

Abstract
For DNA polymerases to proofread a misincorporated nucleotide, the terminal 3–4 nucleotides of the primer strand must be separated from the template strand before being bound in the exonuclease active center. Genetic and biochemical studies of the bacteriophage T4 DNA polymerase revealed that a prominent β-hairpin structure in the exonuclease domain is needed to efficiently form the strand-separated exonuclease complexes. We present here further mutational analysis of the loop region of the T4 DNA polymerase β-hairpin structure, which provides additional evidence that residues in the loop, namely, Y254 and G255, are important for DNA replication fidelity. The mechanism of strand separation was probed in in vitro reactions using the fluorescence of the base analogue 2-aminopurine (2AP) and mutant RB69 DNA polymerases that have modifications to the β hairpin, to the exonuclease active site, or to both. We propose from these studies that the β hairpin in the exonuclease domain of the T4 and RB69 DNA polymerases functions to facilitate strand separation, but residues in the exonuclease active center are required to capture the 3′ end of the primer strand following strand separation.
Strand separation is a critical step in the DNA polymerase proofreading pathway because only single-stranded DNA (ssDNA)1 can be bound in the exonuclease active center of any proofreading DNA polymerase, but the full details of the process are not known. One proposal is that mismatched DNA is inherently unstable and leads to spontaneous strand separation to create the single-stranded primer end that is bound in the exonuclease active center. While some DNA polymerases may rely on this mechanism, another mechanism has been proposed for the phage T4 DNA polymerase: a prominent β-hairpin structure in the exonuclease domain facilitates the strand separation process [Figure 1; (1, 2)]. Facilitated strand separation can explain the exonuclease activity that is observed for the T4 DNA polymerase on correctly base-paired double-stranded DNA (dsDNA) in the absence of dNTPs (3, 4). The proposal that the β hairpin plays a role in strand separation is based on two linked observations. First, a Ser substitution for G255 in the loop of the β hairpin produces a “mutator” (low-fidelity) DNA polymerase (5, 6), and second, the G255S–DNA polymerase has greatly reduced 3′ → 5′ exonuclease activity on dsDNA but near wild-type levels on ssDNA or mismatched DNA (mmDNA), with at least three terminal mismatches (6–8). Thus, even though the G255S–DNA polymerase retains hydrolysis activity, the reduced ability to form strand-separated exonuclease complexes leads to decreased proofreading and low-fidelity DNA replication.
Figure 1.
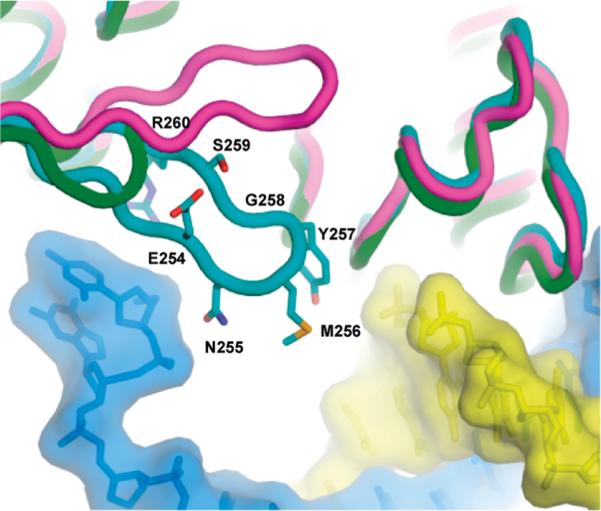
Structures of the RB69 DNA polymerase β hairpin in a polymerase ternary complex [purple, (35)], in a pre-exonuclease binary complex [cyan, (11)], and of the truncated β hairpin in the β− polymerase [green, (12)]. The primer strand is shaded in yellow, and the template strand is shaded in blue. The primer/template is from the binary, pre-exonuclease complex structure (11).
Structural studies of the phage RB69 DNA polymerase, which is closely related to the phage T4 DNA polymerase (9), provides additional evidence that the β hairpin acts as a wedge to separate the end of the primer strand from the template (10–12). However, in one study, the tip of the β hairpin is disordered, which suggests movement (11). To follow up on the apparent dynamic action of the β hairpin, we have used 2-aminopurine (2AP), an adenine base analogue, as a fluorescent reporter to investigate the role of the β hairpin loop in the proofreading pathway in solution.
2AP fluorescence in DNA is highly quenched because of stacking interactions with adjacent bases (13–15), but large increases in fluorescence intensity are observed when T4 DNA polymerase binding perturbs 2AP base-stacking interactions. Changes in 2AP fluorescence intensity in polymerase and exonuclease complexes formed with the T4 DNA polymerase and 2AP-labeled DNA substrates have been used to probe the nucleotide incorporation and proofreading pathways (7,8,16–24). We extend these studies here to the wild-type and several mutant RB69 DNA polymerases with modifications in the β hairpin and the exonuclease active center. While deletion of the loop region of the β hairpin, residues 254–260 (Figure 1), or Ala substitutions for the Mg2+-binding residues D222 and D327 in the exonuclease active site severely reduce the ability of the RB69 DNA polymerase to form exonuclease complexes with dsDNA, the combination of both defects (β−/exo−) nearly eliminates the formation of exonuclease complexes. Thus, we propose that efficient separation of the primer end from the template strand requires action of the β hairpin and residues in the exonuclease active center.
MATERIALS AND METHODS
Genetic Methods
Site-directed mutagenesis was used to introduce amber codons (TAG) into the cloned T4 DNA polymerase gene (g43) for codons 254, 256, and 257. The site-directed mutants were confirmed by DNA sequencing. The amber codons were introduced into g43 of T4 phage by recombination. Bacterial strains, each carrying plasmids with a different cloned, amber g43, were infected with T4 phage amE4306 rUV199. The amE4306 allele encodes an amber codon at position 213 in the T4 DNA polymerase gene. Suppression of the amber codon on strains that insert Ser at the amber codon produces the W213S–DNA polymerase, which is temperature-sensitive. Thus, recombinant phage produced from infections of the plasmid strains that are no longer temperature-sensitive on a Ser-inserting bacterial host could now have a new amber mutation at position 254, 256, or 257 depending upon the plasmid strain infected. Replica plating was used to determine if the recombinant phage carried an amber mutation, and DNA sequencing was used to confirm the location in g43. All of the am g43 phage were viable on the Ser-inserting bacterial host. T4 am g43 phage strains were propagated on different amber suppressing tRNA hosts (25) to produce mutant DNA polymerases with either Ser, Gln, Tyr, Lys, Leu, or Gly insertions at the amber codons 254, 256, or 257. The mutant DNA polymerase phage also carried the rUV199 allele, which can be used to measure spontaneous base substitution revertant frequencies as described previously (26). Mutation frequencies were determined from the median frequency from 10 or more independent cultures.
DNA Polymerases
Expression and purification of the wild-type and mutant RB69 DNA polymerases were described previously (12).
DNA Substrates
The DNA substrates used in the study are described in Table 1. The 2AP-labeled oligonucleotides were prepared as described previously (19) or were purchased from Integrated DNA Technologies (IDT, Coralville, IA). The 3′ terminus of the template strand of the DNA duplexes was protected from enzyme binding by attachment of biotin. The primer and template strands were annealed in buffer containing N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES) (pH 7.6) and 50 mM NaCl, with a 20% excess of the non-2AP-labeled oligomer to ensure complete hybridization of the 2AP-containing strand (16).
Table 1.
Oligonucleotides with 2-Aminopurine (P)a
| 2AP at the 3′ end of the primer strand in single-stranded (ss), double-stranded (ds), and mismatched (mm) DNA substrates | ||
| DNA sequence | fluorescence intensity (counts/s)b | |
|
| ||
| ssDNA | 5′ GGGAAGCACGTCATCGGTAATP | 5800 |
| dsDNA | 3′ B–CCCTTCGTGCAGTAGCCATTATAGATCGATGGTTT | 3500 |
| 5′ GGGAAGCACGTCATCGGTAATP | ||
| mmDNAc | 3′ B–CGTGCAGTAATTGCGCCGGATCGATGGTTT | 2700 |
| 5′ GCACGTCATTAACGGTP | ||
|
| ||
| 2AP at the +1 position in the template strand | ||
|
| ||
| A+T richd | 3′ B–CCCTTCGTGCAGTAGCCATTAAPGATCGATGGTTT | 1750 |
| 5′ GGGAAGCACGTCATCGGTAATT | ||
| G+C riche | 3′ B–CCCTTCGTGCAGTAGCCGCCGCPGATCGATGGTTT | 1720 |
| 5′ GGGAAGCACGTCATCGGCGGCG | ||
2-Aminopurine is indicated by a P. Biotin (B) is placed at the 3′ end of the template strand to prevent DNA polymerase binding.
Fluorescence intensity for a 200 nM solution of the unbound DNA substrate.
There are three terminal mismatches.
There are five AT base pairs in the primer terminal region.
There are eight GC base pairs in the primer terminal region.
Fluorescence Intensity Experiments
The fluorescence emission spectra were recorded using a Photon Technology International (PTI) spectrofluorimeter. All 2AP solutions (2AP as the free base, in DNA, and in protein–DNA complexes) were prepared in buffer containing 25 mM HEPES (pH 7.6), 50 mM NaCl, 1 mM dithiothreitol (DTT), and 0.5 mM ethylenediaminetetraacetic acid (EDTA). DNA–polymerase complexes were formed by mixing 200 nM 2AP-labeled DNA substrate with 500 nM wild-type or mutant RB69 DNA polymerases. Excess enzyme was used to ensure all DNA was bound. The optimal DNA and enzyme concentrations were determined in titration experiments, and maximum fluorescence was obtained when the DNA/enzyme ratio was ~1:2. The sample volume was 200 μL, which was placed in a high-precision, ultramicro fluorescence cell (Hellma) housed in a temperature-controlled cuvette compartment maintained at 20 ( 0.5 °C. The samples were excited at 315 nm, and emission was collected at 368 nm; a 320 nm long-pass filter was placed between the cuvette and emission monochromator. A 2 nm band pass was used for both the excitation and emission monochromators. The absolute fluorescence intensity depends upon several factors. To compare fluorescence intensities of the complexes formed by different polymerases determined with the same instrument and experimental conditions but on different days, the fluorescence intensities were normalized with respect to a stock 2AP solution (200 nM in buffer).
Fluorescence Lifetime Measurements
Solutions of 2AP as the free base, in DNA, or in complexes were prepared the same as for the fluorescence intensity experiments. The temperature in the cuvette compartment was maintained at 20 (0.5 °C. The solutions were excited at 315 nm using the frequency-doubled output from a pulsed dye laser (PTI). A 320 nm long-pass filter was used in the emission side of the cuvette. The PTI Laser Strobe lifetime system is capable of resolving lifetimes as short as 100–200 ps. The instrument response function was collected at 315 nm using light scattered by a dilute suspension of colloidal silica (Ludox). A stroboscopic optical boxcar method was used for the determination of fluorescence lifetimes (27). Decay curves were averaged from seven repetitive acquisitions. The decay curves were fit to a sum of exponentials (eq 1) using the
| (1) |
analysis routine provided by PTI. In eq 1, n is the number of components and τi and αi are the fluorescence lifetime and amplitude values for each component. The quality of curve fits was judged on the basis of the χ2 value (0.9 ≤ χ2 ≤ 1.3) and the randomness of residuals, which was assessed statistically by the Durbin–Watson test and visual inspection. Whenever there was more than one exponential term required to fit the decay curves, the number-averaged fluorescence lifetimes were calculated according to eq 2.
| (2) |
The value τnum is proportional to the quantum yield in the absence of static quenching.
RESULTS
Mutational Analysis of the β-Hairpin Loop of the Bacteriophage T4 DNA Polymerase
Genetic selection schemes for the identification of amino acid residues essential for proofreading by the T4 DNA polymerase identified G255 in the loop of a prominent β-hairpin structure in the exonuclease domain as a critical residue (5, 6). Phage T4 DNA replication fidelity was determined in vivo by measuring the revertant frequency of a rII ochre mutant that reverts to rII+ function by base substitution mutagenesis (26). The revertant frequency was increased >100-fold for phage that expressed the G255S– and G255A–DNA polymerases compared to wild-type phage (Table 2). To assess the role of other loop residues in replication fidelity, the effects of amino acid substitutions for three adjacent residues were examined. Large increases in the revertant frequency were also detected for phage-expressing mutant DNA polymerases with amino acid substitutions for Y254; revertant frequencies increased from ~30- to >70-fold depending upon the amino acid substitution (Table 2). Smaller effects on replication fidelity, however, were observed for phage that expressed DNA polymerases with amino acid substitutions for S256; the S256Q– and S256Y–DNA polymerases replicated DNA with the same fidelity as the wild-type T4 DNA polymerase; and from 11 to ~30-fold increases in revertant frequency were detected for the S256K–, S256L– and S256G–DNA polymerases. Even smaller increases in revertant frequencies were detected for mutant DNA polymerases with amino acid substitutions for K257. Thus, of the loop residues tested, G255 and Y254 appear to be the most critical for proofreading. These residues correspond to G258 and Y257 in the RB69 DNA polymerase (Figure 1 and Table 2).
Table 2.
Mutational Analysis of the β-Hairpin Loop in the Phage T4 DNA Polymerasea
| relative mutant frequencies |
|||||||
|---|---|---|---|---|---|---|---|
| amino acid substitutions |
|||||||
| loop residue | Ser | Gln | Tyr | Lys | Leu | Gly | Ala |
| Y254 (Y257) | 36 | 47 | 1 | 77 | 7 | 72 | – |
| G255 (G258) | 113 | – | – | – | – | 1 | 107 |
| S256 (S259) | 1 | 1 | 1 | 11 | 15 | 34 | – |
| K257 (R260) | 1 | 4 | 3 | 1 | 3 | 6 | – |
Mutation frequencies were determined by measuring reversion of the rIIUV199oc allele as described in the Materials and Methods. The rII+ revertant frequency for wild-type T4 phage is ~1 × 10−6. Strong mutator phenotypes were observed primarily for mutant DNA polymerases with amino acid substitutions for Y254 and G255. Residues in parentheses are the corresponding residues in the RB69 DNA polymerase. The dashes indicate amino acid substitutions that were not tested.
Because structural information exists for binary complexes formed with the exo−-RB69 DNA polymerase, which has Ala substitutions for Mg2+-binding residues D222 and D327 in the exonuclease active site (11), and for the β−–DNA RB69 DNA polymerase, in which the loop of the β hairpin is deleted (amino acid residues E254, N255, M256, Y257, G258, S269, and R260) [Figure 1; (12)], the following biochemical experiments were performed with these and other mutant RB69 DNA polymerases as well as the wild-type enzyme. Although subtle differences have been detected between the T4 and RB69 DNA polymerases (9), control experiments presented here indicate strong similarities in β-hairpin function for both enzymes.
Fluorescent Exonuclease Complexes Formed with dsDNA Substrates Labeled with 2AP at the 3′ End of the Primer Strand
T4 DNA polymerase forms highly fluorescent exonuclease complexes with DNAs labeled with 2AP at the 3′ end of the primer strand [Table 1; (16, 23)]. The same fluorescence enhancement is observed for complexes formed with the wtRB69 DNA polymerase (Table 3). The increase in 2AP fluorescence is caused by base unstacking produced by intercalation of a Phe residue (F123 in the RB69 DNA polymerase) between the 3′ terminal and penultimate bases of the primer strand in the exonuclease active center (10, 11, 18). A >13-fold increase in fluorescence intensity was observed between the unbound dsDNA (~3500 cps) and complexes formed with the wtRB69 DNA polymerase (47 500 cps; Table 3). Exonuclease complexes are formed with dsDNA because the 2AP-T base pair is regarded as a mismatch, particularly if the primer terminal region is rich in AT base pairs as the dsDNA used here [Table 1; (19, 21)].
Table 3.
Formation of Exonuclease Complexes with DNA Labeled with 2AP at the Primer Enda
| fluorescence lifetimes |
|||||
|---|---|---|---|---|---|
| 2AP base or complexes | fluorescence intensity (counts/s) | τ1 [±0.1] (ns) | τ2 [±0.5] (ns) | τ3 [±0.5] (ns) | 〈τnum〉 (ns) |
| 2AP base | 54 200 | 11.5 (1.00) | 11.5 | ||
| wt–dsDNA | 47 500 | 9.5 (1.00) | 9.5 | ||
| G258S–dsDNA | 37 400 | 0.6 (0.32) | 9.8 (0.68) | 6.9 | |
| β−–dsDNA | 16 000 | 0.3 (0.62) | 3.3 (0.11) | 10.0 (0.27) | 3.2 |
| exo−–dsDNA | 12 400 | 0.3 (0.73) | 2.4 (0.10) | 9.5 (0.17) | 2.1 |
| G258S/exo−–dsDNA | 12 000 | 0.3 (0.75) | 2.6 (0.11) | 10.2 (0.13) | 1.8 |
| β−/exo−–dsDNA | 7 000 | 0.3 (0.81) | 2.3 (0.12) | 8.9 (0.07) | 1.1 |
| wt–ssDNA | 49 600 | 9.7 (1.00) | 9.7 | ||
| β−–ssDNA | 49 700 | 9.8 (1.00) | 9.8 | ||
| wt–mmDNA | 49 400 | 9.7 (1.00) | 9.7 | ||
| G258S–mmDNA | 49 200 | 10.1 (1.00) | 10.1 | ||
| β−–mmDNA | 49 400 | 9.8 (1.00) | 9.8 | ||
| exo−–mmDNA | 36 800 | 9.8 (1.00) | 9.8 | ||
| β−/exo−–mmDNA | 36 000 | 9.8 (1.00) | 9.8 | ||
Sequences of the ssDNA, dsDNA, and mmDNA with 2AP at the 3′ primer end are given in Table 1. Fluorescence intensities and lifetimes were determined as described in the Materials and Methods. The fluorescence intensities reported are for 200 nM 2AP either as the base or in DNA. The pre-exponential factors (relative amplitudes) are indicated in parentheses; errors for the amplitudes are ±0.05. Errors in the lifetime are indicated in brackets. 〈τnum〉 indicates the number-averaged fluorescence lifetime.
Less fluorescence enhancement, however, is observed for complexes formed with dsDNA and proofreading-deficient T4 DNA polymerases (16, 21, 23); this was also observed for proofreading-deficient RB69 DNA polymerases. Fluorescence intensity decreased incrementally with the apparent severity of the proofreading defect of the mutant RB69 DNA polymerases (Table 3). A small reduction in fluorescence intensity was observed for the G258S–DNA polymerase (37 400 cps) compared to the wtRB69 DNA polymerase, but a much larger decrease in fluorescence intensity was detected for the β−–DNA polymerase (16 000 cps). The markedly lower level of fluorescence intensity observed for complexes formed with the β−–DNA polymerase provides strong evidence that the β hairpin is required for formation of the strand-separated exonuclease complexes with dsDNA.
Less fluorescence intensity compared to wtRB69 was also observed for complexes formed with the hydrolysis-deficient D222A/D327A (exo−) DNA polymerase (12 400 cps) and the multiply mutant G258S/exo−–DNA polymerase (12 000 cps). However, even less fluorescence intensity was detected for complexes formed with the mutant that lacks both the loop region of the β hairpin and hydrolysis activity, the β−/exo−–DNA polymerase (7000 cps; Table 3). Because the 7000 cps detected for the β−/exo−–DNA polymerase is just 2-fold higher than the fluorescence intensity of unbound dsDNA (Table 1), this mutant RB69 DNA polymerase has almost no ability to form fluorescent exonuclease complexes. The most likely explanation for this result is that both the β hairpin and residues in the exonuclease active center participate in the formation of exonuclease complexes with dsDNA.
Fluorescent Exonuclease Complexes Formed with ssDNA and mmDNA Substrates Labeled with 2AP at the 3′ End of the Primer Strand
The decrease in the formation of fluorescent exonuclease complexes with the β−–DNA polymerase is consistent with the proposal that the β hairpin acts as a wedge that assists in the separation of the primer from the template strand; however, if the β hairpin is needed to form strand-separated exonuclease complexes with dsDNA, then the β hairpin is not expected to be needed to form complexes with ssDNA. This proposal was confirmed by forming binary complexes with either the wtRB69 or β−–DNA polymerase with ssDNA that was labeled at the 3′ end with 2AP and (ssDNA, Table 1); both the wild-type and mutant DNA polymerases formed high levels of fluorescent exonuclease complexes (>49 000 cps; Table 3). This result is consistent with the ability of the β−–DNA polymerase to degrade ssDNA (12). Because the levels of fluorescence intensity observed for the wtRB69 DNA polymerase with ssDNA and dsDNA were nearly the same, dsDNA is not a barrier toward forming exonuclease complexes for the wild-type enzyme; however, dsDNA is a barrier for forming exonuclease complexes for the β−–DNA polymerase because ~3-fold less fluorescence intensity was observed for complexes formed with dsDNA compared to ssDNA.
Because ssDNA could potentially be bound in a different conformation than dsDNA, the formation of fluorescent exonuclease complexes was also tested with dsDNA containing three preformed mismatches at the primer terminus (mmDNA, Table 1), which mimics the extent of strand separation observed in structural studies of binary exonuclease complexes (10, 11). Again, high levels of fluorescence intensity (>49 000 cps) were detected for the wtRB69–, G258S–, and β−–DNA polymerases (Table 3). Thus, the β hairpin is not needed for the RB69 DNA polymerase to form exonuclease complexes if there are preformed mismatches at the primer terminus. A reduced but still high level of fluorescence intensity was detected for the exo−–DNA polymerase (36 800 cps), which suggests that Ala substitutions in the exonuclease active center slightly reduce the ability of this mutant DNA polymerase to form fluorescent exonuclease complexes with mmDNA, but no significant further reduction in fluorescence intensity was detected for the β−/exo−–DNA polymerase. Thus, while the β hairpin and residues in the exonuclease active site are required for the formation of exonuclease complexes with dsDNA, as would occur during proofreading in vivo, these structures are not required if primer strand separation is preformed.
Time-Resolved Studies of 2AP Fluorescence in Complexes Formed with DNA Substrates Labeled with 2AP at the 3′ End of the Primer Strand
Although different levels of fluorescence intensity suggest that fewer exonuclease complexes are formed by the mutant DNA polymerases than by the wtRB69 DNA polymerase, intensity values alone do not provide information about the 2AP environment and, thus, about the type of complex formed. A single fluorescence lifetime of 11.5 ns is observed for the free 2AP base in solution [Table 3; (23, 28)], but short lifetimes (<0.4 ns) predominate for 2AP in unbound DNA (13–15,29). Longer 2AP lifetimes are observed for complexes formed with the T4 and RB69 DNA polymerases and 2AP-labeled DNA, which indicates enzyme-induced base unstacking [Table 3; (21)]. The different lifetimes reflect different emitting populations whose mean lifetimes are represented by individual decay components; thus, each lifetime represents a distribution of 2AP environments with different degrees of 2AP base stacking (14).
Because only exonuclease complexes can be formed with ssDNA and mmDNA, a single fluorescent species is expected. A single fluorescent component was detected with a relatively long 9.7–10.1 ns fluorescence lifetime (Table 3), which approaches the reported lifetime of the 2AP nucleoside in solution [10.5 ns; (13, 29)]. These longer fluorescence lifetime values were expected because the terminal base in the exonuclease active center is unstacked in crystal structures of exonuclease complexes (10, 11) and similar 2AP lifetime values are observed for complexes formed with the phage T4 DNA polymerase (18, 23).
A single, relatively long fluorescence lifetime (9.5 ns) was also observed for complexes formed with the wtRB69 DNA polymerase and dsDNA (Table 3), which indicates that primarily one type of complex was formed – highly fluorescent exonuclease complexes. Less fluorescence intensity was observed for complexes formed with the mutant DNA polymerases and dsDNA (Table 3). A small but significant decrease in the amplitude of the long lifetime component to 0.68 was observed for the G258S–DNA polymerase, and larger decreases in amplitude were observed for the β−– and exo−–DNA polymerases, to 0.27 and 0.17, respectively. A further decrease in amplitude as well as lifetime was observed for the multiply mutant β−/exo−–DNA polymerase. Thus, the reduction in fluorescence intensity for complexes formed with the mutant DNA polymerases is correlated with a decrease in the amplitude of the long lifetime component (Table 3).
The decreases in amplitude of the long lifetime component were accompanied by the appearance of a short lifetime component (~0.3 ns); the amplitude of the short lifetime component increased as the amplitude of the long lifetime component decreased (Table 3). Another lifetime component of about 2–3 ns was also detected. We determined in previous experiments with the T4 DNA polymerase that mutant enzymes with defects in forming exonuclease complexes with dsDNA instead form complexes in which the primer end is bound in the polymerase active center (21); hence, the 2–3 ns lifetime may reflect the environment of 2AP in this type of complex.
Fluorescent Exonuclease Complexes Formed with dsDNA Substrates Labeled with 2AP in the +1 Position of the Template Strand
Formation of polymerase complexes can be detected with dsDNA labeled with 2AP in the +1 position of the template strand [Table 1; (19–21)]. Two types of polymerase complex are detected with the phage T4 DNA polymerase. With A+T-rich DNAs, the major component detected has a fluorescence lifetime of about 10.5–11 ns (21), which indicates a high degree of base unstacking at the +1 template position. Because the amplitude of this component increases for mutant T4 DNA polymerases that have reduced ability to form exonuclease complexes, this “polymerase” complex is proposed to be a pre-exonuclease complex. Thus, fluorescence intensity and lifetime studies for complexes formed with the wild-type and mutant RB69 DNA polymerases with the A+T-rich DNA substrate are expected to provide information on partitioning of DNA between the polymerase and exonuclease active centers.
With G+C-rich DNAs, the longest lifetime component detected for complexes formed with the T4 DNA polymerase is characterized by a shorter fluorescence lifetime of about 8.5–9 ns. The ability to form this complex does not depend upon the ability of the T4 DNA polymerase to proofread. Because G+C richness in the primer-terminal region favors nucleotide incorporation over proofreading (30), complexes characterized by the 8.5–9 ns lifetime are proposed to be pre-polymerase complexes (21). Corresponding results are expected for the wild-type and mutant RB69 DNA polymerases.
High levels of fluorescence intensity and high amplitudes for the long lifetime component (>10.5 ns) were observed for complexes formed with the proofreading-defective RB69 DNA polymerases and the A+T-rich DNA substrate (Table 4). Fluorescence enhancements from 15- to 18-fold were detected for complexes formed with the mutant RB69 DNA polymerases compared to unbound DNA. A smaller 12-fold enhancement was detected for complexes formed with the wtRB69 DNA polymerase. The high level of fluorescence intensity observed for the mutant DNA polymerases was primarily due to the higher amplitude of the long lifetime component. Thus, the β hairpin is not needed to form the highly fluorescent pre-exonuclease complexes that are characterized by the 10.5 ns lifetime. The increased amplitude of this component compared to complexes formed with the wtRB69 DNA polymerase indicates that the reduced ability of the mutant DNA polymerases to form exonuclease complexes (Table 3) increases the formation of pre-exonuclease complexes, as observed for the T4 DNA polymerase. No differences, however, were detected in fluorescence intensity or lifetimes between complexes formed with the wtRB69 and mutant DNA polymerases with the G+C-rich DNA (Table 4); thus, the presence or absence of the β hairpin is not a factor in the formation of pre-polymerase complexes.
Table 4.
Fluorescence Intensities and Fluorescence Lifetimes of Binary Complexes Formed with Wild-Type and Mutant RB69 Polymerases with A+T-or G+C-Rich DNAs Labeled with 2AP in the +1 Position in the Template Stranda
| complexes | fluorescence intensity (counts/s) | fluorescence enhancement (x-fold)b | fluorescence lifetimes |
|||
|---|---|---|---|---|---|---|
| τ1 [±0.1] (ns) | τ2 [±0.5] (ns) | τ3 [±0.5] (ns) | 〈τnum〉 (ns) | |||
| wt–A+T DNA | 21 000 | 12 | 0.2 (0.46) | 3.6 (0.15) | 10.1 (0.39) | 4.6 |
| G258S–A+T DNA | 29 750 | 17 | 0.3 (0.30) | 5.2 (0.19) | 10.7 (0.51) | 6.5 |
| β−–A+T DNA | 31 400 | 18 | 0.3 (0.30) | 5.5 (0.14) | 10.8 (0.56) | 6.9 |
| exo−–A+T DNA | 26 250 | 15 | 0.2 (0.38) | 4.6 (0.16) | 10.4 (0.46) | 5.6 |
| G258S/exo−–A+T DNA | 31 000 | 18 | 0.3 (0.27) | 5.0 (0.20) | 10.5 (0.53) | 6.6 |
| β−/exo−–A+T DNA | 31 600 | 18 | 0.4 (0.30) | 5.1 (0.11) | 10.6 (0.59) | 6.9 |
| wt–G+C DNA | 14 750 | 8.5 | 0.3 (0.43) | 3.3 (0.18) | 9.0 (0.39) | 4.2 |
| β−–G+C DNA | 15 500 | 9 | 0.2 (0.41) | 2.3 (0.20) | 8.8 (0.40) | 4.1 |
| exo−–G+C DNA | 15 600 | 9 | 0.3 (0.40) | 3.1 (0.19) | 9.0 (0.41) | 4.4 |
| β− /exo−–G+C DNA | 15 700 | 9 | 0.3 (0.36) | 2.4 (0.21) | 8.5 (0.43) | 4.3 |
Sequences of A+T- and G+C-rich DNAs with 2AP in the +1 position in the template strand are given in Table 1. Fluorescence intensities and lifetimes were determined as described in the Materials and Methods. The pre-exponential factors (relative amplitudes) are indicated in parentheses; errors for the amplitudes are ±0.05. Errors in the lifetime are indicated in brackets. 〈τnum〉 indicates the number-averaged fluorescence lifetime.
The fluorescence enhancement is with respect to the unbound DNAs (fluorescence intensity of 200 nM A+T- and G+C-rich DNAs are 1750 and 1720 counts/s, respectively).
Where is the Hairpin in Pre-exonuclease Complexes
The ability to form exonuclease complexes with dsDNA labeled at the 3′ end of the primer strand was wtRB69 > G258S > β− > exo− ~ G258S/exo− > β−/exo− (Table 3). We expected the reverse of this trend to be observed for formation of pre-exonuclease complexes with the A+T-rich DNA labeled in the +1 template position with 2AP because reduced formation of exonuclease complexes is correlated with increased formation of pre-exonuclease complexes (21). The reverse trend was observed, except for the exo−–DNA polymerase (Table 4); a lower level of fluorescence intensity was detected for the exo−–DNA polymerase (26 250 cps) than for the β−–DNA polymerase (31 400 cps). The difference in fluorescence intensities for complexes formed with the two mutant DNA polymerases is small, but the difference was reproducible. One possible interpretation is that the full-size β hairpin in the exo−–DNA polymerase but not the truncated β hairpin can partially suppress or quench the long lifetime component. Interactions are reported for the β hairpin and the template strand (11).
DISCUSSION
There are several proposals to explain how the primer terminus migrates the 30–40 Å from the polymerase to the exonuclease active center. One early idea proposed for the Klenow fragment of Escherichia coli DNA polymerase I, a family A DNA polymerase, requires the protein to slide backward along the DNA while melting the terminal 3–4 base pairs (31). Although this proposal provides a mechanism for strand separation, a later study revealed that the Klenow fragment edits its own polymerase errors primarily by an intermolecularprocess,involvingdissociationoftheenzyme–DNA complex and re-association of the DNA with the exonuclease site of a second molecule of Klenow fragment (32). Similar proposals have been made for the T7 DNA polymerase, another family A DNA polymerase (33), and for the T4 DNA polymerase (16, 24); both the T7 and T4 DNA polymerases form polymerase and exonuclease complexes with dsDNA substrates directly from solution. These proposals, however, do not explain how the end of the primer strand is separated from the template strand.
Could strand separation occur spontaneously? Physical conditions that increase denaturation of the primer terminal region, A+T-richness, and high temperature increase proofreading by the T4 DNA polymerase (30). A striking example of this point is that a terminal 2AP-C mismatch that is adjacent to four neighboring GC base pairs is excised 4-fold more slowly by the T4 DNA polymerase than the less profound terminal 2AP-T mismatch with adjacent AT base pairs (16). Because conditions that favor local melting of the primer terminus increase proofreading, there may not be a requirement for the DNA polymerase to facilitate strand separation; however, the ability to do so is expected to increase proofreading over a mechanism that relies solely on spontaneous strand separation. We propose that the high exonuclease activity of the T4 and RB69 DNA polymerases on dsDNA is due in part to the ability of these DNA polymerases to facilitate strand separation.
Previous genetic and biochemical studies of the T4 G255S–DNA polymerase provided evidence that amino acid substitutions for G255 in the loop of the β hairpin reduce replication fidelity and proofreading by slowing the formation of exonuclease complexes (1, 2, 7, 8). We have extended these studies by further mutational analysis of the loop of the T4 DNA polymerases β hairpin. While isolation of the T4 G255S– and G255A–DNA polymerases occurred frequently in genetic selection strategies for mutator DNA polymerases (5, 6), only one other loop mutation was identified, a 15 base pair in-frame direct repeat that encodes a duplication of residues K257, E258, I259, Y260, and S261 (6). To determine the potential importance of any additional loop residues, revertant frequencies for phage-expressing mutant DNA polymerases with six different amino acid substitutions for residues Y254, S256, and K257 were determined (Table 2). Only Y254 appeared to be as important as G255 for proofreading because Lys, Leu, and Gly substitutions for Y254 produced mutator DNA polymerases similar to the low fidelity G255S– and G255A–DNA polymerases. Smaller increases in revertant frequencies were detected for the S259K–, S259L–, and S259G–DNA polymerases, and little change in replication fidelity was observed for phage that expressed mutant DNA polymerases with amino acid substitutions for K257.
T4 DNA polymerase loop residues G255 and Y254 correspond to residues G258 and Y257 in the RB69 DNA polymerase (Figure 1 and Table 2). Given the protein sequence and structural similarities between the T4 and RB69 DNA polymerases in the exonuclease domain (10, 11, 34, 35), we wanted to confirm functional similarities for the formation of exonuclease complexes. The reduced ability of the phage T4 G255S–DNA polymerase to proofread in vivo is correlated with modest decreases in the ability to form exonuclease complexes and to proofread dsDNA substrates labeled at the 3′ end of the primer strand with 2AP (7, 8). A similar reduction in the formation of fluorescent exonuclease complexes was observed for the RB69 G258S–DNA polymerase (Table 3). The reduced ability to form exonuclease complexes was marked by a decrease in the amplitude of the longest lifetime component and the appearance of a short lifetime component, which suggests that the G258S–DNA polymerase is forming polymerase complexes in lieu of exonuclease complexes. This proposal was supported by experiments with the A+T-rich DNA substrate that is labeled with 2AP in the +1 position of the template strand (Table 1). Mutant T4 DNA polymerases that have reduced ability to form exonuclease complexes instead form complexes with DNA bound in the polymerase active center, which are proposed to be pre-exonuclease complexes. This was observed for the RB69 G258S–DNA polymerase (Table 4) and the T4 G255S–DNA polymerase (21). Because the T4 G255S–DNA polymerase displays a strong mutator pheno-type (Table 2), modest changes in partitioning of DNA between the polymerase and exonuclease active centers are correlated with large reductions in proofreading and replication fidelity.
The adjacent tyrosine (Y254 in the T4 DNA polymerase) is also implicated in the formation of exonuclease complexes (Table 2), but while modest decreases in the ability to form exonuclease complexes with dsDNA were observed for the T4 G255S– and RB69 G258–DNA polymerases, a much larger decrease was observed for the β−–RB69 DNA polymerase, in which the entire loop region is deleted (Figure 1 and Table 3). The β−–DNA polymerase also formed more pre-exonuclease complexes with dsDNA (Table 4), which is expected if the formation of exonuclease complexes is hindered. Deletion of the loop region shortens the hairpin structure and also removes residues G258 and Y257 as well as several adjacent residues (Figure 1). While T4 residues G255 and Y254 are important for replication fidelity (Table 2), we cannot rule out the possibility that loop residues not tested, for example, M253, could also play important roles.
Our studies demonstrate that the β hairpin is needed for the formation of complexes with dsDNA, which suggests a role for the β hairpin in separating the template and primer strands or in acting as a wedge to hold the strands apart. However, the β hairpin does not seem to be needed once exonuclease complexes are formed because deletion of the β hairpin does not hinder the formation of exonuclease complexes with ssDNA or mmDNA with three preformed terminal mismatches (Table 3). Thus, the β hairpin acts during strand separation but is not needed to stabilize the strand-separated exonuclease complex once it is formed.
In addition, our studies revealed that the β hairpin is not sufficient for the formation of exonuclease complexes with dsDNA; residues in the exonuclease active center are also necessary. While Ala substitutions for two aspartate residues (D222A/D327A) in the exonuclease active center severely reduced the formation of exonuclease complexes as measured by the formation of fluorescent exonuclease complexes with dsDNA labeled at the 3′ end of the primer strand with 2AP, deleting the β hairpin loop further reduced the formation of exonuclease complexes (Table 3). Thus, strand separation appears to require cooperation between the β hairpin and residues in the exonuclease active center.
Summary
We propose the following model for the proofreading pathway catalyzed by the T4 and RB69 DNA polymerases. When an incorrect nucleotide is incorporated, translocation is prevented and a pre-exonuclease complex is formed. The base in the +1 position in the template strand is unstacked, as determined by the high level of 2AP fluorescence intensity and long fluorescence lifetime [Table 4; (21)]. An apparent nontranslocated, pre-exonuclease structure has also been detected in structural studies of the RB69 DNA polymerase complexed with dsDNA containing a terminal furan-A base pair (11). In this “in-between-polexo” structure, the β hairpin appears to interact with the base in the +2 position of the template strand [Figure 1; (11)].
The next step likely involves the β hairpin, particularly loop residues Y254 and G255 in the T4 DNA polymerase (Y257 and G258 in the RB69 DNA polymerase). The β hairpin could function to tease apart the primer end or to take advantage of local breathing at the primer end by inserting a wedge between the template and primer strands to prevent strand renaturation. These activities are also expected to assist dissociation of contacts between amino acid residues in the polymerase active center with the primer terminal region. Thus, mutant DNA polymerases with altered β hairpins are predicted to dissociate from pre-exonuclease complexes more slowly, which is observed; the dissociation rate for fluorescent pre-exonuclease complexes formed with the T4 G255S-DNA polymerase is >3-fold slower than the dissociation rate for complexes formed the wild-type T4 DNA polymerase (20). Slow dissociation is also predicted to increase the chance for primer extension and, hence, reduced proofreading and replication fidelity. The T4 G255S–DNA polymerase is a mutator DNA polymerase (Table 2). Proofreading by the T4 DNA polymerase appears not to be intrinsically processive because pre-exonuclease complexes are not converted to exonuclease complexes in the presence of a heparin trap (24). Thus, proofreading appears to require dissociation of pre-exonuclease complexes before the formation of exonuclease complexes.
The β hairpin is needed for the T4 and RB69 DNA polymerases to form exonuclease complexes directly with dsDNAs but not with ssDNA or mmDNA with three preformed terminal mismatches (Table 3). Thus, the β hairpin has two proposed roles in the proofreading pathway: dissociation of pre-exonuclease complexes and the formation of exonuclease complexes with the primer end bound in the exonuclease active center. Amino acid residues in the exonuclease active center are also required to form exonuclease complexes, because the ability to form exonuclease complexes is most severely compromised for the −β/exo−RB69 DNA polymerase (Table 3). After hydrolysis, the trimmed primer end is returned to the polymerase active center rapidly and processively (24) in a reaction that is independent of the β hairpin (11).
ACKNOWLEDGMENT
We thank William Konigsberg for providing the RB69 DNA polymerase expression plasmid and D. Zhao for technical assistance.
Supported by grants from the Canadian Institutes of Health Research (L.J.R-K.) and the National Institutes of Health, Grant CA52040 (to S. Wallace). L.J.R-K is a Scientist of the Alberta Heritage Foundation for Medical Research.
Footnotes
Abbreviations: ssDNA, single-stranded DNA; dsDNA, double-stranded DNA; mmDNA, mismatched DNA; 2AP, 2-aminopurine; wtRB69, wild-type RB69 DNA polymerase; exo−, the D222A/D327A-RB69 DNA polymerase, alanine substitutions for aspartates 222 and 327; β−, the I253G,Δ254–260-RB69 DNA polymerase, deletion of the loop in the β hairpin; G258S-RB69 DNA polymerase, serine substitution for glycine 258 in the loop of the β hairpin; β−/exo−, deletion of the β-hairpin loop and exonuclease deficient (D222A/D327A) variant of the RB69 DNA polymerase; G258S/exo−, the D222A/D327A/G258S-RB69 DNA polymerase.
REFERENCES
- 1.Reha-Krantz LJ, Marquez LA, Elisseeva E, Baker RP, Bloom LB, Danford HB, Goodman MF. The proofreading pathway of bacteriophage T4 DNA polymerase. J. Biol. Chem. 1998;273:22969–22976. doi: 10.1074/jbc.273.36.22969. [DOI] [PubMed] [Google Scholar]
- 2.Reha-Krantz LJ. Regulation of DNA polymerase exonucleolytic proofreading activity: Studies of bacteriophage T4 “antimutator” DNA polymerases. Genetics. 1998;148:1551–1557. doi: 10.1093/genetics/148.4.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clayton LK, Goodman MF, Branscomb EW, Galas DJ. Error induction and correction by mutant and wild type T4 DNA polymerases. Kinetic error discrimination mechanisms. J. Biol. Chem. 1979;254:1902–1912. [PubMed] [Google Scholar]
- 4.Capson TL, Peliska JA, Kaboord BF, Frey MW, Lively C, Dahlberg M, Benkovic SJ. Kinetic characterization of the polymerase and exonuclease activities of the gene 43 protein of bacteriophage T4. Biochemistry. 1992;31:10984–10994. doi: 10.1021/bi00160a007. [DOI] [PubMed] [Google Scholar]
- 5.Reha-Krantz LJ. Amino acid changes coded by bacteriophage T4 DNA polymerase mutator mutants. J. Mol. Biol. 1988;202:711–724. doi: 10.1016/0022-2836(88)90552-9. [DOI] [PubMed] [Google Scholar]
- 6.Stocki SA, Nonay RL, Reha-Krantz LJ. Dynamics of bacteriophage T4 DNA polymerase function: Identification of amino acid residues that affect switching between polymerase and 3′ → 5′ exonuclease activities. J. Mol. Biol. 1995;254:15–28. doi: 10.1006/jmbi.1995.0595. [DOI] [PubMed] [Google Scholar]
- 7.Marquez LA, Reha-Krantz LJ. Using 2-aminopurine fluorescence and mutational analysis to demonstrate an active role of bacteriophage T4 DNA polymerase in strand separation required for 3′ → 5′ exonuclease activity. J. Biol. Chem. 1996;271:28903–28911. doi: 10.1074/jbc.271.46.28903. [DOI] [PubMed] [Google Scholar]
- 8.Baker RP, Reha-Krantz LJ. Identification of a transient excision intermediate at the crossroads between DNA polymerase extension and proofreading pathways. Proc. Natl. Acad. Sci. U.S.A. 1998;95:3507–3512. doi: 10.1073/pnas.95.7.3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hogg M, Cooper W, Reha-Krantz L, Wallace SS. Kinetics of error generation in homologous B-family DNA polymerases. Nucleic Acids Res. 2006;34:2528–2535. doi: 10.1093/nar/gkl300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shamoo Y, Steitz TA. Building a replisome from interacting pieces: Sliding clamp complexed to a peptide from DNA polymerase and a polymerase editing complex. Cell. 1999;99:155–166. doi: 10.1016/s0092-8674(00)81647-5. [DOI] [PubMed] [Google Scholar]
- 11.Hogg M, Wallace SS, Doublié S. Crystallographic snapshots of a replication DNA polymerase encountering an abasic site. EMBO J. 2004;23:1483–1493. doi: 10.1038/sj.emboj.7600150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hogg M, Aller P, Konigsberg W, Wallace SS, Doublié S. Structural and biochemical investigation of the role in proofreading of a β hairpin loop found in the exonuclease domain of a replicative DNA polymerase of the B family. J. Biol. Chem. 2007;282:1432–1444. doi: 10.1074/jbc.M605675200. [DOI] [PubMed] [Google Scholar]
- 13.Rachofsky EL, Osman R, Ross JB. Probing structure and dynamics of DNA with 2-aminopurine: Effects of local environment on fluorescence. Biochemistry. 2001;40:946–956. doi: 10.1021/bi001664o. [DOI] [PubMed] [Google Scholar]
- 14.Neely RK, Daujotyte D, Grazulis S, Magennis SW, Dryden DTF, Klimasauskas S, Jones AC. Time-resolved fluorescence of 2-aminopurine as a probe of base flipping in M.HhaI-DNA complexes. Nucleic Acids Res. 2005;33:6953–6960. doi: 10.1093/nar/gki995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jean JM, Hall KB. 2-Aminopurine fluorescence quenching and lifetimes: Role of the base stacking. Proc. Natl. Acad. Sci. U.S.A. 2001;98:37–41. doi: 10.1073/pnas.011442198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bloom LB, Otto MR, Eritja R, Reha-Krantz LJ, Goodman MF, Beechem JM. Pre-steady-state kinetic analysis of sequence-dependent nucleotide excision by the 3′-exonuclease activity of bacteriophage T4 DNA polymerase. Biochemistry. 1994;33:7576–7586. doi: 10.1021/bi00190a010. [DOI] [PubMed] [Google Scholar]
- 17.Frey MA, Sowers LC, Millar DP, Benkovic SJ. The nucleotide analog 2-aminopurine as a spectroscopic probe of nucleotide incorporation by the Klenow fragment of Escherichia coli polymerase I and bacteriophage T4 DNA polymerase. Biochemistry. 1995;34:9185–9192. doi: 10.1021/bi00028a031. [DOI] [PubMed] [Google Scholar]
- 18.Beechem JM, Otto MR, Bloom LB, Eritja R, Reha-Krantz LJ, Goodman MF. Exonuclease–polymerase active site partitioning of primer–template DNA strands and equilibrium Mg2+ binding properties of bacteriophage T4 DNA polymerase. Biochemistry. 1998;28:9095–9103. doi: 10.1021/bi980074b. [DOI] [PubMed] [Google Scholar]
- 19.Mandal SS, Fidalgo da Silva E, Reha-Krantz LJ. Using 2-aminopurine fluorescence to detect base-unstacking in the template strand during nucleotide incorporation by the bacteriophage T4 DNA polymerase. Biochemistry. 2002;41:4399–4406. doi: 10.1021/bi015723p. [DOI] [PubMed] [Google Scholar]
- 20.Fidalgo da Silva E, Mandal SS, Reha-Krantz LJ. Using 2-aminopurine fluorescence to measure incorporation of incorrect nucleotides by wild type and mutant bacteriophage T4 DNA polymerases. J. Biol. Chem. 2002;277:40640–40649. doi: 10.1074/jbc.M203315200. [DOI] [PubMed] [Google Scholar]
- 21.Hariharan C, Reha-Krantz LJ. Using 2-aminopurine fluorescence to detect bacteriophage T4 DNA polymerase–DNA complexes that are important for primer extension and proofreading reactions. Biochemstry. 2005;44:15674–15684. doi: 10.1021/bi051462y. [DOI] [PubMed] [Google Scholar]
- 22.Hariharan C, Bloom LB, Helquist SA, Kool ET, Reha-Krantz LJ. dynamics of nucleotide incorporation: Snapshots revealed by 2-aminopurine fluorescence studies. Biochemistry. 2006;45:2836–2844. doi: 10.1021/bi051644s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tleugabulova D, Reha-Krantz LJ. Probing DNA polymerase–DNA interactions: Examining the template strand in exonuclease complexes using 2-aminopurine fluorescence and acrylamide quenching. Biochemistry. 2007;46:6559–6569. doi: 10.1021/bi700380a. [DOI] [PubMed] [Google Scholar]
- 24.Fidalgo da Silva E, Reha-Krantz LJ. DNA polymerase proofreading: Active site switching catalyzed by the bacteriophage T4 DNA polymerase. Nucleic Acids Res. 2007;35:5452–5463. doi: 10.1093/nar/gkm591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kleina LG, Masson J-M, Normanly J, Abelson J, Miller JH. Construction of Escherichia coli amber suppressor tRNA genes II. Synthesis of additional tRNA genes and improvement of suppressor efficiency. J. Mol. Biol. 1990;213:705–717. doi: 10.1016/S0022-2836(05)80257-8. [DOI] [PubMed] [Google Scholar]
- 26.Reha-Krantz LJ. Learning about DNA polymerase function by studying antimutator DNA polymerases. Trends Biochem. Sci. 1995;20:136–140. doi: 10.1016/s0968-0004(00)88987-2. [DOI] [PubMed] [Google Scholar]
- 27.James DR, Siemiarczuk A, Ware WT. Stroboscopic optical boxcar technique for the determination of fluorescence lifetimes. Rev. Sci. Instrum. 1992;63:1710–1716. [Google Scholar]
- 28.Lakowicz JR. Principles of Fluorescence Spectroscopy. 2nd ed. Plenum Publishers; New York: 1999. [Google Scholar]
- 29.Guest CR, Hochstrasser RA, Sowers LC, Millar DP. Dynamics of mismatched base pairs in DNA. Biochemistry. 1991;30:3271–3279. doi: 10.1021/bi00227a015. [DOI] [PubMed] [Google Scholar]
- 30.Bessman MJ, Reha-Krantz LM. Studies on the biochemical basis of spontaneous mutation. V. Effect of temperature on mutation frequency. J. Mol. Biol. 1977;116:115–123. doi: 10.1016/0022-2836(77)90122-x. [DOI] [PubMed] [Google Scholar]
- 31.Joyce CM, Steitz TA. DNA polymerase I: From crystal structure to function via genetics. Trends Biochem. Sci. 1987;12:288–292. [Google Scholar]
- 32.Joyce CM. How DNA travels between the separate polymerase and 3′–5′-exonuclease sites of DNA polymerase I (Klenow fragment) J. Biol. Chem. 1989;264:10858–10866. [PubMed] [Google Scholar]
- 33.Donlin MJ, Patel SS, Johnson KA. Kinetic partitioning between the exonuclease and polymerase sites in DNA error correction. Biochemistry. 1991;30:538–546. doi: 10.1021/bi00216a031. [DOI] [PubMed] [Google Scholar]
- 34.Wang J, Yu P, Lin TC, Konigsberg WH, Stetiz TA. Crystal structures of an NH2-terminal fragment of the T4 DNA polymerase and its complexes with single-stranded DNA and with divalent metal ions. Biochemistry. 1996;35:8110–8119. doi: 10.1021/bi960178r. [DOI] [PubMed] [Google Scholar]
- 35.Franklin MC, Wang J, Steitz TA. Structure of the replicating complex of a pol α family DNA polymerase. Cell. 2001;105:657–667. doi: 10.1016/s0092-8674(01)00367-1. [DOI] [PubMed] [Google Scholar]