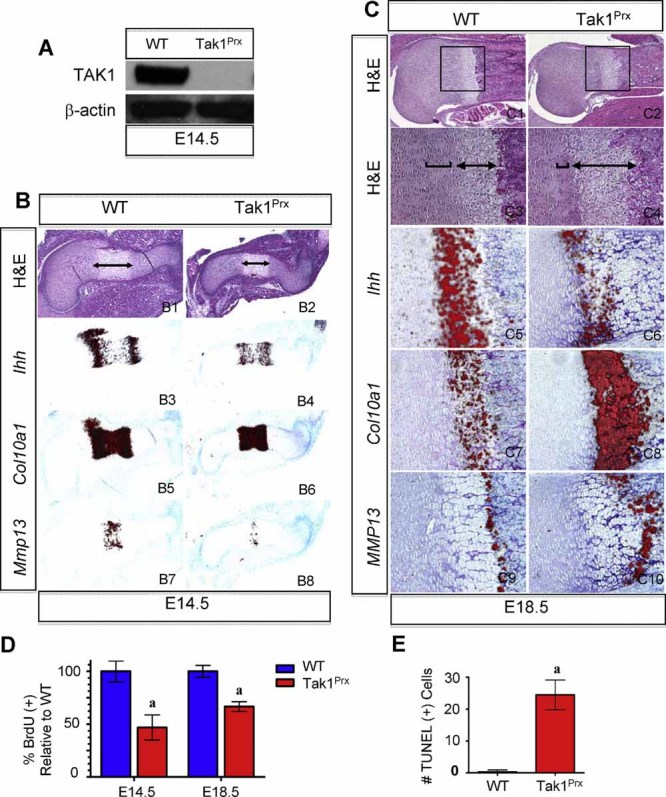
TAK1 indirectly regulates columnar chondrocyte organization and the progression of terminal maturation. (A) Immunoblot for TAK1 protein isolated from humerus cartilage of E14.5 wild-type and Tak1Prx embryos. (B) Histology and in situ hybridization of E14.5 wild-type and Tak1Prx humerus sections for H&E (B1, B2), Ihh (B3, B4), Col10a1 (B5, B6), and Mmp13 (B7, B8). (C1–C4) Double-sided arrows indicate hypertrophic region. H&E staining of proximal humerus from wild-type (C1, C3) and Tak1Prx (C2, C4) embryos at E18.5. Black boxes (C1, C2) indicate region of ×20 magnification. (C3, C4) Brackets denote height of columnar zone of chondrocytes. In situ hybridization of E18.5 wild-type and Tak1Prx humerus sections for Ihh (C5, C6), Col10a1 (C7, C8), and Mmp13 (C9, C10). (D) BrdU incorporation of E14.5 and E18.5 wild-type (blue bars) and Tak1Prx (red bars) embryos. (E) Quantitation of TUNEL+ cells in the proximal humerus at E18.5 (ap < .05 versus wild type).
Loss or inhibition of TAK1 delays normal and BMP-induced chondrocyte maturation
To further define the role of TAK1 in regulating chondrocyte development, we performed in vitro chondrocyte maturation experiments using Tak1f/f sternal chondrocytes infected with Cre and control Gfp adenoviruses (Ad5-CMV-Cre-GFP and Ad5-CMV-GFP) or wild-type sternal chondrocytes treated with a pharmacologic TAK1 inhibitor or inactive homologue. The sternal chondrocyte model is a well-developed in vitro model of chondrocyte maturation(41–43) that was well suited for our study because cartilage in the sternum of Tak1Col2 mutants demonstrates maturational defects similar to those noted in Tak1Col2 developing limbs (Supplemental Fig. 3B). Alkaline phosphatase staining revealed a significant delay in chondrocyte maturation, as evidenced by the decreased staining over an 8-day differentiation period in Tak1-deleted cultures relative to Green Fluorescent Protein (GFP) controls (Fig. 3A). In order to verify that this difference in staining and maturation was not due to an absence of cells or a change in cell phenotype, we also stained the cultures with alcian blue, demonstrating that Tak1-deleted cultures maintained cell viability and the ability to produce matrix (Fig. 3A). The efficacy of the Cre-mediated deletion was approximately 85%, as determined by RT-PCR (Fig. 3B, B1) using primers designed to exon 1 of Tak1. Additionally, immunoblots for total TAK1 on whole-cell lysates following Cre infection demonstrated a near-complete loss of TAK1 protein (Fig. 3B, B2). Similar chondrocyte maturation experiments also were performed using daily treatments of the selective TAK1 inhibitor LLZ1640-2 (TI-2), the inactive homologue LLZ1640-4 (TI-4), or DMSO as an additional negative control. Treatment of sternal chondrocyte cultures with the TI-2 inhibitor (3 µM) displayed a robust suppression in chondrocyte maturation compared with either DMSO or TI-4 inactive homologue (3 µM) treatments (Fig. 3C). Identical effects were observed using primary chondrocytes isolated from E14.5 cartilage elements and treated with the TI-2 TAK1 inhibitor (Supplemental Fig. 4A). Treatment of sternal chondrocyte cultures with 3 µM TI-2 every other day (Fig. 3D) or with daily TI-2 treatments at reduced concentrations (data not shown) resulted in a less severe suppression of chondrocyte maturation.
Fig. 3.
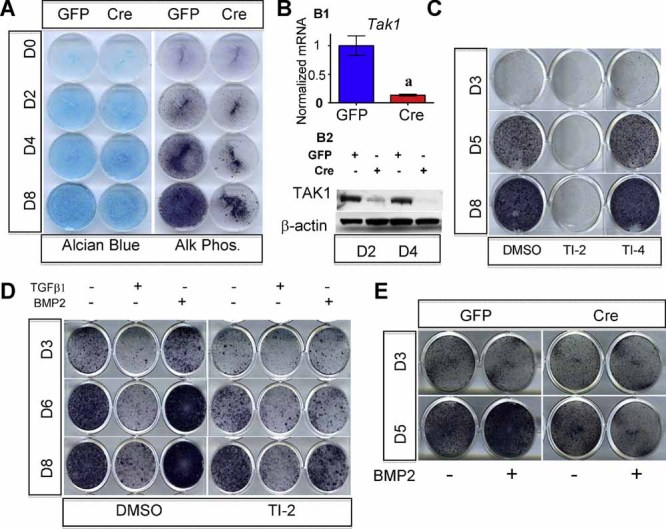
TAK1 regulates normal and BMP-mediated chondrocyte maturation. (A) Alcian blue and alkaline phosphatase (AP) staining of sternal chondrocytes isolated from Tak1f/f pups and infected with Ad5-CMV-GFP (control) or Ad5-CMV-Cre. (B) RT-PCR for exon 1 of the Tak1 gene on mRNA isolated from primary sternal chondrocytes (B1). Immunoblot for total TAK1 in lysates isolated from primary sternal chondrocytes after infection (B2). (C) Wild-type primary sternal chondrocytes treated daily with DMSO, 3 µM TI-2 (LLZ1640-2), or 3 µM TI-4 (LLZ1640-4) and stained for AP on indicated days. (D) AP staining of wild-type primary sternal chondrocytes treated every other day with DMSO or TI-2 (3 µM) and either no treatment, TGF-β (5 ng/mL), or BMP-2 (100 ng/mL). (E) AP staining of Ad-GFP- and Ad-Cre-infected primary sternal chondrocytes in untreated and BMP-2-treated (100 ng/mL) culture.
To determine if TAK1 is required for TGF-β superfamily signaling and the subsequent regulation of chondrocyte maturation, we treated sternal chondrocyte cultures with either TI-2 or DMSO in the presence or absence of TGF-β1 (5 ng/mL) or BMP-2 (100 ng/mL) recombinant proteins. In order to visualize the BMP and TGF-β effects on alkaline phosphatase (AP) staining, we treated cultures with 3 µM TI-2 every other day. AP staining demonstrated that DMSO control cultures underwent normal chondrocyte maturation during the 8-day differentiation period. Treatment with TGF-β1 significantly impaired AP staining and chondrocyte maturation, whereas BMP-2 treatment enhanced or accelerated chondrocyte maturation compared with the DMSO-treated control (Fig. 3D). Treatment of these cultures with TI-2 resulted in decreased AP staining in both the presence and absence of BMP-2 (Fig. 3D). Interestingly, when the same TAK1 inhibitor treatments were performed in the presence of TGF-β1, TI-2 did not affect the ability of TGF-β1 to suppress chondrocyte maturation (Fig. 3D). Similar experiments also were performed using Tak1f/f sternal chondrocytes infected with Cre and control Gfp adenoviruses. Cre-mediated deletion of Tak1 resulted in impaired normal and BMP-2-induced chondrocyte maturation, as measured by AP staining, during a 5-day maturation period (Fig. 3E). All these data indicate that loss of Tak1 or inhibition of TAK1 function disrupts BMP- but not TGF-β-mediated effects on chondrocyte maturation.
Loss or inhibition of TAK1 impairs both MAPK and BMP signaling pathways
To identify the potential molecular mechanisms underlying the impaired BMP responsiveness and chondrocyte maturation observed within TAK1-inhibited or -deficient cultures, we first performed studies analyzing the activation of canonical and noncanonical BMP signaling targets Smad 1/5/8 and p38, respectively. Treatment of sternal chondrocytes with TAK1 inhibitor TI-2 (LLZ1640-2, 3 µM) resulted in decreased Smad 1/5/8 and p38 phosphorylation (Fig. 4A, A1), as well as decreased luciferase activity from the BMP-responsive reporter 12 × -SBE (Fig. 4A, A2). To further support our findings, we verified that loss of Tak1 via Cre-mediated deletion decreased BMP-2-induced phosphorylation of Smads 1/5/8 (Fig. 4B). Additionally, treatment of RCS cells with varying concentrations of TI-2 demonstrated a dose-response for TAK1 regulation of Smad 1/5/8 phosphorylation, noggin expression, and the induction of the 12 × -SBE luciferase reporter (Supplemental Fig. 4C). Collectively, these data suggest that TAK1 affects both canonical and noncanonical BMP signaling in vitro.
Fig. 4.
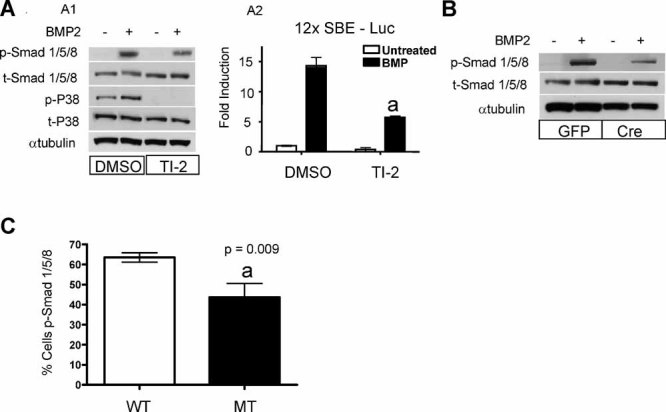
TAK1 regulates both MAPK and BMP signaling. (A1) Immunoblot of whole-cell lysates isolated from control and TI-2 (LLZ1640-2)–pretreated, serum-starved wild-type primary chondrocytes stimulated with BMP-2 (100 ng/mL) for 1 hour. (A2) 12 × -SBE luciferase assay performed on serum-starved wild-type primary sternal chondrocytes pretreated with DMSO or TI-2 and stimulated ± BMP-2 for 24 hours. (B) Immunoblot from primary sternal chondrocyte whole-cell lysates after Ad-Cre-mediated deletion of TAK1. (C) Quantification of pSmad 1/5/8 IHC on humerus of E14.5 wild-type and Tak1Col2 embryos (ap < .05 versus DMSO + BMP-2 control).
To directly assess the effect of Tak1 deletion on canonical BMP signaling in vivo, we performed IHC for p-Smad 1/5/8 on histologic sections of the humerus from E14.5 wild-type and Tak1 mutants. Analyses identified a significant decrease (∼32%) in the number of p-Smad 1/5/8 staining nuclei in the proliferating and prehypertrophic chondrocyte regions of Tak1 mutants (Fig. 4C). Furthermore, in situ hybridization of Tak1 mutant growth plates exhibit reduced levels of the BMP target gene Id1 (data not shown). These data strongly suggest that TAK1 plays an important role in regulating the BMP signaling pathway during chondrocyte maturation in vivo.
Tak1Col2 and Tak1Prx mutants exhibit additional defects in joint and skeletal development
Whole-mount alcian blue and alizarin red staining of E18.5 Tak1Col2 and Tak1Prx embryos revealed additional abnormalities during skeletal development caused by the tissue-specific deletions of Tak1. Targeted deletion of Tak1 in type II collagen–expressing cells resulted in a shorter axial skeleton, a domed-shape skull, a smaller ribcage with delayed chondrocyte maturation, cervical vertebral fusions, and a single joint fusion of the radius and humerus within the appendicular skeleton (Fig. 5A, A1 through A8 and Supplemental Fig. 3A, B). Deletion of Tak1 in the early limb mesenchyme did not alter the size of the ribcage, although it did result in an incomplete fusion of the sternum at midline (Supplemental Fig. 3C). The axial skeleton was not significantly affected in these mutants (Fig. 5A, A9 and A10) because most axial cartilages are not targeted by the Prx1Cre transgene. Tak1Prx embryos did, however, exhibit humeroradial fusions in the forelimbs (Fig. 5A, A15 and A16) identical to those observed in Tak1Col2 embryos. Skeletal staining of Tak1Prx embryos also exhibited an absence of the deltoid tuberosity (Fig. 5A, A11 and A12), patellar fusions of the knee (Fig. 5A, A13 and A14), and multiple other joint fusions in both the forelimbs and hindlimbs (Fig. 5A, A11 through A16, C, C1 through C8, and data not shown). Histologic examination of Tak1Prx limb sections at E18.5 highlights additional fused-cartilage elements, including frequent humeroulnar fusions (Fig. 5B, B1 and B2), patellar knee fusions (Fig. 5B, B3 and B4), occasional lateral knee fusions (Fig. 5B, B5 and B6), fusion of the carpals (Fig. 5B, B7 and B8), and fusion of tarsal elements (Fig. 5B, B9 and B10). Of note, Tak1Prx mutant knees also lacked histologically defined synovial or meniscal tissues at E18.5 (Fig. 5B, B3 and B4, and data not shown). Since the elbow joints for both Tak1Col2 and Tak1Prx embryos displayed cartilaginous fusions, we compared histologic sections from the mutant and wild-type joints at E14.5 and E18.5 to determine the onset and extent of the defects (Fig. 5C, C1 through C8). At E14.5, mutant embryos showed the appearance of loose mesenchymal cells surrounding the joint region, but in place of a well-defined joint containing interzone cells, there existed only disorganized round mesenchymal-like cells that were morphologically distinct from the adjacent epiphyseal chondrocytes. (Fig. 5C, C1, C2, C5, and C6). By E18.5, when the joint is more mature, the Tak1Col2 and Tak1Prx mutants lacked defined synovial tissues within and surrounding the fused elbow joints, whereas intervening round mesenchymal-like cells developed into regions of disorganized chondrocytes exhibiting little histologic staining for proteoglycans (Fig. 5C, C3, C4, C7, and C8, and data not shown).
Fig. 5.
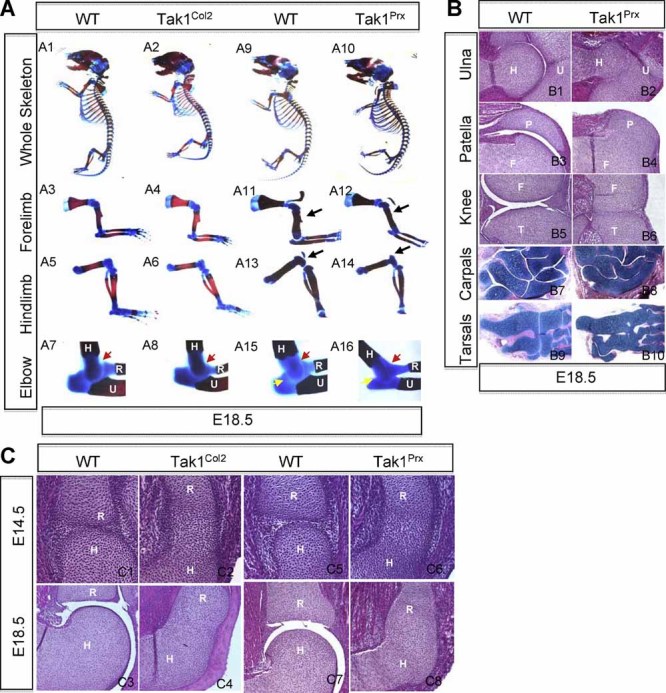
TAK1 is required for normal skeletal development and joint morphogenesis. (A) Alcian blue and alizarin red staining of E18.5 wild-type, Tak1Col2, and Tak1Prx embryos. Black arrow (A11, A12) indicates deltoid tuberosity and fusion of the patella (A13, A14) and humerus (H), radius (R), and ulna (U) at the elbow (A7, A8, A15, A16). Red arrows (A7, A8, A15, A16) indicate fusion of humerus and radius. (A15, A16) Yellow arrows indicate lateral fusion of the humerus (H) and ulna (U). (B) H&E staining of E18.5 wild-type and Tak1Prx embryos at the lateral humeroulnar joint (B1, B2), the femorpatellar joint (B3, B4), and the lateral aspect of the knee joint (B5, B6). (B7–B10) Alcian blue, hematoxylin, and orange G staining of carpals (C5, C6) and tarsals (C7, C8) at E18.5 in wild-type and Tak1Prx embryos. (C) H&E staining of the humeroradial joint of wild-type (C1, C3, C5, C7), Tak1Col2 (C2, C4), and Tak1Prx (C6, C8) embryos at E14.5 (×20 magnification) and E18.5 (×10 magnification).
To more specifically identify the cellular and molecular changes associated with the joint defects observed in Tak1Prx and Tak1Col2 mutants, we analyzed the expression of known regulators of joint and cartilage development and quantified the proliferation and apoptosis of mesenchymal cells within the presumptive joint regions. In situ hybridization for specific regulators of joint development, Wnt9a, Erg, and Gdf5 (Fig. 6A, A1 through A12) were performed on E14.5 sections. Wnt9a, a molecule with antichondrogenic activity, is normally enriched in the most medial layers of the interzone, as well as mesenchymal populations surrounding the joint space (Fig. 6A, A1 and A7). However, Tak1Prx and Tak1Col2 mutant embryos displayed only weak Wnt9a expression in surrounding mesenchymal tissues and a complete loss of expression within the joint forming region (Fig. 6A, A2 and A8). Gdf5 and Erg, molecules with chondrogenic activities, are normally highly expressed in multiple layers of the interzone, the articular cartilage, and within distinct wedges at the sites of cavitation (Fig. 6A, A3, A5, A9, and A11). Interestingly, Tak1Prx and Tak1Col2 mutants displayed dramatically reduced Gdf5 expression throughout the presumptive interzone, whereas Tak1Col2 embryos maintained some expression in the surrounding mesenchyme (Fig. 6A, A4 and A10). On the other hand, Erg expression was maintained at lower levels within cells of both the presumptive interzone and surrounding mesenchyme (Fig. 6A, A5, A6, A11, and A12).
Fig. 6.
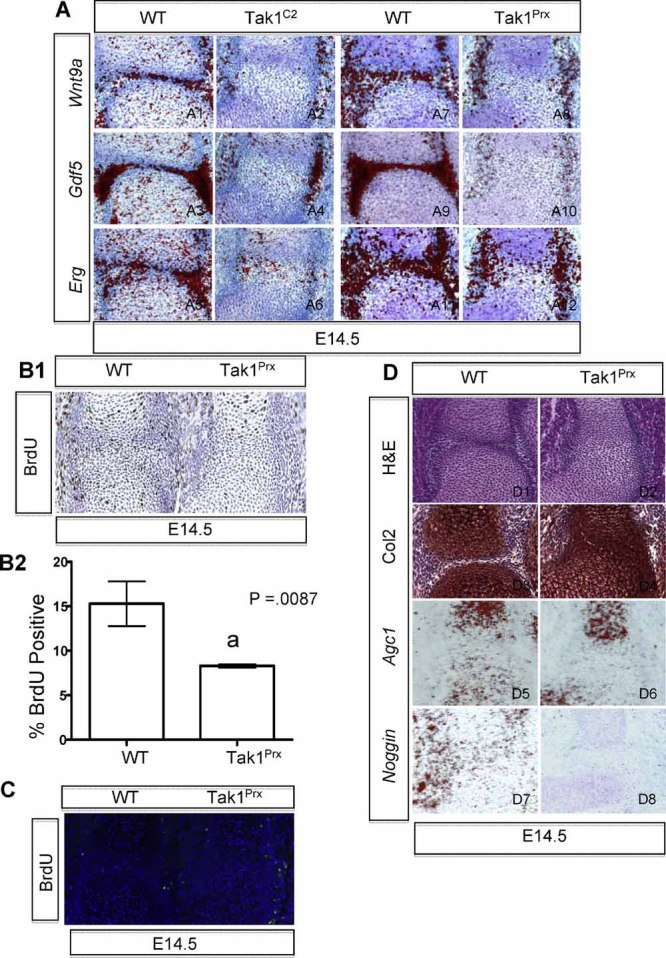
TAK1 regulates proper interzone formation/maintenance during joint development. In situ hybridization for Wnt9a, Gdf5, and Erg in the developing humeroradial joint of E14.5 Tak1Col2 (A1–A6) and Tak1Prx (A7–A12) embryos. BrdU IHC of humeroradial joint of E14.5 Tak1Prx embryos (B1) and quantification from three sections each of wild-type and Tak1Prx embryos (B2). (C) Representative TUNEL staining of humeroradial joint in E14.5 Tak1Prx embryos. (D) E14.5 humeroradial joint of Tak1Prx embryos demonstrating H&E (D1, D2), Col2 IHC (D3, D4), Agc1 in situ hybridization (D5, D6), and noggin in situ hybridization (D7, D8).
To determine the cellular effects caused by misexpression of these joint regulators, we performed proliferation (BrdU) assays, apoptosis (TUNEL) assays, and in situ hybridization or immunohistochemical studies for markers of chondrocyte differentiation on joint sections from Tak1Prx mice. Our data demonstrate that loss of Tak1 in the developing joint results in reduced proliferation of the mesenchyme and chondrocytes within the presumptive joint region (Fig. 6B, B1 and B2), but no significant difference was observed for mesenchymal cell or chondrocyte apoptosis (Fig. 6C). Owing to the aberrant chondrocyte-like cell morphology found within the presumptive interzone of Tak1Prx mice, we assessed the expression of chondrocyte differentiation markers on E14.5 joint sections. Immunohistochemistry for type II collagen demonstrates a continuous expression profile of the immature chondrocyte marker spanning the presumptive joint region in Tak1Prx mutants, whereas wild-type mice exhibit a distinct break in type II collagen expression at the interface of opposing cartilage elements (Fig. 6D, D3 and D4). In situ hybridization analyses for aggrecan1 (Agc1), a marker of maturing chondrocytes, revealed that the immature type II collagen–expressing cells within the presumptive interzone region of Tak1Prx mutants do not express Agc1 (Fig. 6D, D5 and D6). Both wild-type and Tak1Prx mutants demonstrate that Agc1 expression is observed only in maturing chondrocytes adjacent to the joint region at this stage. Finally, since we identified TAK1 as a regulator of BMP signaling in chondrocytes, and since the BMP target gene noggin is important in joint development, we performed in situ hybridization studies to determine the effect that the absence of Tak1 has on noggin expression. Compared with wild-type controls, Tak1Prx mutant sections exhibit a complete absence of noggin expression surrounding the presumptive joint at E14.5 (Fig. 6D, D7 and D8). Therefore, TAK1 represents an essential regulator of interzone formation and/or maintenance, as well as, BMP signaling, during synovial joint development.
Discussion
TAK1 regulates chondrocyte proliferation, maturation, and apoptosis
In our model, cartilage removal of Tak1 (Tak1Col2) resulted in neonatal lethality, reduced chondrocyte proliferation, reduced proliferating chondrocyte survival, and delayed chondrocyte maturation within developing embryonic cartilages. It is of note that a recent study by Shim and colleagues(34) deleted Tak1 in chondrocytes using a different Col2Cre transgenic line that resulted in some similar cartilage phenotypes, except Shim and colleagues(34) observed these defects in only postnatal mutant mice. The general similarities between the two studies include decreased chondrocyte proliferation, general skeletal delay, alterations in BMP and MAPK signaling, and elbow abnormalities. Critical differences between the two models include the ability of their Tak1Col2 mutants to survive postnatally for 2 to 3 weeks, whereas our Tak1Col2 mutants die prior to birth, and the timing for the onset of cartilage phenotypes.(34) Compared with the previous study, it is likely that our mice exhibited an earlier deletion of the Tak1 floxed alleles owing to the use of a different Col2Cre transgenic mouse(36,47) and therefore produced a more profound and slightly different embryonic cartilage phenotype.(34) Interestingly, our Tak1Col2 mutants displayed a dramatic phenotype during early stages of chondrocyte maturation (E14.5) that appeared to diminish progressively at later stages of development (E18.5). This is highlighted by the dramatic decrease in chondrocyte proliferation (∼40%) that was later restored to near-wild-type levels even in the presence of continued TAK1 expression. Tak1Col2 mutants also demonstrated an increase in immature chondrocyte apoptosis that was not evident in maturing chondrocytes. The combination of a transient reduction in chondrocyte proliferation and increased proliferating chondrocyte cell death may serve as an explanation for the apparent temporary delay in chondrocyte maturation. Although Tak1Col2 mutants maintain identifiable cartilage phenotypes until death at P0, it is difficult to ascertain whether the in vivo chondrocyte maturation phenotype is progressive or simply a reflection of the early proliferative and apoptotic defects. Our in vitro studies examining the effects of loss or inhibition of TAK1 on chondrocyte maturation suggest that the delay in chondrocyte maturation is progressive and cannot be accounted for solely by decreased proliferation and increased proliferating chondrocyte apoptosis. In these experiments, cell proliferation is reduced dramatically by days 3 to 4 in culture owing to contact inhibition, and yet the progressive delay in chondrocyte maturation, as evidenced by AP staining, is still observed in cultures well beyond this time point. Cell viability was maintained throughout the culture period and thus cannot account for the dramatically reduced levels of AP staining during the 8-day differentiation period. Collectively, these data argue that TAK1 continuously regulates the various stages of chondrocyte proliferation, maturation, and apoptosis.
In addition to deletion of Tak1 in chondrocytes using the Col2a1Cre transgenic mouse, we further investigated the effects of TAK1's regulation of embryonic chondrocyte maturation via deletion of Tak1 in the developing limb mesenchyme using the Prx1Cre transgenic mouse. Our additional analyses of Tak1Prx mutants revealed that these embryos recapitulated the Tak1Col2 mutant limb cartilage phenotypes, although several additional defects were observed, including a thinner perichondrium, a disorganized and shortened columnar zone of chondrocytes, a persistent reduction in chondrocyte proliferation, and an expanded hypertrophic domain resulting from the delayed progression in chondrocyte terminal maturation. Since the Prx1Cre transgene targets both the chondrocytes and the surrounding perichondrium, it is likely that the additional defects observed in the growth plates of Tak1Prx embryos are due to deletion of Tak1 within perichondrial cells and an imbalance in signaling between these populations of cells. Several studies have reported the importance of the perichondrium in promoting organization of the columnar zone and regulating chondrocyte proliferation and the progression to terminal maturation. In fact, ex vivo organ culture experiments in both chick and mouse have demonstrated that local removal or experimental thinning of the perichondrium adjacent to the prehypertrophic/hypertrophic domains leads to an expansion of the hypertrophic region caused by a delay in the completion of terminal hypertrophic differentiation.(48,49) Additionally, genetic removal of several signaling pathway components, including Ihh/Pthrp,(50–52) Wnt/β-catenin,(39,53,54) and BMP/Smads(15–20) demonstrates that disruption in normal signaling events between the perichondrium and chondrocytes often results in delayed onset of chondrocyte hypertrophy and progression of terminal maturation, as well as disorganization of growth plate chondrocytes. Thus TAK1 appears to be required for normal growth plate organization and terminal chondrocyte maturation via maintenance of normal perichondrial development. The exact function of TAK1 in the perichondrium during osteoblast maturation is of great interest to our group and will be described elsewhere.
TAK1 is required for MAPK and Smad 1/5/8 signaling during cartilage development
In addition to the role for TAK1 in activating MKK3/6 and the subsequent downstream MAPK signaling pathway component p38, recent studies have found that TAK1 is capable of interacting with all R-Smads (Smads 2/3 and Smads 1/5/8).(55) Both BMP and TGF-β pathways also can promote the activation of TAK1 via noncanonical TGF-β/BMP signaling mechanisms.(21) What remains unclear is whether the noncanonical activation of TAK1 works independently of, is required for, or signals in opposition to the TGF-β/BMP canonical signaling pathways. If TAK1 functions in a manner independent of TGF-β/BMP canonical signaling during cartilage development, one would expect that the growth plate phenotypes of our Tak1Col2 and Tak1Prx mutants likely would resemble only those phenotypes generated by inhibition of the MAPK signaling pathway. Alternatively, if TAK1 is required for or signals in opposition to canonical TGF-β/BMP signaling, we would have expected to observe growth plate phenotypes resembling either TGF-β/BMP loss- or gain-of-function mouse models, respectively. Interestingly, we observed primarily growth plate phenotypes that were consistent with disruptions in the BMP signaling pathways during endochondral bone development. Comparison of Tak1 mutant growth plate phenotypes with those found in Col2Cre;Bmpr1a f/f;Bmpr1b+/− and Col2Cre;Smad1f/f;Smad5f/f embryos reveals striking similarities, including the delayed onset of chondrocyte hypertrophy, decreased proliferation, the rarely observed increase in proliferating chondrocyte apoptosis, disorganization of columnar zone chondrocytes, and the delayed progression of terminal maturation(16,17,20) (Fig. 7A). While Col2Cre;Smad1f/f;Smad5f/f mutant phenotypes appear to be significantly more severe than our Tak1 mutant mice, we reason that these discrepancies are likely due to only a partial loss Smad 1/5/8 phosphorylation in Tak1Col2 and Tak1Prx mutants. It is also of note that Col2Cre;Smad1f/f;Smad5f/f mutants show significant decreases in the expression of MAPK components TAK1 and MKK3, leading to the possibility that disruption of canonical BMP signaling also disrupts noncanonical signaling.(20) Many of the Tak1 mutant phenotypes are recapitulated in these BMP pathway mutants, although not all the skeletal phenotypes are consistent. For example, the axial skeletal phenotypes, including fusion of cervical vertebrae in Tak1Col2 mutants and loss of sternal midline fusion in Tak1Prx mutants, strongly resemble the phenotypes of mice deleted for Tgfbr2 using the Col2Cre and Prx1Cre transgenes, respectively.(12,13) Additional roles for TAK1 in mediating TGF-β signals also may occur during postnatal development, although we were unable to determine whether this is the case because our mutants do not survive after birth. Studies analyzing Col2Cre;Tgfbr2f/f and Smad3−/− mice suggest that TGF-β plays a more prominent role in postnatal growth and maintenance of cartilage.(12,56) Finally, Tak1Col2 and Tak1Prx mutants displayed variable MAPK loss-of-function phenotypes similar to the dn-p38 and ATF2−/− mice, although most Tak1 mutant phenotypes were more pervasive and severe, likely owing to the fact that TAK1 is upstream of both these MAPK targets. Therefore, it seems plausible that TAK1 is an important component in the functional crosstalk among the MAPK, BMP, and TGF-β pathways in both temporal and tissue-specific contexts during skeletal development.
Fig. 7.
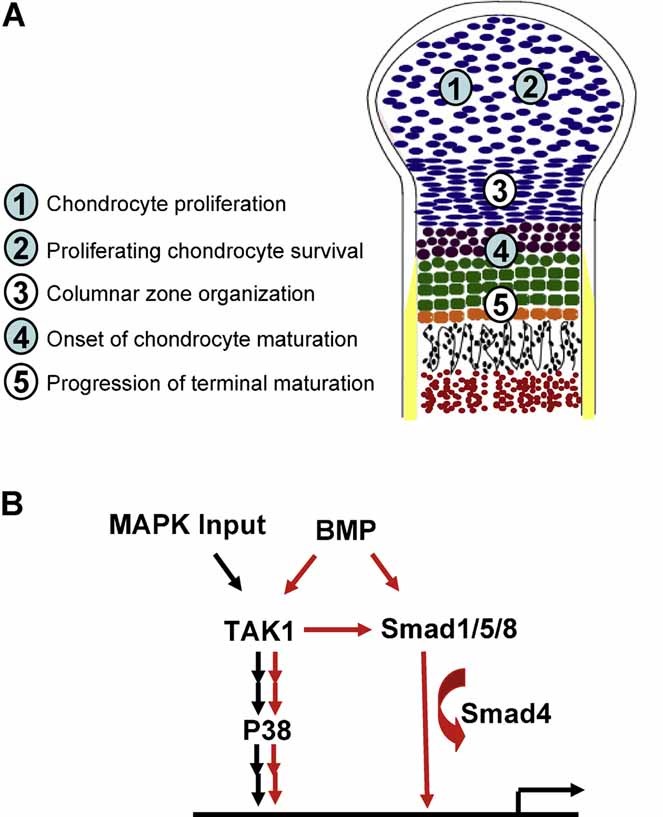
Summary of TAK1 signaling and regulation of cartilage development. (A) Cells are color coded as follows: Round blue cells are immature/slowly proliferating chondocytes; flat blue cells are columnar, highly proliferative chondrocytes; purple cells are prehypertrophic chondrocytes; green cells are hypertrophic chondrocytes; orange cells indicate terminally hypertrophic chondrocytes; black cells indicate trabecular osteoblasts; red cells indicate bone marrow; and the yellow area indicates maturing bone collar. Numbers in light blue circles indicate chondrocyte-specific effects of TAK1, and numbers in white circles are perichondrial effects of TAK1. (B) TAK1 signaling via the MAPK and BMP pathways. Red arrows indicate canonical and noncanonical BMP signaling regulation; black arrows indicate MAPK signaling via TAK1.
Our signaling studies, as well as those performed by Shim and colleagues,(34) provided further support for the importance of TAK1 in mediating BMP activation of Smad 1/5/8 and p38 via noncanonical signaling mechanisms. While the suppression of Smad 1/5/8 activation is incomplete on TAK1 inhibition, the significant decrease in activity suggests that TAK1 is at least one of the critical components by which BMP signals activate R-Smads. Interestingly, p38 activation is completely absent in both normal and BMP-induced chondrocytes following TAK1 inhibition, indicating the importance of TAK1 during normal MAPK signaling and noncanonical BMP signaling (summarized in Fig. 7B). The severely impaired chondrocyte maturation observed in TAK1 mutant cultures supports a role for TAK1 in the promotion of chondrocyte hypertrophy, which is in line with the function of BMP signaling on chondrocyte maturation and not that of the suppressive function for TGF-β. Additionally, TAK1 mutant chondrocytes were unable to respond to BMP-induced maturation (Fig. 3D, E), but the ability of TGF-β to inhibit chondrocyte maturation was well preserved (Fig. 3D). These data suggest that TAK1 mutant chondrocytes are less sensitive to BMP signaling and therefore are impaired in their ability to undergo chondrocyte maturation. Interestingly, studies in cardiomyocytes also have demonstrated the requirement for TAK1 in mediating BMP-induced differentiation.(57) Furthermore, our in vivo data indicate that fewer columnar and hypertrophic chondrocytes displayed significant levels of phosphorylated Smad 1/5/8 proteins in the nuclei of Tak1Col2 and Tak1Prx mutants. It is therefore intriguing to speculate that reduced BMP/Smad signaling in the cartilage of Tak1 mutant mice accounts for the decreased chondrocyte proliferation, increased proliferating chondrocyte apoptosis, and delayed onset of maturation.
TAK1 regulates interzone and joint development
Defects in joint formation observed in Tak1Col2 and Tak1Prx embryos provide direct evidence for the importance of TAK1 signaling during the process of joint formation. While several signaling molecules have been implicated in interzone formation and synovial joint development, the molecular mechanisms underlying these processes remain not well understood. Cells at the site of prospective joints form the mesenchymal interzone. Interzone cells and the surrounding mesenchymal tissues act as signaling centers expressing both antichondrogenic molecules (Wnt9a and noggin) that aid in maintaining the interzone and chondrogenic factors (Gdf5, Erg, and various members of the BMP family) that promote chondrocyte differentiation of the adjacent cartilage. What is clear is that an appropriate balance of these factors must be maintained in order for synovial joints to initiate and develop properly. Molecular analyses of both the Tak1Col2 and Tak1Prx mutant joints revealed a loss of the antichondrogenic gene Wnt9a and Noggin specifically within the developing interzone. Additionally, these mutants lacked significant expression of Gdf5, although chondrogenic factors such as Erg, which is known to promote and maintain an early chondrogenic phenotype, were maintained at low levels in the surrounding joint tissue but also within the presumptive interzone. Therefore, loss of Tak1 may lead to an imbalance of antichondrogenic and chondrogenic factors that favors chondrogenesis, the maintenance of an early chondrogenic phenotype, and the subsequent loss of interzone cells.
Analyses of the Tak1Col2 mutant mice revealed fusions and malformations only at the humeroradial joints, whereas Tak1Prx mutants exhibited joint fusions at nearly all synovial joints except those of the digits. We believe that the elbow joint is fused within Tak1Col2 mutant mice owing to the promiscuity of the Col2Cre transgene in early mesenchymal cells of that particular joint region, as demonstrated previously using a Col2Cre;R26R reporter mouse.(40) Tak1Prx mutant mice have more widespread defects in synovial joints because the Prx1Cre transgene targets nearly all mesenchymal progenitors prior to joint site determination (Supplemental Fig. 1C). These Tak1Prx mutants strongly resemble the joint phenotypes derived from Prx1Cre;Bmp2f/f;Bmp4f/f and Gdf5−/− mutant mice, displaying joint fusions and impaired interzone formation/maintenance at nearly all synovial joints.(14) In addition, our analyses demonstrate a severe reduction in the expression of the BMP target gene noggin in the developing joint region; therefore, it is likely that TAK1 regulates BMP signaling not only in the growth plate but also at sites of joint formation. It is unlikely that the joint defects observed in these Tak1 mutants are due to defective TGF-β or MAPK signaling because mutations in these signaling circuits either do not produce joint fusion abnormalities(33) or the malformations are restricted primarily to joints of the digits,(12) which are not affected in Tak1 mutants.
In this study and through the work of Shim and colleagues,(34) we have provided the first in vivo genetic evidence that TAK1 is a critical component of both the MAPK and BMP signaling pathways during embryonic cartilage and joint development, as well as during early postnatal cartilage regulation.(34) Furthermore, our model extends the developmental analysis via use of the Tak1Prx mouse model. These mice demonstrate that TAK1 signaling is not only critical for the development of nearly all synovial joints but also required in the surrounding limb mesenchyme to control several aspects of chondrocyte proliferation and differentiation. Finally, since our unique observations during skeletal development demonstrate clear similarities to the phenotypes observed in several other models of development in which BMP signaling is deficient, they confirm an in vivo physiologic role of TAK1 in BMP/Smad signaling during limb and joint development. Our continuing studies will aid in elucidating the TAK1-mediated effects on perichondrial or osteoblast maturation, axial skeletal development, and joint formation and the potential role of TAK1 in cartilage maintenance and disease.
Acknowledgments
Special thanks to Dr Michael Schneider for providing the Tak1f/f mice. We also would like to thank Ryan Tierney and Nehal Porecha in the Histology Core of the Center for Musculoskeletal Research, University of Rochester, for their technical expertise in processing tissue samples and immunohistochemistry. This work was supported by NIH Grant R01 AR38945 (RJO) from NIAMS.
Disclosures
MJH and RJO contributed equally to this study. All the authors state that they have no conflicts of interest.
Supplementary material
References
- 1.Rountree RB, Schoor M, Chen H, et al. BMP receptor signaling is required for postnatal maintenance of articular cartilage. PLoS Biol. 2004;2:e355. doi: 10.1371/journal.pbio.0020355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spater D, Hill TP, Gruber M, Hartmann C. Role of canonical Wnt-signalling in joint formation. Eur Cell Mater. 2006;12:71–80. doi: 10.22203/ecm.v012a09. [DOI] [PubMed] [Google Scholar]
- 3.Brunet LJ, McMahon JA, McMahon AP, Harland RM. Noggin, cartilage morphogenesis, and joint formation in the mammalian skeleton. Science. 1998;280:1455–1457. doi: 10.1126/science.280.5368.1455. [DOI] [PubMed] [Google Scholar]
- 4.Storm EE, Kingsley DM. Joint patterning defects caused by single and double mutations in members of the bone morphogenetic protein (BMP) family. Development. 1996;122:3969–3979. doi: 10.1242/dev.122.12.3969. [DOI] [PubMed] [Google Scholar]
- 5.Iwamoto M, Tamamura Y, Koyama E, et al. Transcription factor ERG and joint and articular cartilage formation during mouse limb and spine skeletogenesis. Dev Biol. 2007;305:40–51. doi: 10.1016/j.ydbio.2007.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koyama E, Shibukawa Y, Nagayama M, et al. A distinct cohort of progenitor cells participates in synovial joint and articular cartilage formation during mouse limb skeletogenesis. Dev Biol. 2008;316:62–73. doi: 10.1016/j.ydbio.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo X, Day TF, Jiang X, Garrett-Beal L, Topol L, Yang Y. Wnt/beta-catenin signaling is sufficient and necessary for synovial joint formation. Genes Dev. 2004;18:2404–2417. doi: 10.1101/gad.1230704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Settle SH, Jr, Rountree RB, Sinha A, Thacker A, Higgins K, Kingsley DM. Multiple joint and skeletal patterning defects caused by single and double mutations in the mouse Gdf6 and Gdf5 genes. Dev Biol. 2003;254:116–130. doi: 10.1016/s0012-1606(02)00022-2. [DOI] [PubMed] [Google Scholar]
- 9.Smits P, Li P, Mandel J, et al. The transcription factors L-Sox5 and Sox6 are essential for cartilage formation. Dev Cell. 2001;1:277–290. doi: 10.1016/s1534-5807(01)00003-x. [DOI] [PubMed] [Google Scholar]
- 10.Akiyama H, Chaboissier MC, Martin JF, Schedl A, de Crombrugghe B. The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 2002;16:2813–2828. doi: 10.1101/gad.1017802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stickens D, Behonick DJ, Ortega N, et al. Altered endochondral bone development in matrix metalloproteinase 13-deficient mice. Development. 2004;131:5883–5895. doi: 10.1242/dev.01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baffi MO, Slattery E, Sohn P, Moses HL, Chytil A, Serra R. Conditional deletion of the TGF-beta type II receptor in Col2a expressing cells results in defects in the axial skeleton without alterations in chondrocyte differentiation or embryonic development of long bones. Dev Biol. 2004;276:124–142. doi: 10.1016/j.ydbio.2004.08.027. [DOI] [PubMed] [Google Scholar]
- 13.Seo HS, Serra R. Deletion of Tgfbr2 in Prx1-cre expressing mesenchyme results in defects in development of the long bones and joints. Dev Biol. 2007;310:304–316. doi: 10.1016/j.ydbio.2007.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bandyopadhyay A, Tsuji K, Cox K, Harfe BD, Rosen V, Tabin CJ. Genetic analysis of the roles of BMP2, BMP4, and BMP7 in limb patterning and skeletogenesis. PLoS Genet. 2006;2:e216. doi: 10.1371/journal.pgen.0020216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Storm EE, Kingsley DM. GDF5 coordinates bone and joint formation during digit development. Dev Biol. 1999;209:11–27. doi: 10.1006/dbio.1999.9241. [DOI] [PubMed] [Google Scholar]
- 16.Yoon BS, Ovchinnikov DA, Yoshii I, Mishina Y, Behringer RR, Lyons KM. Bmpr1a and Bmpr1b have overlapping functions and are essential for chondrogenesis in vivo. Proc Natl Acad Sci U S A. 2005;102:5062–5067. doi: 10.1073/pnas.0500031102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoon BS, Pogue R, Ovchinnikov DA, et al. BMPs regulate multiple aspects of growth-plate chondrogenesis through opposing actions on FGF pathways. Development. 2006;133:4667–4678. doi: 10.1242/dev.02680. [DOI] [PubMed] [Google Scholar]
- 18.Yi SE, Daluiski A, Pederson R, Rosen V, Lyons KM. The type I BMP receptor BMPRIB is required for chondrogenesis in the mouse limb. Development. 2000;127:621–630. doi: 10.1242/dev.127.3.621. [DOI] [PubMed] [Google Scholar]
- 19.Kavanagh E, Church VL, Osborne AC, et al. Differential regulation of GDF-5 and FGF-2/4 by immobilisation in ovo exposes distinct roles in joint formation. Dev Dyn. 2006;235:826–834. doi: 10.1002/dvdy.20679. [DOI] [PubMed] [Google Scholar]
- 20.Retting KN, Song B, Yoon BS, Lyons KM. BMP canonical Smad signaling through Smad1 and Smad5 is required for endochondral bone formation. Development. 2009;136:1093–1104. doi: 10.1242/dev.029926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamaguchi K, Shirakabe K, Shibuya H, et al. Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science. 1995;270:2008–2011. doi: 10.1126/science.270.5244.2008. [DOI] [PubMed] [Google Scholar]
- 22.Moriguchi T, Kuroyanagi N, Yamaguchi K, et al. A novel kinase cascade mediated by mitogen-activated protein kinase kinase 6 and MKK3. J Biol Chem. 1996;271:13675–13679. doi: 10.1074/jbc.271.23.13675. [DOI] [PubMed] [Google Scholar]
- 23.Shirakabe K, Yamaguchi K, Shibuya H, et al. TAK1 mediates the ceramide signaling to stress-activated protein kinase/c-Jun N-terminal kinase. J Biol Chem. 1997;272:8141–8144. doi: 10.1074/jbc.272.13.8141. [DOI] [PubMed] [Google Scholar]
- 24.Hanafusa H, Ninomiya-Tsuji J, Masuyama N, et al. Involvement of the p38 mitogen-activated protein kinase pathway in transforming growth factor-beta-induced gene expression. J Biol Chem. 1999;274:27161–27167. doi: 10.1074/jbc.274.38.27161. [DOI] [PubMed] [Google Scholar]
- 25.Nakamura K, Shirai T, Morishita S, Uchida S, Saeki-Miura K, Makishima F. p38 mitogen-activated protein kinase functionally contributes to chondrogenesis induced by growth/differentiation factor-5 in ATDC5 cells. Exp Cell Res. 1999;250:351–363. doi: 10.1006/excr.1999.4535. [DOI] [PubMed] [Google Scholar]
- 26.Jin EJ, Lee SY, Choi YA, Jung JC, Bang OS, Kang SS. BMP-2-enhanced chondrogenesis involves p38 MAPK-mediated down-regulation of Wnt-7a pathway. Mol Cells. 2006;22:353–359. [PubMed] [Google Scholar]
- 27.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 28.Tuli R, Tuli S, Nandi S, et al. Transforming growth factor-beta-mediated chondrogenesis of human mesenchymal progenitor cells involves N-cadherin and mitogen-activated protein kinase and Wnt signaling cross-talk. J Biol Chem. 2003;278:41227–41236. doi: 10.1074/jbc.M305312200. [DOI] [PubMed] [Google Scholar]
- 29.Zuzarte-Luis V, Montero JA, Rodriguez-Leon J, Merino R, Rodriguez-Rey JC, Hurle JM. A new role for BMP5 during limb development acting through the synergic activation of Smad and MAPK pathways. Dev Biol. 2004;272:39–52. doi: 10.1016/j.ydbio.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 30.Namdari S, Wei L, Moore D, Chen Q. Reduced limb length and worsened osteoarthritis in adult mice after genetic inhibition of p38 MAP kinase activity in cartilage. Arthritis Rheum. 2008;58:3520–3529. doi: 10.1002/art.23999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reimold AM, Grusby MJ, Kosaras B, et al. Chondrodysplasia and neurological abnormalities in ATF-2-deficient mice. Nature. 1996;379:262–265. doi: 10.1038/379262a0. [DOI] [PubMed] [Google Scholar]
- 32.Luvalle P, Ma Q, Beier F. The role of activating transcription factor-2 in skeletal growth control. J Bone Joint Surg Am. 2003;85-A(Suppl 2):133–136. doi: 10.2106/00004623-200300002-00018. [DOI] [PubMed] [Google Scholar]
- 33.Zhang R, Murakami S, Coustry F, Wang Y, de Crombrugghe B. Constitutive activation of MKK6 in chondrocytes of transgenic mice inhibits proliferation and delays endochondral bone formation. Proc Natl Acad Sci U S A. 2006;103:365–370. doi: 10.1073/pnas.0507979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shim JH, Greenblatt MB, Xie M, et al. TAK1 is an essential regulator of BMP signalling in cartilage. Embo J. 2009;28:2028–2041. doi: 10.1038/emboj.2009.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu HH, Xie M, Schneider MD, Chen ZJ. Essential role of TAK1 in thymocyte development and activation. Proc Natl Acad Sci U S A. 2006;103:11677–11682. doi: 10.1073/pnas.0603089103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Terpstra L, Prud'homme J, Arabian A, et al. Reduced chondrocyte proliferation and chondrodysplasia in mice lacking the integrin-linked kinase in chondrocytes. J Cell Biol. 2003;162:139–148. doi: 10.1083/jcb.200302066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Logan M, Martin JF, Nagy A, Lobe C, Olson EN, Tabin CJ. Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis. 2002;33:77–80. doi: 10.1002/gene.10092. [DOI] [PubMed] [Google Scholar]
- 38.Hilton MJ, Tu X, Cook J, Hu H, Long F. Ihh controls cartilage development by antagonizing Gli3, but requires additional effectors to regulate osteoblast and vascular development. Development. 2005;132:4339–4351. doi: 10.1242/dev.02025. [DOI] [PubMed] [Google Scholar]
- 39.Hu H, Hilton MJ, Tu X, Yu K, Ornitz DM, Long F. Sequential roles of Hedgehog and Wnt signaling in osteoblast development. Development. 2005;132:49–60. doi: 10.1242/dev.01564. [DOI] [PubMed] [Google Scholar]
- 40.Hilton MJ, Tu X, Long F. Tamoxifen-inducible gene deletion reveals a distinct cell type associated with trabecular bone, and direct regulation of PTHrP expression and chondrocyte morphology by Ihh in growth region cartilage. Dev Biol. 2007;308:93–105. doi: 10.1016/j.ydbio.2007.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li TF, Chen D, Wu Q, et al. Transforming growth factor-beta stimulates cyclin D1 expression through activation of beta-catenin signaling in chondrocytes. J Biol Chem. 2006;281:21296–21304. doi: 10.1074/jbc.M600514200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lefebvre V, Garofalo S, Zhou G, Metsaranta M, Vuorio E, De Crombrugghe B. Characterization of primary cultures of chondrocytes from type II collagen/beta-galactosidase transgenic mice. Matrix Biol. 1994;14:329–335. doi: 10.1016/0945-053x(94)90199-6. [DOI] [PubMed] [Google Scholar]
- 43.Li TF, Darowish M, Zuscik MJ, et al. Smad3-deficient chondrocytes have enhanced BMP signaling and accelerated differentiation. J Bone Miner Res. 2006;21:4–16. doi: 10.1359/JBMR.050911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mukhopadhyay K, Lefebvre V, Zhou G, Garofalo S, Kimura JH, de Crombrugghe B. Use of a new rat chondrosarcoma cell line to delineate a 119-base pair chondrocyte-specific enhancer element and to define active promoter segments in the mouse pro-alpha 1(II) collagen gene. J Biol Chem. 1995;270:27711–27719. doi: 10.1074/jbc.270.46.27711. [DOI] [PubMed] [Google Scholar]
- 45.Ninomiya-Tsuji J, Kajino T, Ono K, et al. A resorcylic acid lactone, 5Z-7-oxozeaenol, prevents inflammation by inhibiting the catalytic activity of TAK1 MAPK kinase kinase. J Biol Chem. 2003;278:18485–18490. doi: 10.1074/jbc.M207453200. [DOI] [PubMed] [Google Scholar]
- 46.Zhao M, Qiao M, Oyajobi BO, Mundy GR, Chen D. E3 ubiquitin ligase Smurf1 mediates core-binding factor alpha1/Runx2 degradation and plays a specific role in osteoblast differentiation. J Biol Chem. 2003;278:27939–27944. doi: 10.1074/jbc.M304132200. [DOI] [PubMed] [Google Scholar]
- 47.Ovchinnikov DA, Deng JM, Ogunrinu G, Behringer RR. Col2a1-directed expression of Cre recombinase in differentiating chondrocytes in transgenic mice. Genesis. 2000;26:145–146. [PubMed] [Google Scholar]
- 48.Long F, Linsenmayer TF. Regulation of growth region cartilage proliferation and differentiation by perichondrium. Development. 1998;125:1067–1073. doi: 10.1242/dev.125.6.1067. [DOI] [PubMed] [Google Scholar]
- 49.Colnot C, Lu C, Hu D, Helms JA. Distinguishing the contributions of the perichondrium, cartilage, and vascular endothelium to skeletal development. Dev Biol. 2004;269:55–69. doi: 10.1016/j.ydbio.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 50.Lanske B, Karaplis AC, Lee K, et al. PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science. 1996;273:663–666. doi: 10.1126/science.273.5275.663. [DOI] [PubMed] [Google Scholar]
- 51.St-Jacques B, Hammerschmidt M, McMahon AP. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999;13:2072–2086. doi: 10.1101/gad.13.16.2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vortkamp A, Lee K, Lanske B, Segre GV, Kronenberg HM, Tabin CJ. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science. 1996;273:613–622. doi: 10.1126/science.273.5275.613. [DOI] [PubMed] [Google Scholar]
- 53.Day TF, Guo X, Garrett-Beal L, Yang Y. Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell. 2005;8:739–750. doi: 10.1016/j.devcel.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 54.Hill TP, Spater D, Taketo MM, Birchmeier W, Hartmann C. Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev Cell. 2005;8:727–738. doi: 10.1016/j.devcel.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 55.Hoffmann A, Preobrazhenska O, Wodarczyk C, et al. Transforming growth factor-beta-activated kinase-1 (TAK1), a MAP3K, interacts with Smad proteins and interferes with osteogenesis in murine mesenchymal progenitors. J Biol Chem. 2005;280:27271–27283. doi: 10.1074/jbc.M503368200. [DOI] [PubMed] [Google Scholar]
- 56.Yang X, Chen L, Xu X, Li C, Huang C, Deng CX. TGF-beta/Smad3 signals repress chondrocyte hypertrophic differentiation and are required for maintaining articular cartilage. J Cell Biol. 2001;153:35–46. doi: 10.1083/jcb.153.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Monzen K, Nagai R, Komuro I. A role for bone morphogenetic protein signaling in cardiomyocyte differentiation. Trends Cardiovasc Med. 2002;12:263–269. doi: 10.1016/s1050-1738(02)00172-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.