

Abstract
The bone morphogenetic protein (BMP) and Wnt signaling pathways both contribute essential roles in regulating bone mass. However, the molecular interactions between these pathways in osteoblasts are poorly understood. We recently reported that osteoblast-targeted conditional knockout (cKO) of BMP receptor type IA (BMPRIA) resulted in increased bone mass during embryonic development, where diminished expression of Sost as a downstream effector of BMPRIA resulted in increased Wnt/β-catenin signaling. Here, we report that Bmpr1a cKO mice exhibit increased bone mass during weanling stages, again with evidence of enhanced Wnt/β-catenin signaling as assessed by Wnt reporter TOPGAL mice and TOPFLASH luciferase. Consistent with negative regulation of the Wnt pathway by BMPRIA signaling, treatment of osteoblasts with dorsomorphin, an inhibitor of Smad-dependent BMP signaling, enhanced Wnt signaling. In addition to Sost, Wnt inhibitor Dkk1 also was downregulated in cKO bone. Expression levels of Dkk1and Sost were upregulated by BMP2 treatment and downregulated by Noggin. Moreover, expression of a constitutively active Bmpr1a transgene in mice resulted in the upregulation of both Dkk1 and Sost and partially rescued the Bmpr1a cKO bone phenotype. These effectors are differentially regulated by mitogen-activated protein kinase (MAPK) p38 because pretreatment of osteoblasts with SB202190 blocked BMP2-induced Dkk1 expression but not Sost. These results demonstrate that BMPRIA in osteoblasts negatively regulates endogenous bone mass and Wnt/β-catenin signaling and that this regulation may be mediated by the activities of Sost and Dkk1. This study highlights several interactions between BMP and Wnt signaling cascades in osteoblasts that may be amenable to therapeutic intervention for the modification of bone mass density. © 2010 American Society for Bone and Mineral Research
Keywords: BMP receptor type IA (BMPRIA), Wnt, osteoblasts, Sost, Dkk1
Introduction
Bone morphogenetic proteins (BMPs), originally discovered as inducers of ectopic bone,(3) are members of the transforming growth factor β (TGF-β) superfamily.(1,2) BMP signals, like those of other TGF-β family members, are mediated by the concerted activation of type I and type II serine-threonine kinase receptors.(4) On engaging BMP ligands in a heteromeric complex, type II receptors activate the kinase function of type I receptors to initiate signaling via intracellular Smads 1, 5, and 8. Of the three type I receptors that recognize BMPs (BMPRIA or ALK3, BMPRIB or ALK6, and ACVRI or ALK2), BMPRIA is the most effective receptor for transducing canonical BMP ligands BMP2(5) and BMP4,(6) which are abundantly expressed in bone.(7)
The osteogenic function of BMPs has been documented extensively, with numerous in vitro studies supporting a critical role of BMP signaling in osteoblastogenesis.(8) While constitutive activation of BMP signaling in muscle appears to induce ectopic ossification in patients with fibrodysplasia ossificans progressiva(9) and in a mouse model,(10) the physiologic effects of BMP signaling on endogenous bone formation in vivo have not been fully elucidated. The early embryonic lethality of conventional knockout mice deficient in BMP ligands and receptors prior to bone development has precluded these studies.(11–13) To circumvent this lethality, we recently generated conditional knockout (cKO) mice in which BMP signaling through BMPRIA was disrupted in a bone-specific and age-dependent manner using a tamoxifen-inducible Cre/lox P system under the control of a 3.2 kb type I collagen promoter. In these Bmpr1a cKO mice, we unexpectedly observed increased bone mass in embryos, weanlings, and adult animals.(14,15) In cKO adult bones, increased bone mass resulted from severely suppressed bone resorption owing to reduced RANKL-OPG pathway-induced osteoclastogenesis despite a simultaneous small reduction in the rate of bone formation.(15) These findings suggest that BMP signaling in osteoblasts regulates the balance between bone formation and resorption to control bone mass.
Wnt signaling in osteoblasts also plays an important role in regulating bone formation and mass.(16–20) Experiments using pluripotent mesenchymal cell lines to test the interaction between BMP and Wnt signaling in osteoblasts have yielded somewhat contradictory results. BMP2 has been reported to induce both Wnt3a and Wnt/β-catenin signaling,(21–23) whereas Wnt3a, in turn, enhances BMP4 expression.(24) However, Wnt3a also has been reported to repress BMP2-dependent Id1 expression.(25) In contrast, we recently demonstrated that loss of BMPRIA signaling in osteoblasts downregulates sclerostin/Sost and upregulates Wnt/β-catenin signaling, resulting in increased bone mass during embryonic stages.(14) Our results provide a potential mechanism by which BMP signaling in osteoblasts negatively regulates Wnt signaling to control fetal bone mass.
Since BMPs are used clinically to improve fracture healing,(26) our previous findings of increased bone mass in Bmpr1a-deficient mice is unexpected and contrary to the rationale behind the application of BMPs in orthopedics. This study seeks to elucidate further the molecular mechanism by which BMP signaling regulates bone mass. Here we report that loss of BMPRIA in postnatal osteoblasts increases bone mass via upregulation of Wnt/β-catenin signaling. In addition to Sost, we find that Wnt inhibitor Dkk1 is also a downstream target of BMPRIA signaling in osteoblasts, lending further support for a negative regulation of Wnt signaling via BMPRIA.
Materials and Methods
Mice and tamoxifen administration
A transgenic mouse line expressing the tamoxifen (TM)–inducible Cre fusion protein Cre-ER™ under the control of a 3.2 kb mouse procollagen α1(I) promoter (Col1-CreERTM) was generated by pronuclear injection(14,15) and crossed with floxed Bmpr1a mice.(27) TM (T5648, Sigma, St. Louis, MO, USA) was dissolved in a small volume of ethanol, diluted with corn oil at a concentration of 10 mg/mL, and stored at −20°C until use. To generate Bmpr1a cKO mice (Col1-CreERTM+:Bmpr1afx/fx) and littermate controls (Col1-CreERTM–:Bmpr1afx/fx) in weanling stages, TM (75 mg/kg) was injected intraperitoneally (i.p.) into nursing females every 3 days from P2 until euthanasia at P10, P14, or P21.(15) Mice that conditionally express a constitutively active form of Bmpr1a (caBmpr1a), which has a mutation in the glycine and serine residue (GS) regulatory domain of BMPRIA (Q227D) resulting in ligand-independent activation of Smad signaling after Cre recombination, were generated as reported previously(14) and crossed with Col1-CreERTM mice. After injection of TM into nursing females every 3 days from P2 to P21, caBmpr1a mutant mice (Col1-CreERTM+:caBmpr1a+) were compared with littermate controls (Col1-CreERTM–:caBmpr1a+). Both received TM through milk until sacrifice. ROSA26 Cre reporter (R26R)(28) and TOPGAL(29) mice were obtained from Dr. Philippe Soriano and the Jackson Laboratory, respectively. C57BL/6 and CD1 mice were used for isolation of wild-type osteoblasts and calvaria culture. The animal protocol was approved by the Institutional Animal Care and Use Committee (IACUC) at the NIEHS/NIH.
Histology
For hematoxylin and eosin (H&E) staining, rib cages and skulls were fixed in 4% paraformaldehyde, decalcified with 10% EDTA, and embedded in paraffin. Paraffin sections were cut at 8 µm and stained using a standard protocol. For β-galactosidase (β-gal) staining, decalcified bones were prepared in 30% sucrose before frozen sectioning. Sections were stained with X-gal for β-gal activity and counterstained with eosin. Whole rib bones and skulls also were stained with X-gal as previously.(14,15) For immunofluorescence, osteoblasts from newborn cKO and littermate controls were fixed with 4% paraformaldehyde for 10 minutes, and immunostaining was performed using rabbit polyclonal antibodies against β-gal (1:100, Cappel Laboratories, Inc., Cochranville, PA, USA) and antirabbit IgG labeled with Alexa Fluor 488 (1:1000, Invitrogen, Carlsbad, CA, USA). Nuclei were stained with 4,6-diamidino-2-phenylindole (DAPI) (10 µg/mL, Sigma) for 10 minutes. Then β-gal-positive cells per DAPI-positive cells were calculated (%) in the same field.
X-ray and DXA analyses
X-ray images of rib cages from P21 mice were taken using a Faxitron X-ray system (Faxitron X-Ray, Lincolnshire, IL, USA). Bone mineral density (BMD) was determined by dual-energy X-ray absorptiometry (DXA) using the Lunar PIXImus2 densitometer (GE, Fairfield, CT, USA).
Quantitative real-time RT-PCR (qRT-PCR)
RNA was isolated from P21 rib bones using Trizol (Invitrogen) and from primary osteoblasts using Picopure (Arcturus, Sunnyvale, CA, USA). cDNA was synthesized using the SuperScript Preamplification System (Invitrogen). PCR reactions, data quantification, and analysis were performed according to the manufacturer's standard protocol of TaqMan gene expression assays: Axin2: Mm01265783_m1, Bmpr1a: Mm00477650_m1, Ctgf: Mm01192931_g1, Dkk1: Mm00438422_m1, Lef1: Mm00550265_m1, Lrp5: Mm01227476_m1, and Sost: Mm03024247_g1 (Applied Biosystems, Rotkreuz, Switzerland). Values were normalized to Gapdh using TaqMan Rodent GAPDH Control Reagents (Applied Biosystems). All measurements were performed in triplicate and analyzed using the 2−ΔΔC(t method.(30)
Primary osteoblast and calvaria culture
Newborn and P10 calvariae were digested with type I collagenase (Sigma) and dispase II (Roche, Indianapolis, IN, USA) to isolate osteoblasts, as described previously.(14) Primary osteoblasts were maintained in α-MEM containing 10% fetal bovine serum (FBS) and ascorbic acid (50 µg/mL, Sigma). Primary osteoblasts from wild-type mice were treated with BMP2 for 3 hours at varied concentrations (10, 50, and 100 ng/mL, R&D, Minneapolis, MN, USA). Wild-type osteoblasts also were pretreated with dorsomorphin (10 µM), p38 mitogen-activated protein kinase (MAPK) inhibitor SB202190 (10 µM, Calbiochem, Gibbstown, NJ, USA), and DMSO in the absence of serum for 1 hour before BMP2 treatment (100 ng/mL). For primary osteoblasts from Bmpr1a cKO mice or caBmpr1a mutant mice, 4-hydroxyl tamoxifen (4OH TM, 100 ng/mL, Sigma) was added in culture every other day. For ex vivo bone culture, newborn calvariae from wild-type mice were dissected at the sagittal suture and cultured in modified BGJ (Invitrogen) supplemented with 5% FBS and ascorbic acid (50 µg/mL) for the first 24 hours in culture. Hemicalvariae were treated with 4OH TM (100 ng/mL) and Noggin (100 ng/mL, R&D) in the absence of serum for 5 days.
Dual luciferase reporter assays
Primary osteoblasts from Bmpr1a cKO newborn mice and their littermate controls were plated onto six-well plates at a density of 2 × 105 cells/well containing 10% FBS in α-MEM and grown to 50% to 60% confluence. Cells were transfected with plasmid mixtures containing 2 µg TOPFLASH luciferase construct and 0.05 µg Renilla luciferase driven by the Drosophila actin 5C promoter(31) (kindly provided by Dr. Paul A. Wade) using FuGENE 6 Transfection Reagent (Roche) according to the manufacturer's protocol. After 48 hours of transfection, the cells were lysed, and luciferase activity was measured. The values of TOPFLASH luciferase activity were normalized to those of Renilla activity using a dual luciferase assay kit (Promega, Madison, WI, USA). For dorsomorphin treatment, primary osteoblasts from wild-type mice were transfected as described, cultured for 40 hours, treated with DMSO or dorsomorphin (10 µM) and further incubated for 8 hours. The dual luciferase assay was performed in the same manner as described previously. No obvious toxicity was detected in the experiments based on Renilla levels.
Statistical analysis
All statistical analyses were performed using a two-tailed Student's t test.
Results
Tissue specificity and efficiency of Cre recombinase in Col1-CreER™ mice
TM-inducible Col1-CreERTM mice were mated to ROSA26 reporter mice (R26R) to assess Cre activity by staining for β-galactosidase (β-gal) at P21. TM was injected intraperitoneally into nursing females every 3 days from P2 to P20. Cre transgenic mice administered TM (Col1-CreERTM+, ROSA+, TM+) stained for β-gal in rib bones (Fig. 1A, a), where many osteoblasts and osteocytes showed evidence of recombination (see Fig. 1A, b). No staining was observed in cartilage (see Fig. 1A, a). At this stage, no recombination was detected in chondrocytes and osteoclasts, as described previously.(15) These results demonstrate that the TM administration delivered through milk specifically and efficiently induces Cre recombination in osteoblasts and osteocytes in weanlings.
Fig. 1.
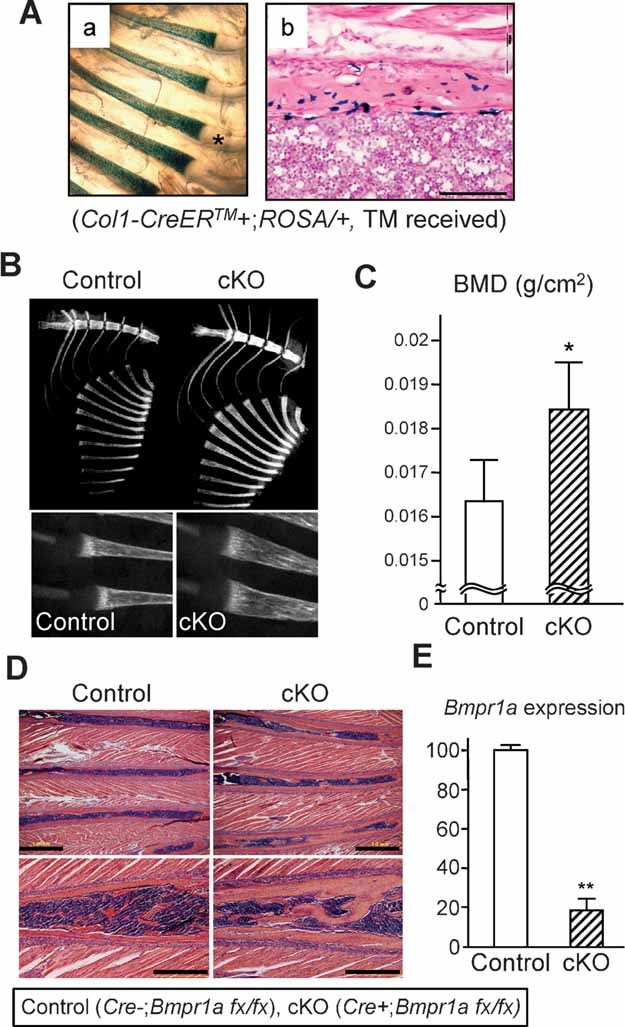
Increased bone mass in Bmpr1a cKO weanling mice. Tamoxifen was injected intraperitoneally into nursing females every 3 days from P2 to P20. (A) ROSA26 Cre reporter mice showed Cre activity on rib bones stained with β-galactosidase (β-gal) at P21 (a). Cre-dependent DNA recombination was detected in osteoblasts in rib bones by β-gal staining (b). The asterisk indicates negative staining for β-gal on rib cartilage. Bar: 100 µm. (B) Radiodensity of rib bones and sternum was increased in cKO mice (Cre+, Bmpr1afx/fx) compared with controls (Cre–, Bmpr1afx/fx) when assessed by X-ray imaging at P21. Rib flaring in cKO mice is shown (right panel). (C) BMD was determined in ribs using DXA at P21. Values are expressed as mean ± SD (control: n = 10; cKO: n = 12). The asterisk indicates a statistically significant difference between control and cKO (mean ± SD; t test; *p < .04). (D) H&E staining of P21 rib bones. Rib bones were thicker in cKO mice, and bone mass was increased compared with littermate controls in both the diaphysis (upper panels) and metaphysis (lower panels). Bars: 1 mm (upper panels), 500 µm (lower panels). (E) qRT-PCR analysis for Bmpr1a using P21 rib bones (mean ± SD; t test; **p < .02).
Increased bone mass in BMPRIA cKO rib bones
Col1-CreERTM mice were bred with mice homozygous for floxed Bmpr1a to generate Bmpr1a cKO mice (cKO, Cre+:Bmpr1afx/fx) and wild-type controls (Cre–:Bmpr1a fx/fx) after maternal TM administration. X-ray analysis demonstrated a modest increase in radiodensity in the rib bones and sternum (see Fig. 1B, upper panel) and rib flaring (see Fig. 1B, lower panels) in cKO mice at P21. BMD measured by DXA was significantly increased in rib bones at P21 (see Fig. 1C). Consistent with X-ray imaging and DXA results, H&E staining showed that rib bones in the diaphysis and metaphysis of cKO mice were thicker and had higher bone mass compared with control mice at P21 (see Fig. 1D). Bmpr1a expression was 80% reduced in cKO rib bones at P21, as assessed by qRT-PCR (see Fig. 1E). Bone mass in other bones (i.e., femur, tibia, humerus, tail, and calvaria) also was increased by histology, as previously described,(15) but BMD was unchanged in other bones at this stage (data not shown). Thus we focused on rib bones to investigate the mechanism by which BMP signaling regulates bone mass.
Upregulation of canonical Wnt signaling in cKO rib bones
Whereas BMP and Wnt signaling are known to independently contribute to bone mass regulation,(17–20) the interaction between these two pathways is incompletely defined and has not been demonstrated previously in vivo. We recently reported that loss of BMPRIA in osteoblasts during embryogenesis upregulates canonical Wnt signaling in mice.(14) To assay canonical Wnt signaling in situ, cKO mice were mated with TOPGAL Wnt reporter mice, which express a β-galactosidase transgene driven by a T-cell factor (TCF) β-catenin-responsive promoter.(29) cKO TOPGAL mice demonstrated increased Wnt signaling activity in cKO rib bones at P14 compared with controls when assessed by whole mount (Fig. 2A, upper panels) and sections (see Fig. 2A, lower panels) via β-gal staining, as were calvariae at P10 and P14 (see Fig. 2B; data not shown). Consistent with P21 cKO calvariae,(15) P10 cKO calvariae also were thicker than controls (see Fig. 2B, lower panels). Upregulation of canonical Wnt signaling in cKO rib bones and calvariae thus was found in conjunction with increased bone mass.
Fig. 2.
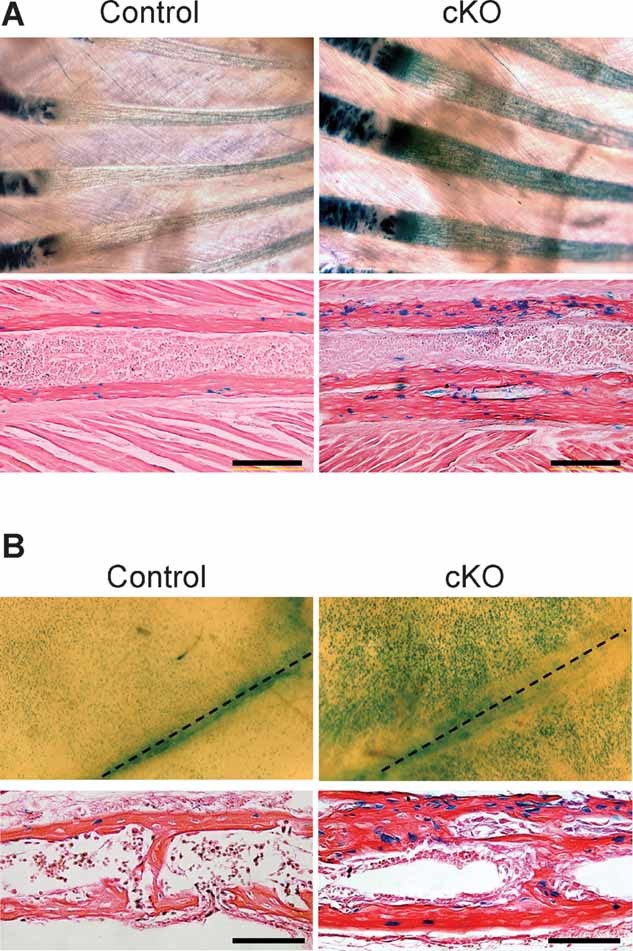
Upregulation of canonical Wnt signaling in Bmpr1a cKO mice. (A) Canonical Wnt signaling in P14 rib bones assessed using TOPGAL mice. Whole-mount β-gal staining showed increased staining in cKO rib bones compared with controls (upper panels). Histologic analysis showed an increased number of β-gal-positive osteoblasts in cKO rib bones compared with controls (lower panels). Bar: 200 µm. (B) Canonical Wnt signaling in P10 skull bones assessed using TOPGAL mice. Whole-mount β-gal staining showed increased staining in cKO calvariae compared with controls (upper panels). Histologic analysis showed an increased number of β-gal-positive osteoblasts in cKO calvariae compared with controls (lower panels). Dotted lines indicate the sagittal suture. Bar: 100 µm.
Consistent with findings in cKO rib bones, canonical Wnt signaling activity was enhanced in primary osteoblasts isolated from P10 calvariae from cKO mice compared with control osteoblasts (see Fig. 2B). The frequency of β-gal-positive cells was increased significantly in cKO primary osteoblasts compared with control osteoblasts (control: 2.8%, cKO: 20.8%; Fig. 3A). When Lef/Tcf-dependent transcriptional activity was assayed using the TOPFLASH reporter system, canonical Wnt signaling was significantly increased in cKO osteoblasts (see Fig. 3B). Corroborating the effects of targeted disruption of BMPRIA in osteoblasts, Wnt signaling activity also was increased significantly (see Fig. 3C) after treating wild-type primary osteoblasts with dorsomorphin, a selective BMP type I receptor inhibitor that effectively blocks BMPRIA-mediated Smad activation under these conditions.(32) Taken together, these findings in primary osteoblasts, calavarial explants, and in situ in transgenic reporter mice confirm that osteoblast BMPRIA signaling negatively regulates canonical Wnt signaling during early postnatal development and that its disruption is associated with increased bone mass.
Fig. 3.
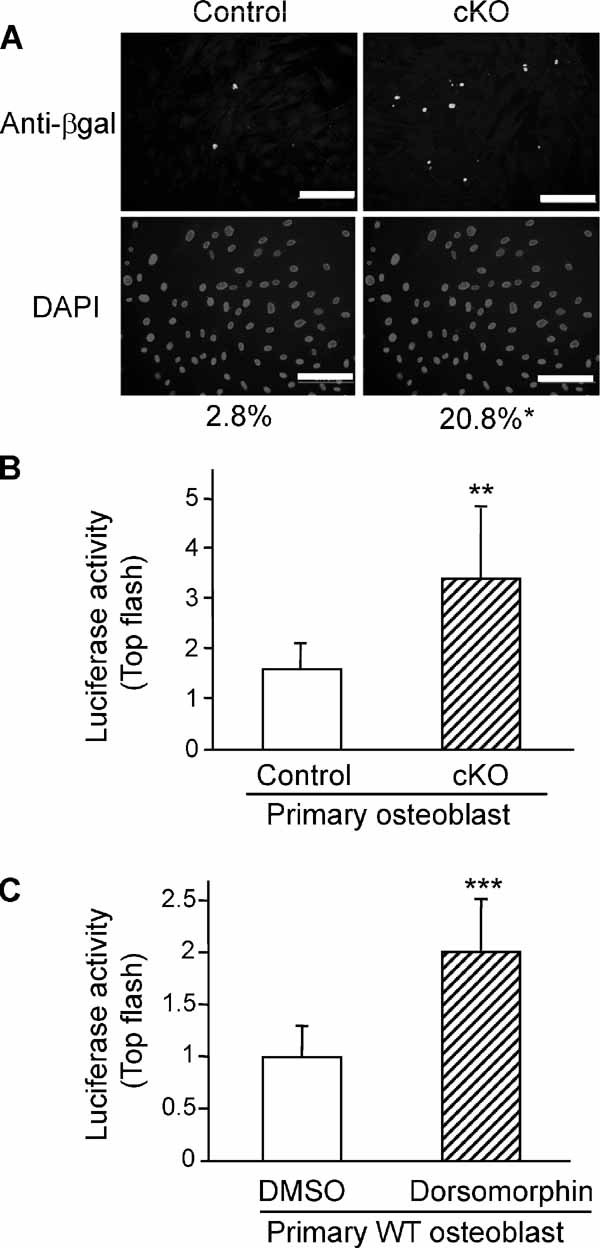
Upregulation of canonical Wnt signaling by loss of BMP signaling in vitro. (A) Detection of canonical Wnt signaling in primary osteoblasts from P10 Bmpr1a cKO and control calvariae (upper panels) using TOPGAL mice assessed by immunocytochemical staining for β-gal (green). Nuclei were stained with DAPI (blue, lower panels). Relative ratio of β-gal-positive cell number to DAPI-positive cell number was determined in the same field (mean from 50 fields; t test; *p < .01; n > 4 per each genotype). Bars: 200 µm. (B) TOPFLASH plasmid was cotransfected with pRL-actin vector into primary osteoblasts from newborn Bmpr1a cKO and wild-type control calvariae using FuGENE 6 for 48 hours. Transfection efficiency for luciferase activity was normalized to Renilla luciferase (pRL-actin vector) activity. The y axis shows relative luciferase units (RLUs, luciferase activity/Renilla units). Promoter activities for Lef/Tcf-dependent transcription were increased significantly in cKO osteoblasts (mean ± SD; t test; **p < .02). (C) Effects of BMP signaling inhibitor dorsomorphin on canonical Wnt signaling in vitro. TOPFLASH plasmid was cotransfected with pRL-actin vector into primary osteoblasts from newborn wild-type calvariae using FuGENE 6. After 40 hours of transfection, these cells were treated with dorsomorphin (10 µM) or DMSO without serum for 8 hours, and then cells were harvested. The Lef/Tcf-dependent transcriptional activities were increased significantly by the treatment with dorsomorphin, as measured by dual luciferase activity (mean ± SD; t test; ***p < .05).
Downregulation of Dkk1 and Sost in cKO bones
To confirm the findings of enhanced canonical Wnt signaling in cKO tissues and primary cultures, expression of Wnt-related target genes was examined by qRT-PCR using rib bones and primary osteoblasts isolated from calvariae. In P21 cKO rib bones, expression levels of Wnt target genes Axin2, Ctgf, and Lef1 were increased significantly (Fig. 4A). It has been reported that osteoblasts secrete several proteins into bone extracellular matrix (ECM) that inhibit Wnt signaling and that these ECM molecules can regulate bone mass along with coreceptors.(33–38) We reported that sclerostin/Sost, a Wnt inhibitor, plays an important role in regulating bone mass as a downstream target molecule of BMP signaling during embryonic bone development.(14) In P21 cKO rib bones, expression levels of Sost and Dkk1 mRNAs were reduced significantly, whereas levels of Wnt ligands (Wnt3a, Wnt5a, Wnt7a, Wnt7b, and Wnt9a), other Wnt inhibitors including Dkk2, secreted frizzled-related proteins (sFrp), and coreceptor Lrp5 were unchanged (see Fig. 4B; data not shown). In cKO primary osteoblasts, where Bmpr1a expression was reduced significantly, Sost and Dkk1 mRNA expression levels also were reduced compared with controls (see Fig. 4C), whereas Wnt target genes were increased (data not shown). Consistent with a positive regulatory effect of BMP signaling on Wnt antagonists, treatment of wild-type calvariae with Noggin, a BMP2 and BMP4 antagonist, inhibited both Sost and Dkk1 expression ex vivo (see Fig. 4D), upregulated Wnt signaling,(14) and increased the expression of Wnt target genes (data not shown). These results obtained from two different types of bone (i.e., rib bone and calvaria) and primary cultures suggest that loss of BMPRIA signaling enhances canonical Wnt signaling during postnatal bone development in osteoblasts via decreased expression of Dkk1 and Sost.
Fig. 4.
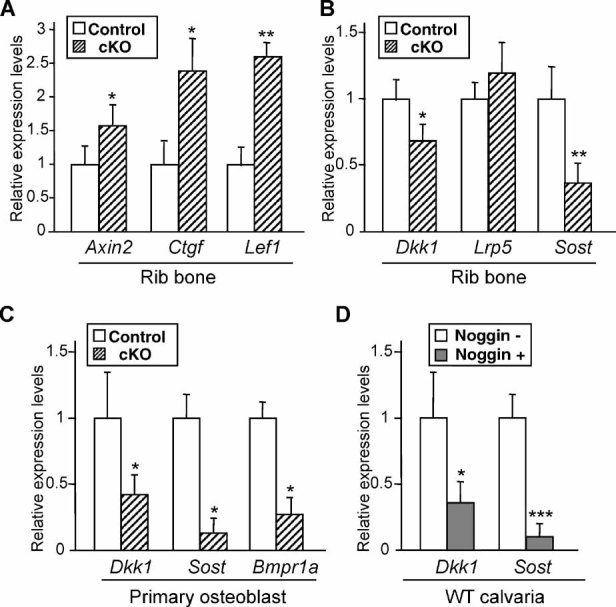
Downregulation of Dkk1 and Sost in Bmpr1a cKO rib bones. (A) qRT-PCR analysis for Wnt target genes Axin2, Ctgf, and Lef1 using P21 rib bones. Expression levels of Axin2, Ctgf, and Lef1 were increased significantly in cKO rib bones. Values in cKO bones (striped bar) are expressed relative to controls (open bar). (B) qRT-PCR analysis for Dkk1, Lrp5, and Sost using P21 rib bones. Expression levels of Wnt inhibitors (Dkk1 and Sost) were reduced significantly in cKO rib bones (striped bar), whereas expression of coreceptor Lrp5 was unchanged. (C) qRT-PCR analysis for Dkk1, Sost, and Bmpr1a using primary osteoblasts from P10 Bmpr1a cKO and control calvariae. Expression levels of Dkk1, Sost, and Bmpr1a were reduced significantly in cKO osteoblasts (striped bar) compared with control osteoblasts (open bar). (D) Suppressed expression of Dkk1 and Sost by Noggin treatment ex vivo, as assessed by qRT-PCR. Newborn calvariae from wild-type mice were treated with Noggin (100 ng/mL) for 5 days. The asterisk indicates a statistically significant difference between Noggin treatment (Noggin+) and nontreatment (Noggin–) from three independent experiments. (A–D) Mean ± SD; t test; *p < .05; **p < .02; ***p < .01.
Positive regulation of Dkk1 and Sost by BMP2
We next investigated a potential link between expression of Wnt inhibitors Sost and Dkk1 and BMP signaling using wild-type primary osteoblasts. In primary osteoblasts treated with canonical BMPRIA ligand BMP2,(5) levels of Dkk1 and Sost increased up to 8- and 20-fold, respectively, after 3 hours, as assessed by qRT-PCR (Fig. 5A). Peak expression levels of Dkk1 and Sost were detected at 3 hours after BMP2 treatment (100 ng/mL) (see Fig. 5B). Upregulation of Dkk1 and Sost expression by BMP2 treatment (100 ng/mL) was blocked by pretreatment with dorsomorphin (see Fig. 5C), which selectively inhibits Smad activation without inhibiting BMP-induced activation of MAPK p38.(32) These results suggest that Dkk1 and Sost expression are regulated by BMP signaling, at least in part via Smad-dependent signaling.
Fig. 5.

Positive regulation of Dkk1 and Sost expression by BMP2. Primary osteoblasts were isolated from wild-type newborn calvariae. (A) Dose-dependent effects of BMP2 on Dkk1 and Sost expression as assessed by qRT-PCR. mRNA was isolated from wild-type osteoblasts treated with BMP2 at the indicated concentration for 3 hours. Values are expressed relative to nontreated osteoblasts (mean ± SD; t test; *p < .05; **p < .01). (B) Time course of Dkk1 and Sost expression levels using wild-type osteoblasts treated with BMP2 (100 ng/mL). (C) Effects of pretreatments of primary osteoblasts with Smad-dependent inhibitor dorsomorphin. mRNA was isolated from wild-type osteoblasts pretreated with dorsomorphin (10 µM) or DMSO in the absence of serum for 1 hour prior to the addition of BMP2 (100 ng/mL) or PBS for 3 hours. Upregulation of Dkk1 and Sost expression by BMP2 treatment was restored by dorsomorphin pretreatment, as assessed by qRT-PCR. Values are expressed relative to osteoblasts without dorsomorphin and BMP2 (mean ± SD; t test; *p < .05).
Upregulation of Dkk1 and Sost by Smad-dependent BMPRIA signaling
On ligand binding, BMPRIA phosphorylates BMP-responsive Smads (Smad1, Smad5, and Smad8) to induce nuclear translocation.(39) The introduction of certain mutations in the GS regulatory domain of BMPRIA is thought to render the receptor protein active independent of ligand, resulting in “constitutively active” Bmpr1a (caBmpr1a).(40) To confirm whether our observations on the effects of Smad-dependent BMPRIA signaling on Dkk1 and Sost expression were recapitulated in vivo, we generated inducible transgenic mice that conditionally express caBmpr1a, as we described previously.(14) These mice were crossed with Col1-CreERTM mice, primary osteoblasts were obtained from newborn calvariae, and then Smad-dependent BMPRIA signaling was induced by administration of 4OH TM into the culture medium. Both Dkk1 expression and Sost expression assessed by qRT-PCR were significantly increased, 5- and 10-fold, respectively, in caBmpr1a osteoblasts (Cre+, caBmpr1a+) compared with controls (Cre–, caBmpr1a+) (Fig. 6A). We next generated compound transgenic-knockout mice expressing caBmpr1a on a Bmpr1a cKO background (rescued: Cre+, caBmpr1a+, Bmpr1afx/fx) and compared them to littermate Bmpr1a cKO mice (cKO: Cre+, caBmpr1a–, Bmpr1afx/fx) or controls (Cre–, caBmpr1a+, Bmpr1afx/fx). X-ray imaging demonstrated a moderate reduction in radiodensity of the rib bones and sternum obtained from compound transgenic-knockout mice compared with Bmpr1a cKO mice at P21 (see Fig. 6B). In addition, the observation of increased BMD in cKO ribs and sternum was abrogated by enhanced Smad-dependent BMPRIA signaling at P21 (see Fig. 6C). These results using inducible compound caBmpr1a transgenic and Bmpr1a cKO mice demonstrate that both Dkk1 expression and Sost expression are positively regulated by BMPRIA-mediated Smad signaling in vivo, with effects on bone mineral density that are most concordant with effects on Wnt pathway regulation.
Fig. 6.
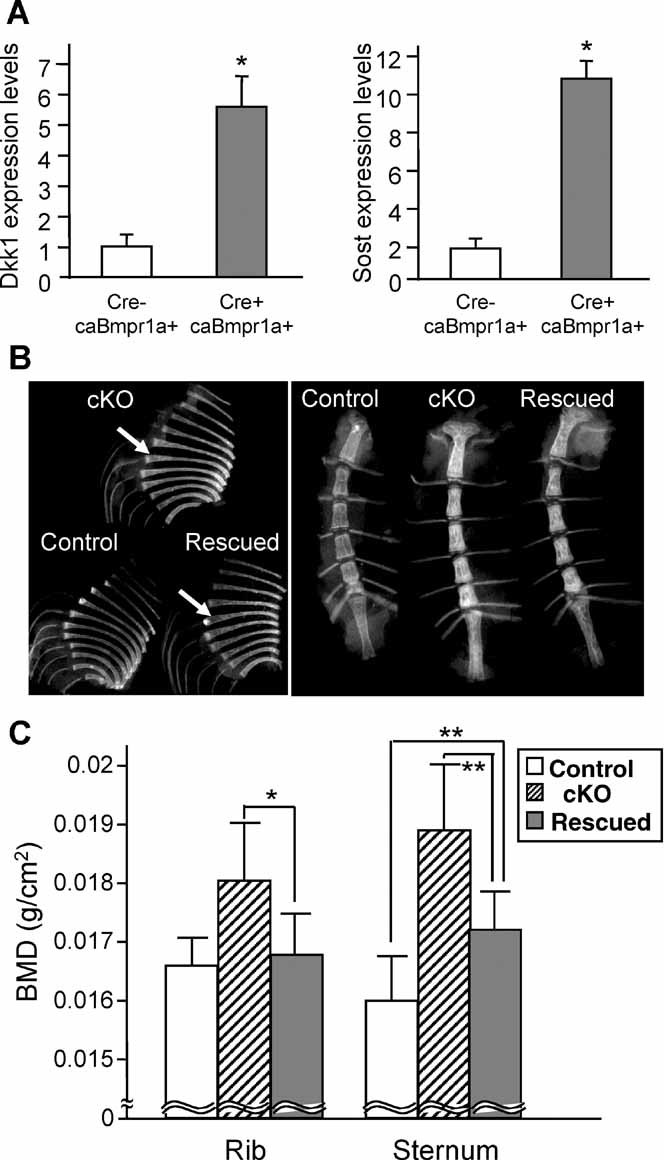
Effects of Smad-dependent Bmpr1a signaling on Dkk1 and Sost expression. (A) Positive regulation of Dkk1 and Sost expression by overexpressing Smad-dependent Bmpr1a signaling. Primary osteoblasts were isolated from constitutively active Bmpr1a (caBmpr1a) newborn mice or littermate controls (Cre–, caBmpr1a+) and were treated with 4-hydroxyl tamoxifen (100 ng/mL). Both Dkk1 expression and Sost expression were increased significantly more than 5- and 10-fold, respectively, in the caBmpr1a mutant osteoblasts (Cre+, caBmpr1a+) compared with controls (Cre–, caBmpr1a+), as assessed by qRT-PCR. Values of the mutant osteoblasts (striped bar) are expressed relative to controls (open bar) (mean ± SD; t test; *p < 0.01). (B) Radiodensity of rib bones and sternum from rescued mice expressing caBmpr1a on a Bmpr1a cKO background (Cre+, caBmpr1a+, Bmpr1afx/fx), littermate Bmpr1a cKO mice (Cre+, caBmpr1a–, Bmpr1afx/fx) and littermate controls (Cre–, caBmpr1a+, or Bmpr1afx/fx) at P21 when assessed by X-ray imaging. White arrows indicate rib flaring observed in cKO and rescued mice. (C) BMD as determined by DXA using ribs from control (open bar), cKO (striped bar), and rescued mice (gray bar) at P21. BMD of rescued sternums was reduced significantly by 10% compared with cKO sternum and increased by 6% compared with controls. Values are expressed as mean ± SD (n > 6; t test, *p = .07, **p < .05).
Effects of activation of a non-Smad pathway on Dkk1 and Sost expression
Rib flaring was still observed in the compound transgenic knockout mice (see Fig. 6B), and the BMD of the rescued sternum was significantly higher than that of control sternum (see Fig. 6C). The partial rescue of the cKO phenotype by the constitutively active transgene led us to postulate that loss of BMP signaling through Smad-independent pathways in addition to the Smad-dependent pathway may contribute to the cKO bone phenotype. BMP signaling can activate intracellular effectors, in particular p38 MAPK, via Smad-dependent pathways both in vitro and in vivo.(41–45) To investigate whether regulation of Dkk1 and Sost expression by BMPRIA signaling requires activation of p38 MAPK, we pretreated primary wild-type osteoblasts with a p38 MAPK inhibitor SB202190 (10 µM) before BMP2 treatment (100 ng/mL). Upregulation of Dkk1 expression by BMP2 treatment was blocked by SB202190 pretreatment, as assessed by qRT-PCR (Fig. 7A). In contrast, Sost expression was increased significantly by BMP2 treatment even after SB202190 pretreatment (see Fig. 7A). These results suggest that Dkk1 expression is regulated by both Smad-mediated and p38 MAPK signaling, whereas Sost expression is regulated mainly by Smad signaling (see Fig. 7B).
Fig. 7.
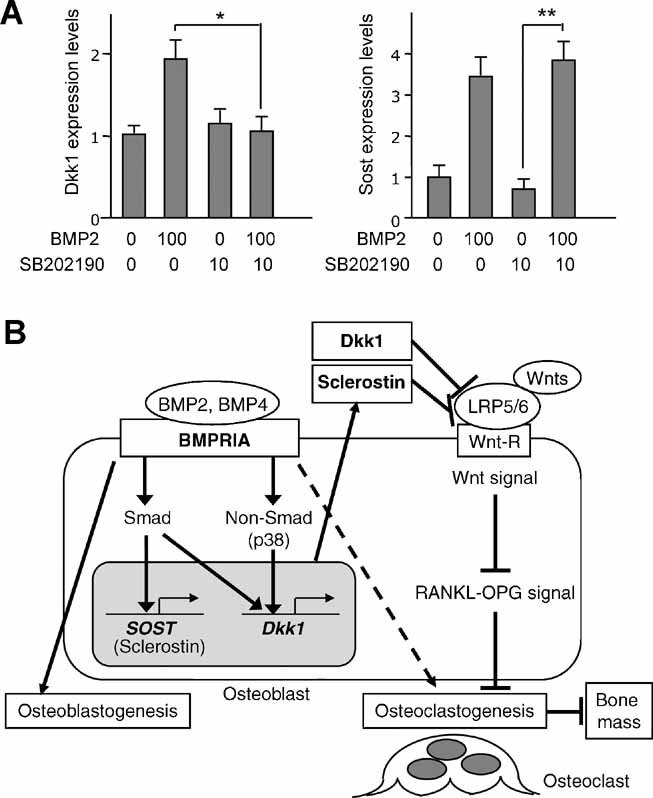
Effects of non-Smad signaling on Dkk1 and Sost expression in osteoblasts. (A) Effects of p38 MAPK inhibitor SB202190 on Dkk1 and Sost expression using primary osteoblasts from newborn wild-type mice as assessed by qRT-PCR. mRNA was isolated from wild-type osteoblasts pretreated with SB202190 (10 µM) or DMSO in the absence of serum for 1 hour prior to the addition of BMP2 (100 ng/mL) or PBS for 3 hours. Upregulation of Dkk1 expression by BMP2 treatment was restored by SB202190 pretreatment. Sost expression was increased significantly by BMP2 treatment with SB202190 pretreatment. Values are expressed relative to untreated osteoblasts (neither SB202190 nor BMP2) (mean ± SD; t test; *p < .05; **p < .01). (B) A proposed model of the relationship between BMPRIA and canonical Wnt signaling through Dkk1 and sclerostin/Sost in bone. BMPRIA signaling upregulates Sost expression primarily through Smad-dependent signaling while upregulating Dkk1 expression through both Smad and non-Smad signaling (p38 MAPK). Both Dkk1 and sclerostin/Sost inhibit canonical Wnt signaling, leading to a decrease in bone mass. Dkk1 and sclerostin/Sost play an important role in regulating bone mass as downstream effectors of BMPRIA signaling.
Discussion
We previously reported that loss of BMPRIA-mediated signaling resulted in increased bone mass in embryos and adults(14,15) and discovered that sclerostin/Sost is a downstream target of BMPRIA signaling during embryogenesis that inhibits Wnt to regulate bone mass.(14) In this study we found that Dkk1, another Wnt inhibitor, is also a downstream target of BMPRIA signaling at the weanling stage. Based on our findings, we propose a mechanism by which Dkk1 is regulated by signaling through both Smad-dependent and Smad-independent pathways in osteoblasts during postnatal development, whereas Sost is regulated predominantly by the Smad pathway (see Fig. 7).
There is a growing body of evidence to suggest that the BMP and Wnt signaling pathways regulate one another synergistically or antagonistically in a context and age-dependent manner.(46–49) The interplay of Wnt/β-catenin and BMP signaling pathways has not been demonstrated previously during bone development in vivo. However, we recently reported that BMP signaling in osteoblasts downregulates canonical Wnt signaling during embryonic bone development.(14) Consistent with the effects of BMP signaling during embryogenesis, we presently found evidence for negative regulation of canonical Wnt signaling by BMPRIA signaling in osteoblasts postnatally (see Fig. 2). Our findings that embryonic and postnatal BMPRIA signaling negatively regulate canonical Wnt signaling in osteoblasts agree with other studies that have shown an inhibitory effect of BMP signaling on Wnt signaling in lungs,(50) intestines,(49) hair,(51) and joints.(48) Although many mouse models and human mutations modifying the Wnt signaling pathway have been described, no studies to date have addressed the in vivo interaction of BMP and Wnt signaling in osteoblasts.
Recent studies using pluripotent mesenchymal cell lines have suggested that BMP signaling may upregulate Wnt signaling to synergistically regulate osteoblastogenesis,(23,52) possibly through an autocrine or paracrine loop.(21) This is in contrast with our mouse studies showing that BMP signaling downregulates Wnt signaling. The discrepancy may be due to the use of mesenchymal cell lines rather than primary osteoblasts or the supraphysiologic levels of in vitro BMP treatment or reflect the coordinated action of multiple cell types including osteoclasts in a physiologic setting compared with cell monoculture. The potential importance of the physiologic context of these pathways may be reflected in the finding that canonical Wnt signaling was markedly upregulated in cKO mice (see Fig. 2B, C) but only modestly increased in isolated Bmpr1a-deficient osteoblasts (see Fig. 3A, B).
In mice, both Sost and Dkk1 expression levels were downregulated by disrupting Bmpr1a (see Fig. 4) and upregulated by enhancing BMP signaling via caBmpr1a (see Fig. 6). These results, which were replicated in cell culture (see Fig. 5A), suggest that both Dkk1 and sclerostin/Sost are downstream targets of BMPRIA. Previous reports support this notion because BMPRIA ligands BMP2 and BMP4 induce Dkk1 expression during limb development in mice and chickens,(53,54) as well as Sost expression in mouse and human osteoblasts.(55,56) In parallel, inhibition of BMP signaling with dorsomorphin suppressed the expression of Dkk1 and Sost (see Fig. 5B), resulting in upregulated canonical Wnt signaling (see Fig. 3C). Noggin also inhibited Dkk1 and Sost expression (see Fig. 4D), consistent with previous reports that Noggin inhibited Sost expression with upregulation of Wnt signaling in mice(14) and suppressed Dkk1 expression in chickens.(54) Although Dkk1 has been proposed to be a target of BMP signaling in mouse limb development,(53) this study provides the first evidence to our knowledge that BMPRIA regulates Dkk1 expression in osteoblasts. The finding that both Smad-dependent and Smad-independent pathways appear to contribute to Dkk1 expression, whereas Sost requires only Smad-dependent signaling suggests differential regulation of these genes by BMPRIA.
Both Dkk1 and sclerostin/Sost are expressed by osteoblasts as secreted proteins that inhibit Wnt/β-catenin signaling by binding to coreceptors low-density lipoprotein receptor–related protein 5 and 6 (LRP5 and LRP6).(36,37,57) Conventional knockouts of Dkk1 die in utero from defective head induction and limb formation.(53) Mice heterozygous for Dkk1 (Dkk1+/− mice) exhibit a high-bone-mass (HBM) phenotype,(58) whereas overexpression of Dkk1 in osteoblasts causes osteopenia.(59) In addition, increased DKK1 expression in bone marrow also has been associated with lytic bone lesions in patients with multiple myeloma.(60) Collectively, these results support the hypothesis that Dkk1 functions as a potent negative regulator of bone mass. Sost KO mice are viable and exhibit increased bone mass,(61) similar to Dkk1+/− mice. In humans, loss-of-function and hypomorphic mutations in SOST cause sclerosteosis(62,63) and Van Buchem disease,(64,65) respectively, with an HBM phenotype. Consistent with these observations, Bmpr1a cKO mice, which are deficient in Dkk1 and Sost expression, show an HBM phenotype. Furthermore, increased expression of Dkk1 and Sost in osteoblasts by constitutively activated BMPRIA signaling is associated with partial rescue of the Bmpr1a cKO bone phenotype. Our present data support the interpretation that Dkk1 and sclerostin/Sost act physiologically as inhibitors of canonical Wnt signaling and therefore as negative regulators of bone mass in mice.
Both Smad-dependent and Smad-independent pathways are important for BMP signaling. p38 MAPK plays a central role in Smad-independent pathways of TGF-β/BMP signaling(41–45) by accelerating osteoblastogenesis,(66,67) which can be repressed by p38 MAPK inhibitors.(68,69) This study demonstrated that the p38 MAPK inhibitor SB202190 prevented the upregulation of Dkk1 by BMP2 but did not alter Sost expression in osteoblasts (see Fig. 7A). This is consistent with a previous report showing that upregulation of Dkk by BMP5 was severely impaired after cotreatment with p38 MAPK inhibitors in chick limb buds.(42) The p38 MAPK pathway thus appears to contribute at least part of the regulation of Wnt signaling by BMPRIA. Given the demonstrated importance of p38 MAPK signaling in osteoblastogenesis, it is highly plausible that additional Smad-independent mechanisms may link BMP signaling and Wnt signaling in this lineage.
The Food and Drug Administration (FDA) has approved BMP2 and BMP7 for clinical use in long bone open fractures, nonunion fractures, and spinal fusion. However, despite significant evidence of their potential for bone regeneration in animal and preclinical studies, the current clinical data supporting the effectiveness of BMP therapy is somewhat unconvincing.(70,71) The lack of a clear benefit from BMPs might stem from an incomplete understanding of the variable and context-sensitive effects BMPs exert on diverse cell types in bone, including chondrocytes, osteoblasts, and osteoclasts. Studies focusing on BMP receptors in chondrocytes including mesenchymal cells suggest that these cells respond to BMP signaling by increasing bone mass during the endochondral formation process.(72–75) In contrast, when we examined function of BMPRIA in osteoblasts with respect to bone mass determination, BMP signals consistently inhibited canonical Wnt signaling and bone mass while exerting concordant effects on Dkk1 and Sost. Our current evidence adds an important nuance to the conventional notion of BMPs as inducers of osteogenesis and positive regulators of bone mass owing to their inverse effects on Wnt signaling in osteoblasts. The functions of BMPRIA signaling in osteoclasts remain largely unknown and merit future study, although we previously observed a significant decrease in osteoblast-dependent osteoclastogenesis in cKO mice.(14,15) This revision of our understanding of the BMP signaling pathway in clinical therapeutics might suggest that in some circumstances BMP inhibition would be desirable for promoting bone mass. Understanding the complex roles of the BMP signaling pathway in chondrocytes, osteoblasts, osteoclasts, and other cell types that contribute to bone development, homeostasis, and remodeling not only will help to improve our knowledge of the dynamic processes that are perturbed in settings of bone fracture, ovariectomy, orchiectomy, mechanical loading, and aging but also may provide novel therapeutically useful strategies.
Acknowledgments
We gratefully acknowledge Tomokazu Fukuda and Greg Scott for generation of the caBmpr1a mouse line and Laurie K McCauley and Donald J Lucas for critical reading of this manuscript. This work was supported by National Institutes of Health (NIH) Grants P01 DK56246 (HK) and the Intramural Research Program of the NIEHS/NIH ES071003-11 (Y. Mi). The Lilly Fellowship Foundation supported NK.
Disclosures
The authors state that they have no conflicts of interest.
References
- 1.Massague J. Receptors for the TGF-β family. Cell. 1992;69:1067–1070. doi: 10.1016/0092-8674(92)90627-o. [DOI] [PubMed] [Google Scholar]
- 2.Wrana JL, Attisano L, Wieser R, Ventura F, Massague J. Mechanism of activation of the TGF-β receptor. Nature. 1994;370:341–347. doi: 10.1038/370341a0. [DOI] [PubMed] [Google Scholar]
- 3.Urist MR. Bone: formation by autoinduction. Science. 1965;150:893–899. doi: 10.1126/science.150.3698.893. [DOI] [PubMed] [Google Scholar]
- 4.Shi Y, Massague J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 5.Keller S, Nickel J, Zhang JL, Sebald W, Mueller TD. Molecular recognition of BMP-2 and BMP receptor IA. Nat Struct Mol Biol. 2004;11:481–488. doi: 10.1038/nsmb756. [DOI] [PubMed] [Google Scholar]
- 6.Hatta T, Konishi H, Katoh E, et al. Identification of the ligand-binding site of the BMP type IA receptor for BMP-4. Biopolymers. 2000;55:399–406. doi: 10.1002/1097-0282(2000)55:5<399::AID-BIP1014>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 7.Wozney JM. The bone morphogenetic protein family and osteogenesis. Mol Reprod Dev. 1992;32:160–167. doi: 10.1002/mrd.1080320212. [DOI] [PubMed] [Google Scholar]
- 8.Chen D, Zhao M, Mundy GR. Bone morphogenetic proteins. Growth Factors. 2004;22:233–241. doi: 10.1080/08977190412331279890. [DOI] [PubMed] [Google Scholar]
- 9.Shore EM, Xu M, Feldman GJ, et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet. 2006;38:525–527. doi: 10.1038/ng1783. [DOI] [PubMed] [Google Scholar]
- 10.Yu PB, Deng DY, Lai CS, et al. BMP type I receptor inhibition reduces heterotopic [corrected] ossification. Nat Med. 2008;14:1363–1369. doi: 10.1038/nm.1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang H, Bradley A. Mice deficient for BMP2 are nonviable and have defects in amnion/chorion and cardiac development. Development. 1996;122:2977–2986. doi: 10.1242/dev.122.10.2977. [DOI] [PubMed] [Google Scholar]
- 12.Winnier G, Blessing M, Labosky PA, Hogan BL. Bone morphogenetic protein-4 is required for mesoderm formation and patterning in the mouse. Genes Dev. 1995;9:2105–2116. doi: 10.1101/gad.9.17.2105. [DOI] [PubMed] [Google Scholar]
- 13.Mishina Y, Suzuki A, Ueno N, Behringer RR. Bmpr encodes a type I bone morphogenetic protein receptor that is essential for gastrulation during mouse embryogenesis. Genes Dev. 1995;9:3027–3037. doi: 10.1101/gad.9.24.3027. [DOI] [PubMed] [Google Scholar]
- 14.Kamiya N, Ye L, Kobayashi T, et al. BMP signaling negatively regulates bone mass through sclerostin by inhibiting the canonical Wnt pathway. Development. 2008;135:3801–3811. doi: 10.1242/dev.025825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kamiya N, Ye L, Kobayashi T, et al. Disruption of BMP signaling in osteoblasts through type IA receptor (BMPRIA) increases bone mass. J Bone Miner Res. 2008;23:2007–2017. doi: 10.1359/JBMR.080809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harada S, Rodan GA. Control of osteoblast function and regulation of bone mass. Nature. 2003;423:349–355. doi: 10.1038/nature01660. [DOI] [PubMed] [Google Scholar]
- 17.Baron R, Rawadi G, Roman-Roman S. Wnt signaling: a key regulator of bone mass. Curr Top Dev Biol. 2006;76:103–127. doi: 10.1016/S0070-2153(06)76004-5. [DOI] [PubMed] [Google Scholar]
- 18.Krishnan V, Bryant HU, Macdougald OA. Regulation of bone mass by Wnt signaling. J Clin Invest. 2006;116:1202–1209. doi: 10.1172/JCI28551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hartmann C. A Wnt canon orchestrating osteoblastogenesis. Trends Cell Biol. 2006;16:151–158. doi: 10.1016/j.tcb.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 20.Glass DA, 2nd, Karsenty G. Molecular bases of the regulation of bone remodeling by the canonical Wnt signaling pathway. Curr Top Dev Biol. 2006;73:43–84. doi: 10.1016/S0070-2153(05)73002-7. [DOI] [PubMed] [Google Scholar]
- 21.Rawadi G, Vayssiere B, Dunn F, Baron R, Roman-Roman S. BMP-2 controls alkaline phosphatase expression and osteoblast mineralization by a Wnt autocrine loop. J Bone Miner Res. 2003;18:1842–1853. doi: 10.1359/jbmr.2003.18.10.1842. [DOI] [PubMed] [Google Scholar]
- 22.Bain G, Muller T, Wang X, Papkoff J. Activated β-catenin induces osteoblast differentiation of C3H10T1/2 cells and participates in BMP2-mediated signal transduction. Biochem Biophys Res Commun. 2003;301:84–91. doi: 10.1016/s0006-291x(02)02951-0. [DOI] [PubMed] [Google Scholar]
- 23.Mbalaviele G, Sheikh S, Stains JP, et al. β-Catenin and BMP-2 synergize to promote osteoblast differentiation and new bone formation. J Cell Biochem. 2005;94:403–418. doi: 10.1002/jcb.20253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Winkler DG, Sutherland MS, Ojala E, et al. Sclerostin inhibition of Wnt-3a-induced C3H10T1/2 cell differentiation is indirect and mediated by bone morphogenetic proteins. J Biol Chem. 2005;280:2498–2502. doi: 10.1074/jbc.M400524200. [DOI] [PubMed] [Google Scholar]
- 25.Nakashima A, Katagiri T, Tamura M. Cross-talk between Wnt and bone morphogenetic protein 2 (BMP-2) signaling in differentiation pathway of C2C12 myoblasts. J Biol Chem. 2005;280:37660–37668. doi: 10.1074/jbc.M504612200. [DOI] [PubMed] [Google Scholar]
- 26.Simpson AH, Mills L, Noble B. The role of growth factors and related agents in accelerating fracture healing. J Bone Joint Surg Br. 2006;88:701–705. doi: 10.1302/0301-620X.88B6.17524. [DOI] [PubMed] [Google Scholar]
- 27.Mishina Y, Hanks MC, Miura S, Tallquist MD, Behringer RR. Generation of Bmpr/Alk3 conditional knockout mice. Genesis. 2002;32:69–72. doi: 10.1002/gene.10038. [DOI] [PubMed] [Google Scholar]
- 28.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 29.DasGupta R, Fuchs E. Multiple roles for activated LEF/TCF transcription complexes during hair follicle development and differentiation. Development. 1999;126:4557–4568. doi: 10.1242/dev.126.20.4557. [DOI] [PubMed] [Google Scholar]
- 30.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(–ΔΔC(t)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 31.Fujita N, Kajita M, Taysavang P, Wade PA. Hormonal regulation of metastasis-associated protein 3 transcription in breast cancer cells. Mol Endocrinol. 2004;18:2937–2949. doi: 10.1210/me.2004-0258. [DOI] [PubMed] [Google Scholar]
- 32.Yu PB, Hong CC, Sachidanandan C, et al. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat Chem Biol. 2008;4:33–41. doi: 10.1038/nchembio.2007.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ai M, Holmen SL, Van Hul W, Williams BO, Warman ML. Reduced affinity to and inhibition by DKK1 form a common mechanism by which high bone mass-associated missense mutations in LRP5 affect canonical Wnt signaling. Mol Cell Biol. 2005;25:4946–4955. doi: 10.1128/MCB.25.12.4946-4955.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li X, Liu P, Liu W, et al. Dkk2 has a role in terminal osteoblast differentiation and mineralized matrix formation. Nat Genet. 2005;37:945–952. doi: 10.1038/ng1614. [DOI] [PubMed] [Google Scholar]
- 35.Patel MS, Karsenty G. Regulation of bone formation and vision by LRP5. N Engl J Med. 2002;346:1572–1574. doi: 10.1056/NEJM200205163462011. [DOI] [PubMed] [Google Scholar]
- 36.Li X, Zhang Y, Kang H, et al. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem. 2005;280:19883–19887. doi: 10.1074/jbc.M413274200. [DOI] [PubMed] [Google Scholar]
- 37.Semenov M, Tamai K, He X. SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J Biol Chem. 2005;280:26770–26775. doi: 10.1074/jbc.M504308200. [DOI] [PubMed] [Google Scholar]
- 38.Bodine PV, Zhao W, Kharode YP, et al. The Wnt antagonist secreted frizzled-related protein-1 is a negative regulator of trabecular bone formation in adult mice. Mol Endocrinol. 2004;18:1222–1237. doi: 10.1210/me.2003-0498. [DOI] [PubMed] [Google Scholar]
- 39.Miyazono K. Signal transduction by bone morphogenetic protein receptors: functional roles of Smad proteins. Bone. 1999;25:91–93. doi: 10.1016/s8756-3282(99)00113-1. [DOI] [PubMed] [Google Scholar]
- 40.Fujii M, Takeda K, Imamura T, et al. Roles of bone morphogenetic protein type I receptors and Smad proteins in osteoblast and chondroblast differentiation. Mol Biol Cell. 1999;10:3801–3813. doi: 10.1091/mbc.10.11.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu X, Han J, Ito Y, Bringas P, Jr, Deng C, Chai Y. Ectodermal Smad4 and p38 MAPK are functionally redundant in mediating TGF-β/BMP signaling during tooth and palate development. Dev Cell. 2008;15:322–329. doi: 10.1016/j.devcel.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zuzarte-Luis V, Montero JA, Rodriguez-Leon J, Merino R, Rodriguez-Rey JC, Hurle JM. A new role for BMP5 during limb development acting through the synergic activation of Smad and MAPK pathways. Dev Biol. 2004;272:39–52. doi: 10.1016/j.ydbio.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 43.Tseng YH, Kokkotou E, Schulz TJ, et al. New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature. 2008;454:1000–1004. doi: 10.1038/nature07221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Inagaki K, Otsuka F, Miyoshi T, et al. p38-Mitogen-activated protein kinase stimulated steroidogenesis in granulosa cell-oocyte cocultures: role of bone morphogenetic proteins 2 and 4. Endocrinology. 2009;150:1921–1930. doi: 10.1210/en.2008-0851. [DOI] [PubMed] [Google Scholar]
- 45.Nohe A, Keating E, Knaus P, Petersen NO. Signal transduction of bone morphogenetic protein receptors. Cell Signal. 2004;16:291–299. doi: 10.1016/j.cellsig.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 46.Huelsken J, Vogel R, Erdmann B, Cotsarelis G, Birchmeier W. β-Catenin controls hair follicle morphogenesis and stem cell differentiation in the skin. Cell. 2001;105:533–545. doi: 10.1016/s0092-8674(01)00336-1. [DOI] [PubMed] [Google Scholar]
- 47.Barrow JR, Thomas KR, Boussadia-Zahui O, et al. Ectodermal Wnt3/β-catenin signaling is required for the establishment and maintenance of the apical ectodermal ridge. Genes Dev. 2003;17:394–409. doi: 10.1101/gad.1044903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guo X, Day TF, Jiang X, Garrett-Beal L, Topol L, Yang Y. Wnt/β-catenin signaling is sufficient and necessary for synovial joint formation. Genes Dev. 2004;18:2404–2417. doi: 10.1101/gad.1230704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.He XC, Zhang J, Tong WG, et al. BMP signaling inhibits intestinal stem cell self-renewal through suppression of Wnt-β-catenin signaling. Nat Genet. 2004;36:1117–1121. doi: 10.1038/ng1430. [DOI] [PubMed] [Google Scholar]
- 50.Dean CH, Miller LA, Smith AN, Dufort D, Lang RA, Niswander LA. Canonical Wnt signaling negatively regulates branching morphogenesis of the lung and lacrimal gland. Dev Biol. 2005;286:270–286. doi: 10.1016/j.ydbio.2005.07.034. [DOI] [PubMed] [Google Scholar]
- 51.Zhang J, He XC, Tong WG, et al. Bone morphogenetic protein signaling inhibits hair follicle anagen induction by restricting epithelial stem/progenitor cell activation and expansion. Stem Cells. 2006;24:2826–2839. doi: 10.1634/stemcells.2005-0544. [DOI] [PubMed] [Google Scholar]
- 52.Chen Y, Whetstone HC, Youn A, et al. Beta-catenin signaling pathway is crucial for bone morphogenetic protein 2 to induce new bone formation. J Biol Chem. 2007;282:526–533. doi: 10.1074/jbc.M602700200. [DOI] [PubMed] [Google Scholar]
- 53.Mukhopadhyay M, Shtrom S, Rodriguez-Esteban C, et al. Dickkopf1 is required for embryonic head induction and limb morphogenesis in the mouse. Dev Cell. 2001;1:423–434. doi: 10.1016/s1534-5807(01)00041-7. [DOI] [PubMed] [Google Scholar]
- 54.Grotewold L, Ruther U. The Wnt antagonist Dickkopf-1 is regulated by BMP signaling and c-Jun and modulates programmed cell death. EMBO J. 2002;21:966–975. doi: 10.1093/emboj/21.5.966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sutherland MK, Geoghegan JC, Yu C, et al. Sclerostin promotes the apoptosis of human osteoblastic cells: a novel regulation of bone formation. Bone. 2004;35:828–835. doi: 10.1016/j.bone.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 56.Ohyama Y, Nifuji A, Maeda Y, Amagasa T, Noda M. Spaciotemporal association and bone morphogenetic protein regulation of sclerostin and osterix expression during embryonic osteogenesis. Endocrinology. 2004;145:4685–4692. doi: 10.1210/en.2003-1492. [DOI] [PubMed] [Google Scholar]
- 57.Semenov MV, Tamai K, Brott BK, Kuhl M, Sokol S, He X. Head inducer Dickkopf-1 is a ligand for Wnt coreceptor LRP6. Curr Biol. 2001;11:951–961. doi: 10.1016/s0960-9822(01)00290-1. [DOI] [PubMed] [Google Scholar]
- 58.Morvan F, Boulukos K, Clement-Lacroix P, et al. Deletion of a single allele of the Dkk1 gene leads to an increase in bone formation and bone mass. J Bone Miner Res. 2006;21:934–945. doi: 10.1359/jbmr.060311. [DOI] [PubMed] [Google Scholar]
- 59.Li J, Sarosi I, Cattley RC, et al. Dkk1-mediated inhibition of Wnt signaling in bone results in osteopenia. Bone. 2006;39:754–766. doi: 10.1016/j.bone.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 60.Tian E, Zhan F, Walker R, et al. The role of the Wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. N Engl J Med. 2003;349:2483–2494. doi: 10.1056/NEJMoa030847. [DOI] [PubMed] [Google Scholar]
- 61.Li X, Ominsky MS, Niu QT, et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res. 2008;23:860–869. doi: 10.1359/jbmr.080216. [DOI] [PubMed] [Google Scholar]
- 62.Balemans W, Ebeling M, Patel N, et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST) Hum Mol Genet. 2001;10:537–543. doi: 10.1093/hmg/10.5.537. [DOI] [PubMed] [Google Scholar]
- 63.Brunkow ME, Gardner JC, Van Ness J, et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet. 2001;68:577–589. doi: 10.1086/318811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Balemans W, Patel N, Ebeling M, et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet. 2002;39:91–97. doi: 10.1136/jmg.39.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Staehling-Hampton K, Proll S, Paeper BW, et al. A 52-kb deletion in the SOST-MEOX1 intergenic region on 17q12-q21 is associated with van Buchem disease in the Dutch population. Am J Med Genet. 2002;110:144–152. doi: 10.1002/ajmg.10401. [DOI] [PubMed] [Google Scholar]
- 66.Suzuki A, Guicheux J, Palmer G, et al. Evidence for a role of p38 MAP kinase in expression of alkaline phosphatase during osteoblastic cell differentiation. Bone. 2002;30:91–98. doi: 10.1016/s8756-3282(01)00660-3. [DOI] [PubMed] [Google Scholar]
- 67.Yuge L, Okubo A, Miyashita T, et al. Physical stress by magnetic force accelerates differentiation of human osteoblasts. Biochem Biophys Res Commun. 2003;311:32–38. doi: 10.1016/j.bbrc.2003.09.156. [DOI] [PubMed] [Google Scholar]
- 68.Yuge L, Hide I, Kumagai T, et al. Cell differentiation and p38(MAPK) cascade are inhibited in human osteoblasts cultured in a three-dimensional clinostat. In Vitro Cell Dev Biol Anim. 2003;39:89–97. doi: 10.1290/1543-706x(2003)039<0089:cdapca>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 69.Lee DY, Yeh CR, Chang SF, et al. Integrin-mediated expression of bone formation-related genes in osteoblast-like cells in response to fluid shear stress: roles of extracellular matrix, Shc, and mitogen-activated protein kinase. J Bone Miner Res. 2008;23:1140–1149. doi: 10.1359/jbmr.080302. [DOI] [PubMed] [Google Scholar]
- 70.Wingerter S, Tucci M, Bumgardner J, Benghuzzi H. Evaluation of short-term healing following sustained delivery of osteoinductive agents in a rat femur drill defect model. Biomed Sci Instrum. 2007;43:188–193. [PubMed] [Google Scholar]
- 71.Gautschi OP, Frey SP, Zellweger R. Bone morphogenetic proteins in clinical applications. Aust NZ J Surg. 2007;77:626–631. doi: 10.1111/j.1445-2197.2007.04175.x. [DOI] [PubMed] [Google Scholar]
- 72.Yoon BS, Ovchinnikov DA, Yoshii I, Mishina Y, Behringer RR, Lyons KM. Bmpr1a and Bmpr1b have overlapping functions and are essential for chondrogenesis in vivo. Proc Natl Acad Sci USA. 2005;102:5062–5067. doi: 10.1073/pnas.0500031102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bandyopadhyay A, Tsuji K, Cox K, Harfe BD, Rosen V, Tabin CJ. Genetic analysis of the roles of BMP2, BMP4, and BMP7 in limb patterning and skeletogenesis. PLoS Genet. 2006;2:e216. doi: 10.1371/journal.pgen.0020216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tsuji K, Bandyopadhyay A, Harfe BD, et al. BMP2 activity, although dispensable for bone formation, is required for the initiation of fracture healing. Nat Genet. 2006;38:1424–1429. doi: 10.1038/ng1916. [DOI] [PubMed] [Google Scholar]
- 75.Rountree RB, Schoor M, et al. BMP receptor signaling is required for postnatal maintenance of articular cartilage. PLoS Biol. 2004;2:e355. doi: 10.1371/journal.pbio.0020355. [DOI] [PMC free article] [PubMed] [Google Scholar]