

Abstract
Age-dependent changes in skeletal growth are important for regulating skeletal expansion and determining peak bone mass. However, how G protein–coupled receptors (GPCRs) regulate these changes is poorly understood. Previously, we described a mouse model expressing Rs1, an engineered receptor with high basal Gs activity. Rs1 expression in osteoblasts induced a dramatic age-dependent increase in trabecular bone with features resembling fibrous dysplasia. To further investigate how activation of the Gs-GPCR pathway affects bone formation at different ages, we used the tetracycline-inducible system in the ColI(2.3)+/Rs1+ mouse model to control the timing of Rs1 expression. We found that the Rs1 phenotype developed rapidly between postnatal days 4 and 6, that delayed Rs1 expression resulted in attenuation of the Rs1 phenotype, and that the Rs1-induced bone growth and deformities were markedly reversed when Rs1 expression was suppressed in adult mice. These findings suggest a distinct window of increased osteoblast responsiveness to Gs signaling during the early postnatal period. In addition, adult bones encode information about their normal shape and structure independently from mechanisms regulating bone expansion. Finally, our model provides a powerful tool for investigating the effects of continuous Gs-GPCR signaling on dynamic bone growth and remodeling. © 2010 American Society for Bone and Mineral Research.
Keywords: Fibrous Dysplasia, Rassl, G Protein–coupled Receptor, GS Signaling, Age-dependent GS Activation, Osteoblasts, Inbred Mice
Introduction
Osteoporosis affects over 10 million people in the United States and contributes to 1.5 million fractures each year.(1) A crucial determinant of osteoporosis and fracture risk in adults is thought to be the acquisition of peak bone mass during childhood and puberty.(2–4) Osteoblasts and their precursors play key roles in regulating bone development and acquisition of peak bone mass.(5) Although certain G protein–coupled receptors (GPCRs) expressed in osteoblasts [e.g., parathyroid hormone receptor (PTHR1)(6) and HTR1b serotonin receptor(7)] are involved in regulating bone mass, the exact in vivo roles of GPCR signaling during skeletal growth have not been clearly elucidated.
GPCRs signal through a select number of pathways, including the Gs and Gi pathways that influence intracellular cyclic AMP (cAMP) levels.(8) We described a synthetic biology approach(9) for activating the Gs pathway that uses the strong basal Gs signaling activity of Rs1, an engineered receptor activated solely by a synthetic ligand (RASSL).(10,11) RASSLs are engineered receptors that no longer respond to endogenous hormones but can be activated by synthetic small-molecule drugs. They have proven useful for studying the roles of G protein signaling in complex systems.(12–15)
We found that when Rs1 was expressed continuously in mouse osteoblasts from gestation until adulthood, skeletal bone mass and trabecular bone formation were dramatically greater than in wild-type mice.(11) Cortical bone was lost, and serum markers of bone turnover were greatly increased. In addition, the normal bone marrow space was obliterated and replaced with small uniform cells morphologically resembling young osteoblasts. In contrast, when Rs1 was expressed beginning at 4 weeks of age, no grossly abnormal skeletal phenotype was evident through 30 weeks of age. These findings suggested that receptor-mediated Gs signals in osteoblasts have age-dependent effects on bone formation. In addition, evidence from human diseases and animal models suggest that osteoblast function is markedly different in children than in adults.(16–21)
In this study, we asked how the timing of Gs signaling affects the formation of skeletal bone at different ages. We used Rs1 to activate the Gs signaling pathway in osteoblasts in mice at different ages in four experiments (Fig. 1). First, we identified a window of osteoblast responsiveness to Rs1 expression by examining when the Rs1-induced trabecular bone phenotype begins. Second, we narrowed the window of Rs1 expression necessary for developing the trabecular overgrowth phenotype by delaying Rs1 expression until after birth. Third, we assessed whether expression of Rs1 after 4 weeks of age could lead to long-term attenuation of the Rs1 phenotype in adult mice. Finally, we determined whether the Rs1-induced bone phenotype could be reversed if Rs1 expression was discontinued after formation of the abnormal trabecular bone in adult mice. Our findings demonstrate the utility of the Rs1 mouse model for controlling the timing and tissue-specific activation of Gs signaling as a way to understand the age-dependent effects of continuous Gs signaling by GPCRs.
Fig. 1.
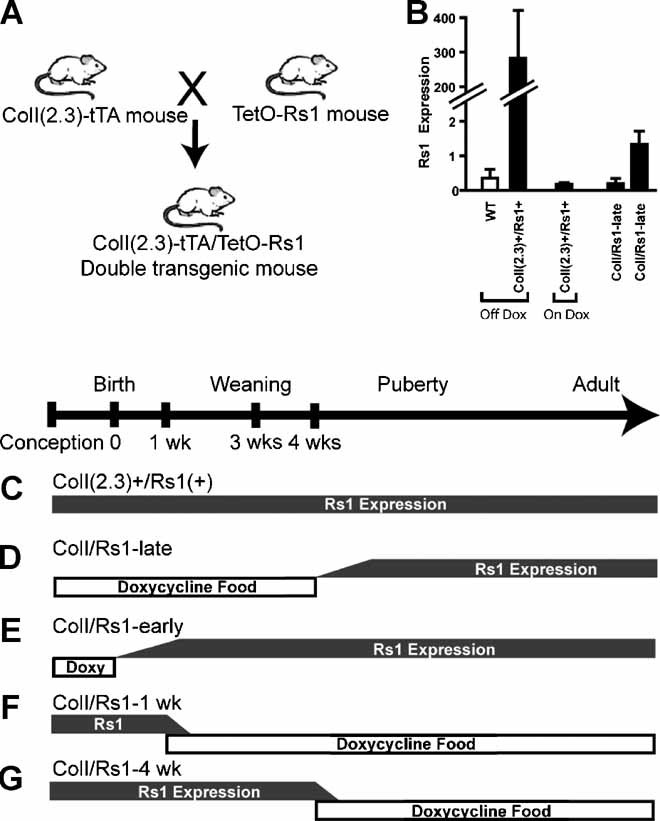
Timing of Rs1 expression in the ColI(2.3)-tTA/TetO-Rs1 mice. (A) We used a doxycycline-regulated expression system to focus on the effects of basal Gs signaling induced by Rs1 expression without ligand activation. (B) Rs1 expression was allowed by feeding the mice regular doxycycline-free food. Six-week-old ColI(2.3)+/Rs1+ mice raised on regular food (Off Dox) had high levels of Rs1 expression. We suppressed Rs1 expression by giving doxycycline in the mouse chow (On Dox). Mice born and raised on doxycycline-containing food and subsequently switched to doxycycline-free food at 4 weeks of age show low but variable levels of Rs1 expression at 6 weeks of age (ColI/Rs1-late). Expression levels from femora of representative mice are shown. Portions of this figure use RNA samples from mice used in Fig. S5 as published.(11) Values are mean ± 1 SD of technical triplicates. (C–G) The pattern of doxycycline food administration and Rs1 expression for each experimental manipulation is indicated in gray. (C) ColI(2.3)+/Rs1+ mice: Rs1 expression was allowed from gestation through adulthood by feeding the pregnant mothers and their pups regular chow. (D) ColI/Rs1-late mice: Rs1 expression was suppressed through the first 4 weeks of development by feeding doxycycline chow to mothers during mating and to their pups after weaning. We switched the mice to regular chow at 4 weeks of age. (E) ColI/Rs1-early mice: Rs1 expression was allowed after birth by feeding the mother doxycycline chow from mating until delivery. The nursing mothers were switched to regular chow at delivery. (F) ColI/Rs1–1 week mice: Rs1 expression was allowed until 1 week of age by feeding the mothers regular chow from mating until 1 week after birth. The nursing mothers were switched to doxycycline chow to suppress Rs1 expression in the nursing pups. Weaned animals were maintained on doxycycline chow. (G) ColI/Rs1–4 week mice: Rs1 expression was allowed from gestation through 4 weeks of age by feeding mothers regular chow from mating through weaning. The pups were given regular chow until 4 weeks of age. Rs1 expression then was suppressed by switching the pups to doxycycline chow.
Methods
Animal studies
All transgenic mouse studies were approved by and performed in accord with the Institutional Animal Care and Use Committee and the Laboratory Animal Research Center at the University of California, San Francisco. The ColI(2.3)-tTA/TetO-Rs1 double transgenic mice were generated by heterozygote crosses of mice carrying the TetO-Rs1 transgene with mice carrying the ColI(2.3)-tTA transgene as described previously.(11) Transgene expression was suppressed by continuous administration of doxycycline-impregnated mouse chow (DoxDiet 200 mg/kg; BioServ, Frenchtown, NJ, USA) during the times indicated in Fig. 1. Transgene expression was activated by switching the mice to regular mouse chow without doxycycline (LabDiet 5053, PMI Nutrition, St. Louis, MO, USA) at the times indicated in Fig. 1. Full transgene expression was expected to occur within 1.5 to 2 weeks of doxycycline withdrawal.(12,22) Transgene expression was verified by quantitative polymerase chain reaction (qPCR) in representative animals from each cohort (see Figs. 1B, 3C, 5C, and 7F). Neonatal pups analyzed in Fig. 3 were not manipulated until the day of tissue collection to minimize any risk of maternal distress or cannibalism. Genotyping and selection of the appropriate ColI(2.3)+/Rs1+ and control pups were done after tissue collection, or after 4 weeks of age for adult animals. No alterations in litter size, pup health, or maternal health were observed in our experiments. Both males and females were analyzed together in our experiments because no sex-dependent differences were observed.
Fig. 3.
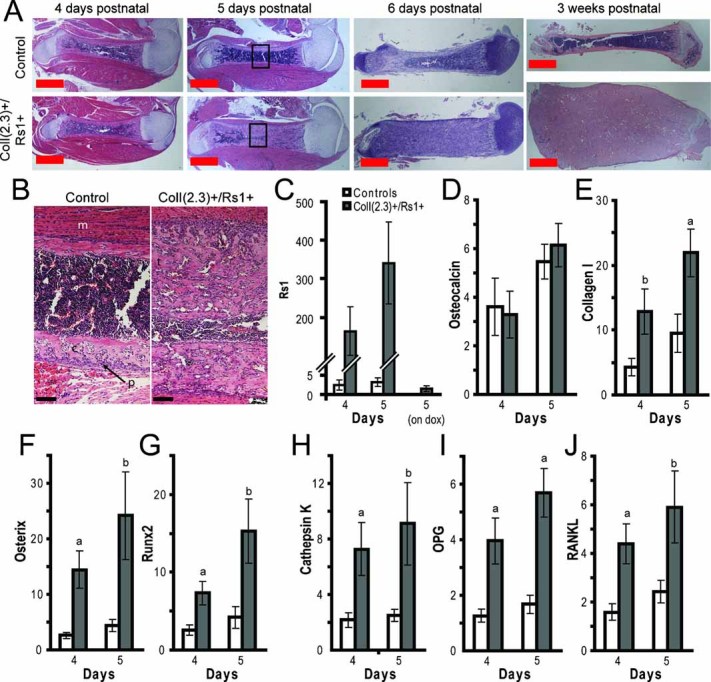
Rs1-induced bone formation occurs between days 4 to 6 in postnatal skeletal growth. (A) Representative hematoxylin and eosin histology of paraffin-embedded decalcified femur sections of 4-, 5-, and 6-day-old and 3-week-old ColI(2.3)+/Rs1+ or control (wild-type or single-transgenic) mice. Note the increased trabecular bone, decreased cortical bone, and narrowed bone marrow space in the day 5 and 6 femora reminiscent of the changes seen in the 3-week-old mice. Skeletal muscle was left in place for 4- and 5-day-old samples to preserve adjacent bone morphology during processing. p = periosteum; s = bone marrow space; c = cortical bone; t = trabecular bone; m = skeletal muscle. Scale bar = 1 mm. (B) High-magnification images of day 5 control and ColI(2.3)+/Rs1+ femora (boxed regions in panel A) showing the increased trabecular bone resulting from Rs1 expression. (C–J) Relative expression levels by quantitative PCR of whole humeri from ColI(2.3)+/Rs1+ (filled bars) and control littermates (open bars). For the quantitative PCR, n = 6 control and 6 mutant 4-day-old mice; n = 6 control and 5 mutant 5-day-old mice. Expression levels were determined in technical triplicates for each mouse and normalized to GAPDH. Values are mean ± 1 SEM. a: p < .05 versus controls; b: p < .1 versus controls. (C) Rs1 expression in 4- and 5-day-old ColI(2.3)+/Rs1+ pups and littermate controls. Five-day-old ColI(2.3)+/Rs1+ pups from mothers fed doxycycline-containing food (day 5, on dox) demonstrate suppression of Rs1 expression, indicating that doxycycline fed to nursing mothers crossed adequately into breast milk. n = 4 ColI(2.3)+/Rs1+ pups maintained on doxycycline. (D, E) Osteocalcin and collagen I expression, markers of mature osteoblasts. (F, G) Osterix and runx2 expression, markers of immature osteoblasts. (H–J) Cathepsin K, OPG, and RANKL expression, markers of osteoclastogenesis.
Fig. 5.
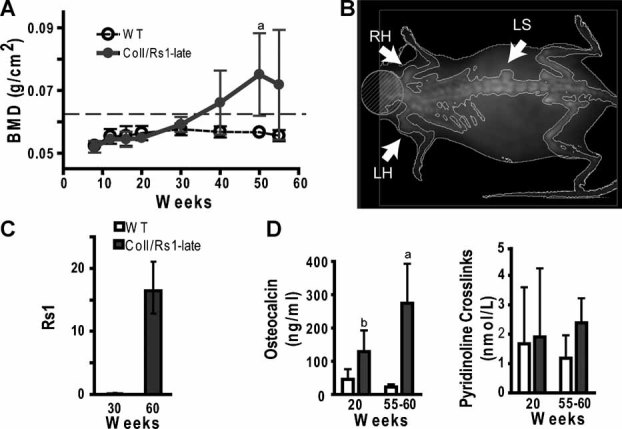
Expression of Rs1 after 4 weeks of age leads to long-term attenuation of the Rs1 phenotype. (A) Longitudinal whole-body aBMD showed a significant increase in average BMD as the ColI/Rs1-late mice aged. The dotted line indicates 2 SD above the WT BMD at 30 weeks (0.0613 g/cm2). n ≥ 4 mice for each time point. Values are mean ± 1 SD. a: p < .05 versus WT. (B) DXA image of a representative 60-week-old ColI/Rs1-late mouse showing isolated regions of subtle increased bone formation. RH = right humerus; LH = left humerus; LS = lumbar spine. (C) Rs1 expression in RNA isolated from whole humeri from representative ColI/Rs1-late mice showing low expression at 30 weeks (before the phenotype is evident) and higher expression at 60 weeks (when bone overgrowth is evident). Values represent mean ± 1 SD of technical triplicates. (D) Serum analysis of osteocalcin (a marker of bone formation) and pyridinoline cross-links (a marker of bone resorption) in ColI/Rs1-late mice. At 20 weeks, n = 8 mutants, 6 WT; at 55 to 60 weeks, n = 4 mutants, 4 WT. Values are mean ± 1 SD. a: p < .05 versus WT; b: p < .01 versus WT.
Fig. 7.
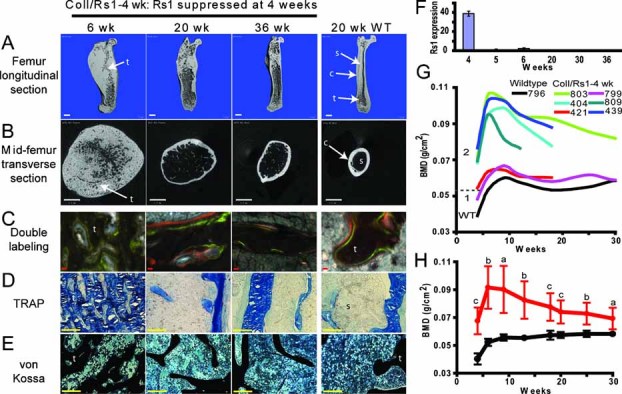
The Rs1-induced trabecular bone phenotype is reversible. (A, B) Representative longitudinal (A) and transverse (B) µCT images of femora from ColI/Rs1–4 week mice expressing Rs1 from gestation until 4 weeks of age, showing the loss of accumulated trabecular bone and return of the bone marrow space and cortical shell after Rs1 expression has been discontinued. A femur from a 20-week-old WT mouse is shown for comparison. t = trabecular bone; c = cortical bone; s = bone marrow space. Scale bar = 1 mm. (C) Fluorescent imaging of double-labeled bone showing the return of linear, orderly bone mineral apposition (green = calcein; orange = xylenol orange) similar to that seen in WT animals. Scale bar = 10 µm. (D) TRAP staining shows a significant decrease in osteoclast lineage cells (pink) as the trabecular bone overgrowth resolves. Scale bar = 100 µm. (E) von Kossa staining showing the return of normal-appearing trabeculi and bone marrow cells, similar to that seen in WT animals. Scale bar = 100 µm. (F) Rs1 expression levels in whole humeri isolated from ColI/Rs1–4 week mice shows rapid suppression after doxycycline is started at 4 weeks of age. Representative mice are shown for each timepoint. Values are mean ± 1 SD of technical triplicates. (G) Plots of whole-body BMD of six representative ColI/Rs1-4 week mice (colored lines) and one WT mouse (black, bottom line) indicate variation in the degree of bone accumulation at the start of the study at 4 weeks. All mutant mice showed decreases in their BMD after Rs1 expression was discontinued. At 4 weeks, mice were stratified into two groups: BMD < 3 SD above the average WT BMD (0.0521 g/cm2, as indicated by the dotted line; WT average = 0.0403 ± 0.0039 g/cm2) and BMD > 3 SD above the average WT BMD. Mice in group 2 were selected for pooled analysis in panel H. (H) Average whole-body BMD for group 2 mice (as determined in panel G) showed a continuous decline after Rs1 expression was discontinued. The number of animals meeting the criteria for group 2 at 4, 6, 9, 13, 18, 20, 25, and 30 weeks: WT: 16, 10, 9, 11, 12, 7, 4, and 4; ColI/Rs1–4 week: 7, 6, 5, 6, 10, 8, 6, and 5. Values are mean ± 1 SD. a: p < .05; b: p < .005; c: p < .001 versus WT.
Gene expression analysis was performed on RNA isolated from the right femur or right humerus of adult experimental animals, as indicated in each figure legend. For the experiments in Fig. 3, both humeri from the same 4- or 5-day-old animal were combined to maximize RNA yield. Briefly, whole bones were dissected away from surrounding soft tissue and frozen in liquid nitrogen. Bones for each experiment were batch processed by crushing (multisample Bio-Pulverizer, Research Products International, Mt. Prospect, IL, USA) followed by homogenization (4.5 mm Tissue Tearor, Research Products International) in RNAStat-60 (Iso-Tex Diagnostics, Friendswood, TX, USA). cDNA was generated using the SuperScript III First Strand Synthesis kit (Invitrogen, Carlsbad, CA, USA) as directed by the manufacturer. Expression was assayed using SybrGreen primers as described previously,(11) Sybr green primers for RANKL (forward = TTGCACACCTCACCATCAAT; reverse = TCCGTTGCTTAACGTCATGT) and OPG (forward = CAGAGACCAGGAAATGGTGAA; reverse = AAGCTGCTCTGTGGTGAGGT) or TaqMan primers (Mm99999913_g1 for GAPDH, Mm00484036_m1 for cathepsin K, Mm0081666_g1 for collagen 1a1, Mm00504574_m1 for osterix, and Mm00501578_m1 for runx2). All samples were assayed in technical triplicate, and expression levels were normalized to GAPDH. All qPCR reactions were run on an Applied Biosystems (Foster City, CA, USA) 7900HT real-time thermocycler.
Bone densitometry and imaging
Mice identified for dual-energy X-ray absorptiometry (DXA) to measure whole-body areal bone mineral density (aBMD) were anesthetized with inhaled isofluorane (1.5% to 2% in oxygen) and scanned on a GE Lunar Piximus2 (Madison, WI, USA) at predetermined times. Mice that underwent whole-mouse or ex vivo femur micro-computed tomographic (µCT) scans were sacrificed before scanning on a Scanco vivaCT-40 µCT scanner, (SCANCO, Waynes, PA, USA). Femora for ex vivo scanning were fixed in 10% neutral buffered formalin (Fisher Scientific, Fairlawn, NJ, USA) for 24 hours and stored in 70% ethanol. Images were obtained at an X-ray energy of 55 kV, with a voxel size of 76 or 10.5 µm and integration times of 200 or 1000 ms for whole-body images or ex vivo femur images, respectively. Because of age-dependent variations in bone mineralization, segmentation values for whole-animal µCT were determined empirically as 0.1/1/100 (gauss sigma/gauss support/threshold in mg HA/ccm) for newborns and 1.5-week-old animals. Segmentation values of 0.7/1/250 were used for the ex vivo femur CT scans.
Bone histology
Mice identified for histomorphometry were injected with 15 mg/kg calcein (Sigma-Aldrich, St. Louis, MO, USA) 7 days before harvesting and with 90 mg/kg xylenol orange (Sigma-Aldrich) 2 days before harvesting to label areas of active bone mineralization. Harvested bones were processed for von Kossa staining, tartrate-resistant acid phosphatase (TRAP) immunostaining, and fluorescence microscopy as described previously.(11) Bones for hematoxylin and eosin staining were decalcified for 12 to 48 hours in 10% EDTA/PBS before paraffin embedding, sectioning, and staining by the Gladstone Histology Core.
Serum analysis
Blood was collected from euthanized mice by cardiac puncture and processed in MicroTainer serum separator tubes according to the manufacturer's instructions (BD Biosciences, San Jose, CA, USA). Routine blood analysis was carried out by Antech Diagnostics (Irvine, CA, USA). Serum osteocalcin and pyridinoline measurements were carried out using the mouse osteocalcin EIA Kit BT-470 from BTI (Stoughton, MA, USA) and the MetraBiosystems SerumPYD Kit 8019 (Palo Alto, CA, USA) according to manufacturers' directions.
Statistical analysis
Two-tailed Student's t tests with unequal variances were performed using Microsoft Excel 2003. ANOVA analysis was performed using JMP8 (Version 8.0, SAS Institute, Cary, NC, USA).
Results
Rs1-induced bone formation in early postnatal skeletal growth
We previously reported that Rs1-induced basal Gs signaling in osteoblasts of ColI(2.3)+/Rs1+ mice increased trabecular bone and decreased cortical bone in lesions reminiscent of fibrous dysplasia of the bone.(11) In addition, increases in the whole-body BMD of ColI(2.3)+/Rs1+ mice expressing Rs1 were detectable only after 3 weeks of age.(11) To determine if Rs1-induced trabecular bone formation occurs earlier in postnatal growth and if Rs1 affects the patterning of the normal bone structures, we used µCT, histology, and gene expression to analyze bone formation in young mice expressing Rs1 from gestation.
µCT imaging showed no gross differences in skeletal patterning or overall bone shape before 1.5 weeks of age (see Fig. 2). At 1.5 weeks of age, small changes in bone structure were observed, primarily in the long bones of the mice. Since µCT analysis in prepubertal mice is complicated by rapidly changing bone mineral content during postnatal growth, we assessed bone structure by performing histology on femora from ColI(2.3)+/Rs1+ mice at 4 to 6 days after birth.
Fig. 2.
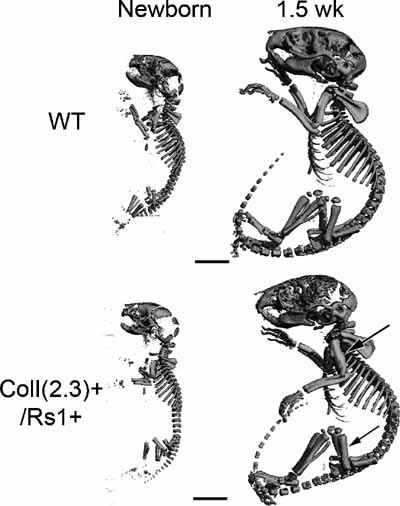
Mice expressing Rs1 from gestation showed no gross skeletal patterning defect. Note the subtle changes in bone size in the 1.5-week-old ColI(2.3)+/Rs1+ mice with mild thickening in the diaphyses of the femora and humeri (arrows). Scale bar = 5 mm.
Histology revealed that femora of the ColI(2.3)+/Rs1+ mice had normal periosteum, bone collar, and bone marrow cells through 4 days of postnatal growth (see Fig. 3A) despite detectable Rs1 expression (see Fig. 3C). However, by 5 days of postnatal growth, femora from the ColI(2.3)+/Rs1+ mice showed increased trabecular bone, decreased cortical bone, and narrowed bone marrow space (see 3A, B). By 6 days, the femora fully recapitulated the trabecular bone replacement of the entire bone as described in 3-week-old ColI(2.3)+/Rs1+ mice(11) (see 3A).
Gene expression analysis from 4 day old ColI(2.3)+/Rs1+ and control mice was used to examine the expression of genes marking mature (osteocalcin and collagen I) and immature osteoblasts (osterix and runx2) (see Fig. 3D–G). Osteocalcin levels were unchanged in the 4-day-old ColI(2.3)+/Rs1+ mice. However, collagen I, osterix, and runx2 all showed increased levels in the ColI(2.3)+/Rs1+ mice despite the lack of an obvious histologic phenotype. By day 5, expression of all these genes was greater in the bones of ColI(2.3)+/Rs1+ mice than in those of wild-type mice. In addition, expression levels of cathepsin K (a marker of osteoclasts) as well as osteoprotegerin (OPG) and RANKL (both regulators of osteoclast activity) were increased in the ColI(2.3)+/Rs1+ mice at both 4 and 5 days of age. The RANKL/OPG ratios, used as a measure of osteolytic balance,(23) were not significantly different in the ColI(2.3)+/Rs1+ mice between days 4 and 5 (1.25 versus 1.11, respectively).
These results show that continuous Gs signaling induced by Rs1 in osteoblasts leads to increased trabecular bone formation starting after day 4 of postnatal growth and increased expression of early osteoblast markers. In contrast, the increased bone formation was not accompanied by a change in the RANKL/OPG ratios.
Delayed timing of Rs1 expression
The prior results show that Rs1 expression occurs before development of the trabecular bone phenotype at day 5 of postnatal growth. In addition, Rs1 expression after 4 weeks of age (ColI/Rs1-late mice, 1D) led to no grossly abnormal bone phenotype in adult mice,(11) suggesting a window of sensitivity to Rs1 signaling between postnatal day 5 and 4 weeks of age. We used the tetracycline transactivator system to limit Rs1 expression to narrow the window of maximal osteoblast sensitivity to Rs1 signaling.
To determine if Rs1 expression during the late prenatal to early postnatal period is required to increased trabecular bone formation in adult mice, we suppressed Rs1 expression through birth (ColI/Rs1-early mice, 1E) by feeding pregnant mice doxycycline-containing chow. Pups and their nursing mothers were removed from doxycycline on the day of delivery to allow Rs1 expression. The pups were followed by serial whole-body BMD scans through the first 9 weeks of postnatal life. The ColI/Rs1-early mice developed the trabecular overgrowth phenotype, but to a milder degree than in the ColI(2.3)+/Rs1+ mice expressing Rs1 from gestation (see 4A), as assessed by whole-body BMD.
Fig. 4.
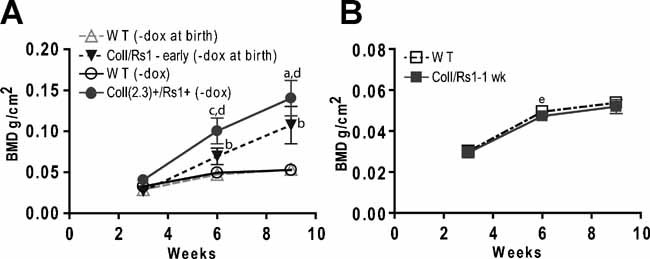
Delayed Rs1 expression leads to a milder bone overgrowth phenotype. (A) ColI/Rs1-early mice in which Rs1 expression began after birth have intermediate whole-body BMD as compared to ColI(2.3)+/Rs1+ mice expressing Rs1 from gestation and similarly treated wild-type littermates. n = 4 WT (–dox at birth), 10 WT (–dox), 10 ColI(2.3)+/Rs1+ (–dox), and 6 ColI/Rs1-early (–dox at birth) at each time point. Values are mean ± 1 SD. a: p < .05 versus ColI/Rs1-early; b: p < .01 versus WT; c: p < .001 versus ColI/Rs1-early; d: p < .001 versus WT. (B) BMD in ColI/Rs1–1 week mice and wild-type littermates indicate that the abnormal bone phenotype is not maintained once Rs1 expression is discontinued. n = 9 WT and 5 mutant mice per time point. Values are mean ± 1 SD. e: p < .05 versus WT.
To further define the timing of Rs1 expression necessary to develop and maintain the bone-overgrowth phenotype, we examined ColI/Rs1–1 week mice in which Rs1 expression was limited to gestation through the first week of postnatal life (see 1F). These mice did not develop the increased trabecular bone as adults or show whole-body BMDs different from wild-type littermate controls (see 4B).
These results indicate that maximal Rs1-induced bone formation in an adult mouse is strongly influenced by early Rs1 expression during prenatal gestation and early postnatal development. Furthermore, discontinuing Rs1 expression early in postnatal growth but after the trabecularization has been initiated resulted in normal BMD in adult animals, suggesting that growing bone could recover from the Rs1-induced abnormal bone formation if Rs1 expression was discontinued early.
Long-term expression of Rs1 in adult mice
The results for the ColI/Rs1-early mice and our prior observation that ColI/Rs1-late mice (see 1D) did not develop the grossly abnormal bone through 30 weeks of age(11) suggested that the ability of Rs1 to induce trabecular bone formation decreased as the mice aged despite detectable Rs1 expression in bone. To identify whether the ColI/Rs1-late mice remained free of the bone overgrowth phenotype as the mice aged, we preformed long-term follow-up on these mice through 60 weeks of age.
The ColI/Rs1-late mice showed no significant increases in whole-body BMD until 40 weeks of age. Between 40 and 60 weeks of age, all the ColI/Rs1-late mice showed increased trabecular bone, but only 50% of the mice developed a whole-body BMD greater than 2 SD above the peak wild-type BMD (see 5A). Sites of abnormal bone formation often were adjacent to regions of normal bone and occurred as distinct nodules (see 5B). Serum osteocalcin (a marker for bone formation) was increased slightly at 20 and 55 weeks, but serum pyridinoline cross-links (a marker for bone resorption) did not change (see 5D). These results suggest that Rs1 activity in adult bone primarily increases bone formation over bone resorption. No significant differences in serum calcium or phosphate levels were detected at either 20 or 55 weeks (data not shown).
µCT analysis on femora from 20-week-old ColI/Rs1-late mice showed a subtle expansion of trabecular bone starting at the femoral metaphyses. The increase in trabecular bone was accompanied by a loss of endocortical bone and narrowing of the bone marrow space in the ColI/Rs1-late mice (Fig. 6). Double fluorochrome labeling to assess bone mineralization showed an increase in disorganized trabecular bone formation in the 20 and 55-week-old ColI/Rs1-late mice. In addition, TRAP staining showed a localized increase in osteoclast-like cells within the regions of disorganized trabecular bone in the ColI/Rs1-late mice at both ages despite the normal systemic levels of pyridinoline crosslinks (see 5D). Von Kossa staining showed that the regions of disordered trabecular bone in the ColI/Rs1-late mice shared similar histologic features with the abnormal bones in the ColI(2.3)+/Rs1+ mice expressing Rs1 from gestation, including a decrease in bone marrow cells and an increase in small osteoblast-like cells by morphology.(11)
Fig. 6.
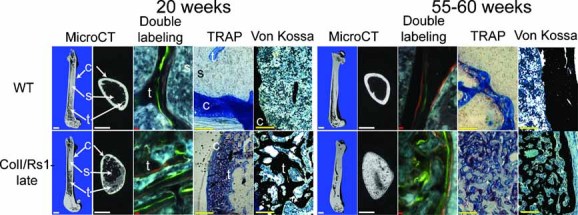
Representative µCT images of femora in longitudinal and transverse mid-diaphysis sections of age- and sex-matched mice show a delayed but gradual increase in trabecular bone and loss of cortical bone in the ColI/Rs1-late mice. Scale bar = 1 mm. Double fluorochrome labeling (middle panels) with calcein (green) and xylenol orange (orange) demonstrated disordered bone formation in the areas of increased trabecular bone. Scale bar = 10 µm. TRAP staining (pink), indicative of osteoclast-lineage cells, is increased within the regions of expanded trabecular bone in ColI/Rs1-late mice. Scale bar = 100 µm. Von Kossa staining shows the increased trabecular bone and loss of normal bone marrow cells (scale bar = 100 µm). c = cortical bone; t = trabecular bone; s = bone marrow space.
These results indicate that delaying Rs1 expression until 4 weeks of age leads to long-term attenuation of the Rs1-induced phenotype. However, a subtle increase in bone formation could be detected as early as 20 weeks of age. Although the bone-overgrowth phenotype was clearly evident by 40 to 60 weeks of age, bone formation was significantly slower in the ColI/Rs1-late mice than in the ColI(2.3)+/Rs1+ mice expressing Rs1 from gestation. In addition, the new trabecular bone appeared to originate from sites of existing trabecular bone, such as at the femoral metaphyses, rather than as de novo formation of trabecular bone at a nontrabecular site, such as at the mid-diaphysis.
Reversibility of the Rs1-induced trabecular bone formation
The absence of an increased BMD in the ColI/Rs1–1 week mice indicated that the Rs1-induced trabecular bone growth might be reversible if Rs1 expression were discontinued before weaning. To determine if established Rs1-induced bone lesions in adult mice also could be reversed and if continued activation of the Gs signaling pathway by Rs1 was required to maintain the abnormal increase in trabecular bone, we assessed mice in which Rs1 expression was suppressed after 4 weeks of age (ColI/Rs1–4 week mice, see 1G). The ColI/Rs1–4 week mice developed the abnormal increase in trabecular bone, decrease in cortical bone, and loss of bone marrow space during the first 4 weeks of life. After this bone phenotype was established, we suppressed Rs1 expression by feeding the mice doxycycline-containing chow.
ColI/Rs1-4 week femora examined over the next 32 weeks showed a gradual decrease in trabecular bone and a restoration of cortical bone and the bone marrow space (see 7A, B). Double fluorochrome labeling showed that the normal linear bone mineralization pattern gradually returned as the trabecular bone disappeared (see 7C). TRAP staining also indicated a decrease in osteoclast lineage cells within the bones (see 7D). Although the overall shape of the bone did not fully return to normal by 36 weeks of age, the gross deformation originally present in the 4- to 6-week-old mice was significantly reversed. Histology on undecalcified bone sections (see 7E) indicated the return of a normal hematopoietic bone marrow space. In addition, we observed a rapid decrease in the number of morphologically small osteoblast-like cells previously identified in the ColI(2.3)+/Rs1+ trabecular overgrowth bones.
BMDs of individual mice at the start of this experiment at 4 weeks of age were variable (see 7G), but all BMDs declined or stabilized after Rs1 expression was suppressed. In mice with a starting 4 week BMD of greater than 3 SD above the mean wild-type BMD, whole-body BMDs gradually decreased over 26 weeks, correlating with the reversal of the trabecular bone phenotype observed by µCT imaging (see 7H). Serum calcium and phosphate levels at 20 and 36 weeks were similar between wild-type and ColI/Rs1–4 week mice (data not shown). Three-week-old ColI/Rs1–4 week mice (before starting doxycycline) showed higher levels of serum osteocalcin but not pyridinoline cross-links than age-matched control littermates (359.20 ± 45.91 versus 257.50 ± 50.15 ng/mL, p = .01, and 1.42 ± 0.31 versus 1.53 ± 0.42 nmol/L, p = .6, respectively; n = 5 mutants and n = 5 controls). Osteocalcin and pyridinoline cross-link levels were both normal in the 20- to 25-week-old ColI/Rs1–4 week mice compared with age-matched control littermates (78.36 ± 44.78 versus 62.91 ± 35.22 ng/mL, p = .5, and 1.53 ± 0.63 versus 1.81 ± 0.89 nmol/L, p = .6, respectively; n = 6 mutants and n = 5 controls).
These results indicate that the abnormal trabecular bone formation and loss of cortical bone observed in the ColI(2.3)+/Rs1+ mice expressing Rs1 from gestation can be reversed in adult mice by suppressing the expression of Rs1. Although the bone did not return fully to a normal structure at the end of the 36 week experiment, our results demonstrate that adult bone maintains a high degree of plasticity for bone remodeling.
Discussion
In this study, we applied an engineered receptor approach and mouse model of temporally regulated osteoblast-specific Gs activation by the Rs1 RASSL to identify how Gs signals regulate skeletal bone formation in mice of different ages. We focused our experiments on the effects of Rs1 expression without ligand activation because Rs1 has a high degree of basal Gs signaling activity.(10,11) The double-transgenic “Tet-Off” strategy allowed doxycycline-dependent regulation of Rs1 expression and hence control of the basal Gs signaling activity of Rs1. Our studies are especially relevant to the long-term effects of continuous Gs signaling by GPCRs in osteoblasts. We found that the Rs1 phenotype developed rapidly between postnatal days 4 and 6, that progressively delayed Rs1 expression resulted in decreased Rs1-induced bone formation, and that the Rs1-induced bone growth and deformities were dramatically reversed when Rs1 expression was suppressed in adult mice.
Our findings with the ColI/Rs1-early and ColI/Rs1-late mice indicate that osteoblasts from young animals and osteoblasts from older animals both form trabecular bone in response to Gs activation by Rs1. However, delaying Rs1 expression leads to quantitatively different amounts of bone formation. Prior results suggested that Rs1 expression may be lower in adult animals,(11) possibly as a result of age-dependent changes in collagen I promoter methylation.(24) Age-dependent decreases in sensitivity to Gs signaling or cAMP also may contribute because osteoblast adenylyl cyclase activity decreases with age.(25)
A number of GPCRs are expressed in bone, including the parathyroid hormone/parathyroid hormone–releasing peptide (PTH/PTHrP) receptor,(26) endothelin receptor,(27) prostaglandin E2 receptor,(28) adrenaline receptor,(29,30) thyroid-stimulating hormone receptor,(31) and the HTR1b serotonin receptor.(7) Age-dependent activity of these receptors or their ligands may allow other GPCR signaling pathways to modulate the bone-formation effects of Rs1. Activation of the Gi signaling pathway by the Ro1 engineered RASSL(22) or other GPCRs(7,32) is associated with decreased trabecular bone formation, suggesting that the Gi pathway could dampen the bone-formation effects of Gs signaling in older animals. In addition, activation of the Gq pathway leads to osteopenia and may desensitize osteoblasts to Gs signaling.(33,34) Since the PTH receptor signals through Gs, Gi, and Gq, this suggests that the non-Gs pathways balance the Gs-mediated trabecular bone-formation process. The interactions between the different GPCRs expressed in an osteoblast also may account for the phenotypic variability we observed between individual mice.
Age-dependent increases in osteoclast formation(39) also may contribute to the decreased Rs1-induced trabecular bone formation we observed in adult mice. Although increased markers of osteoclasts are evident in the ColI(2.3)+/Rs1+ bone lesions, our results indicate that osteoblast-induced osteoclast formation does not occur during the initial formation of abnormal trabecular bone between days 4 and 5 of postnatal growth. Continued exposure to Rs1 activation eventually may result in increased osteoclastogenesis driven by more mature osteoblasts, similar to what has been reported after PTH treatment of cultured bone marrow stromal cells.(40) As a result, a net increase in osteoclast formation or activity may protect adult bones from the early effects of Rs1.
The bone-formation pattern in the ColI(2.3)+/Rs1+ mice expressing Rs1 from gestation resembles the lesions in fibrous dysplasia patients(35) but to a more severe degree. Clinically, fibrous dysplasia lesions are heterogeneous with abnormal and normal trabecular bone, often at distinct anatomic sites.(36) The ColI/Rs1-late mice in our study particularly resemble this type of lesion, suggesting that the heterogeneous nature of fibrous dysplasia may be a result of changes in osteoblast sensitivity to Gs signaling. This idea that osteoblasts in fibrous dysplasia lesions may have reacquired the characteristics of a perinatal osteoblast correlates with suggestions that fibrous dysplasia may be a stem cell disease.(37) Furthermore, a non-cell autonomous mechanism may contribute to the development of fibrous dysplasia lesions(38) and may similarly modify the osteoblast response to Rs1-mediated Gs activation in an age-dependent manner.
Our finding that Rs1-induced trabecular bone formation occurs during a discrete window of days 4 to 5 of early postnatal growth was surprising because the ColI(2.3) promoter is active starting before birth,(41,42) and Rs1 expression was detectable before any bone changes were evident on histology. This result suggests that the early developmental processes that establish bone patterning and structure prior to postnatal day 4(43) are not significantly affected by Gs signaling. Since the distinct transition at days 4 to 5 of postnatal growth is associated with early weight-bearing activity and the appearance of the secondary spongiosum in mice,(44,45) Gs signaling may act at this point to promote trabecular bone expansion. In contrast, Gi signaling appears to have roles in neonatal bone formation. Earlier results using the ColI(2.3) promoter to express a Gi coupled engineered receptor in osteoblasts before birth resulted in embryonic lethality.(22) In addition, cre-mediated inactivation of the osteoblast GNAS gene encoding the Gsα subunit resulted in severe neonatal skeletal malformations and decreased trabecular bone.(32) These findings suggest that the roles of Gs signaling may shift during early skeletal development from maintenance of the osteoblast niche to driving expansion of trabecular bone.
Our result that the Rs1-induced trabecular bone formation is largely reversible has several important implications. First, bone formation during prepubertal growth can have long-term effects on adult bone, but that bone remodeling may temper these effects. Recent animal studies(46) and clinical studies in children(2) suggest that exercise during childhood can lead to an increase in bone mass that persists with age. The unique nature of the prepubertal skeletal system is further emphasized in that a number of human diseases affecting G protein signaling in the bone have a tendency to present during childhood with long-term sequela in adults.(47–49) However, very early changes in bone formation, as indicated by the ColI/Rs1–1 week mice, may not have long-term effects on the skeleton. Second, continuous activation of the Gs signaling pathway is required to maintain the increase in trabecular bone induced by Rs1. Clinical studies show that bone formed by recombinant PTH (teriparatide) is lost rapidly unless an antiresorptive medication is used when PTH is discontinued.(50,51) Our results indicate that the changes in bone mass are more likely due to changes in Gs pathway activation rather than to the signaling properties of a specific receptor (e.g., PTHR1). Finally, our results show that normal bone has a dramatic ability to repair itself once an insult is discontinued. Since the ColI(2.3)+/Rs1+ mice share strong histologic resemblance to polyostotic fibrous dysplasia(35) and the bone lesions in McCune-Albright syndrome,(35) our results suggest that inhibiting the Gs signaling pathway may be an effective strategy for treating these patients. Our findings also suggest that bones retain information about their normal shape and structure independent of the capacity for skeletal expansion, which could be used during bone remodeling to restore normal bone structure.
In conclusion, bone formation is a highly plastic process with a complex array of signaling inputs that ultimately determine peak bone mass and normal bone architecture. Understanding how bone tissue can be grown and re-formed may reveal basic properties of bone that will be useful in bone regeneration and repair. Our model provides a dramatic example of how osteoblast-specific hormone signaling can completely reshape bone architecture in a reversible fashion. Insight into how hormone signals can alter morphology will help in the development of new ways to modulate the signaling of endogenous GPCRs to reshape bone in a therapeutic setting.
Acknowledgments
We thank Carlota Manalac, Alyssa Louie, Mark Scott, Gary Howard, Mimi Zeiger, and the Gladstone Histology Core (Jo-dee Fish and Caroline Miller) for valuable technical assistance and discussions. We also thank Scott Kogan and the Center for Genomic Pathology (http://ctrgenpath.net/) slide rounds for valuable histology input. This work was supported by National Institutes of Health (NIH) Grants R01 HL60664-07 (to BRC) and R01 DK072071 (to RAN). ECH received support from an NIH Training Grant (2T32DK07418-26), an NIH career development award (7 K08 AR056299-02), from the California Institute of Regenerative Medicine/Gladstone Institute CIRM Fellowship Program (Grant T2-00003), and from the San Francisco Veterans Affairs Research Enhancement Awards Program. RAN and BPH are senior research career scientists of the Department of Veterans Affairs and received support from the Veterans Affairs Merit Review Program. The J. David Gladstone Institutes received support from National Center for Research Resources Grant RR18928-01.
Disclosures
The authors state that they have no conflicts of interest.
References
- 1.National Osteoporosis Foundation. America's Bone Health: The State of Osteoporosis and Low Bone Mass in Our Nation. Washington: National Osteoporosis Foundation; 2002. [Google Scholar]
- 2.MacKelvie KJ, Khan KM, Petit MA, Janssen PA, McKay HA. A school-based exercise intervention elicits substantial bone health benefits: a 2-year randomized controlled trial in girls. Pediatrics. 2003;112:e447. doi: 10.1542/peds.112.6.e447. [DOI] [PubMed] [Google Scholar]
- 3.Cooper C, Harvey N, Javaid K, Hanson M, Dennison E. Growth and bone development. Nestle Nutr Workshop Ser Pediatr Program. 2008;61:53–68. doi: 10.1159/000113170. [DOI] [PubMed] [Google Scholar]
- 4.Cooper C, Westlake S, Harvey N, Javaid K, Dennison E, Hanson M. Developmental origins of osteoporotic fracture. Osteoporos Int. 2006;17:337–347. doi: 10.1007/s00198-005-2039-5. [DOI] [PubMed] [Google Scholar]
- 5.Aubin JE, Lian JB, Stein GS. Bone Formation: Maturation and Functional Activities of Osteoblast Lineage Cells. In: Favus MJ, editor. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 6th ed. Washington: American Society for Bone and Mineral Research; 2006. pp. 20–29. [Google Scholar]
- 6.Lanske B, Karaplis AC, Lee K, et al. PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science. 1996;273:663–666. doi: 10.1126/science.273.5275.663. [DOI] [PubMed] [Google Scholar]
- 7.Yadav VK, Ryu JH, Suda N, et al. Lrp5 controls bone formation by inhibiting serotonin synthesis in the duodenum. Cell. 2008;135:825–837. doi: 10.1016/j.cell.2008.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gether U. Uncovering molecular mechanisms involved in activation of G protein–coupled receptors. Endocr Rev. 2000;21:90–113. doi: 10.1210/edrv.21.1.0390. [DOI] [PubMed] [Google Scholar]
- 9.Conklin BR, Hsiao EC, Claeysen S, et al. Engineering GPCR signaling pathways with RASSLs. Nat Methods. 2008;5:673–678. doi: 10.1038/nmeth.1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang WC, Ng J, Nguyen T, et al. Modifying ligand-induced and constitutive signaling of the human 5-HT4 receptor. PLoS One. 2007;2:e1317. doi: 10.1371/journal.pone.0001317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hsiao EC, Boudignon BM, Chang WC, et al. Osteoblast expression of an engineered Gs-coupled receptor dramatically increases bone mass. Proc Natl Acad Sci USA. 2008;105:1209–1214. doi: 10.1073/pnas.0707457105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Redfern CH, Coward P, Degtyarev MY, et al. Conditional expression and signaling of a specifically designed Gi-coupled receptor in transgenic mice. Nat Biotechnol. 1999;17:165–169. doi: 10.1038/6165. [DOI] [PubMed] [Google Scholar]
- 13.Scearce-Levie K, Lieberman MD, Elliott HH, Conklin BR. Engineered G protein–coupled receptors reveal independent regulation of internalization, desensitization and acute signaling. BMC Biol. 2005;3:3. doi: 10.1186/1741-7007-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sweger EJ, Casper KB, Scearce-Levie K, Conklin BR, McCarthy KD. Development of hydrocephalus in mice expressing the G(i)-coupled GPCR Ro1 RASSL receptor in astrocytes. J Neurosci. 2007;27:2309–2317. doi: 10.1523/JNEUROSCI.4565-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao GQ, Zhang Y, Hoon MA, et al. The receptors for mammalian sweet and umami taste. Cell. 2003;115:255–266. doi: 10.1016/s0092-8674(03)00844-4. [DOI] [PubMed] [Google Scholar]
- 16.Nieminen S, Nurmi M, Satokari K. Healing of femoral neck fractures: influence of fracture reduction and age. Ann Chir Gynaecol. 1981;70:26–31. [PubMed] [Google Scholar]
- 17.Nilsson BE, Edwards P. Age and fracture healing: a statistical analysis of 418 cases of tibial shaft fractures. Geriatrics. 1969;24:112–117. [PubMed] [Google Scholar]
- 18.Skak SV, Jensen TT. Femoral shaft fracture in 265 children: log-normal correlation with age of speed of healing. Acta Orthop Scand. 1988;59:704–707. doi: 10.3109/17453678809149430. [DOI] [PubMed] [Google Scholar]
- 19.Lu C, Miclau T, Hu D, et al. Cellular basis for age-related changes in fracture repair. J Orthop Res. 2005;23:1300–1307. doi: 10.1016/j.orthres.2005.04.003.1100230610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meyer RA, Jr, Desai BR, Heiner DE, Fiechtl J, Porter S, Meyer MH. Young, adult, and old rats have similar changes in mRNA expression of many skeletal genes after fracture despite delayed healing with age. J Orthop Res. 2006;24:1933–1944. doi: 10.1002/jor.20124. [DOI] [PubMed] [Google Scholar]
- 21.Tonna EA, Cronkite EP. Changes in the skeletal cell proliferative response to trauma concomitant with aging. J Bone Joint Surg Am. 1962;44:1557–1568. [Google Scholar]
- 22.Peng J, Bencsik M, Louie A, et al. Conditional expression of a Gi-coupled receptor in osteoblasts results in trabecular osteopenia. Endocrinology. 2008;149:1329–1337. doi: 10.1210/en.2007-0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grimaud E, Soubigou L, Couillaud S, et al. Receptor activator of nuclear factor κB ligand (RANKL)/osteoprotegerin (OPG) ratio is increased in severe osteolysis. Am J Pathol. 2003;163:2021–2031. doi: 10.1016/s0002-9440(10)63560-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ohi T, Uehara Y, Takatsu M, Watanabe M, Ono T. Hypermethylation of CpGs in the promoter of the COL1A1 gene in the aged periodontal ligament. J Dent Res. 2006;85:245–250. doi: 10.1177/154405910608500308. [DOI] [PubMed] [Google Scholar]
- 25.Donahue HJ, Zhou Z, Li Z, McCauley LK. Age-related decreases in stimulatory G protein–coupled adenylate cyclase activity in osteoblastic cells. Am J Physiol. 1997;273:E776–781. doi: 10.1152/ajpendo.1997.273.4.E776. [DOI] [PubMed] [Google Scholar]
- 26.Juppner H, Abou-Samra AB, Freeman M, et al. A G protein–linked receptor for parathyroid hormone and parathyroid hormone–related peptide. Science. 1991;254:1024–1026. doi: 10.1126/science.1658941. [DOI] [PubMed] [Google Scholar]
- 27.Kasperk CH, Borcsok I, Schairer HU, et al. Endothelin-1 is a potent regulator of human bone cell metabolism in vitro. Calcif Tissue Int. 1997;60:368–374. doi: 10.1007/s002239900245. [DOI] [PubMed] [Google Scholar]
- 28.Suzawa T, Miyaura C, Inada M, et al. The role of prostaglandin E receptor subtypes (EP1, EP2, EP3, and EP4) in bone resorption: an analysis using specific agonists for the respective EPs. Endocrinology. 2000;141:1554–1559. doi: 10.1210/endo.141.4.7405. [DOI] [PubMed] [Google Scholar]
- 29.Moore RE, Smith CK, 2nd, Bailey CS, Voelkel EF, Tashjian AH., Jr Characterization of beta-adrenergic receptors on rat and human osteoblast-like cells and demonstration that beta-receptor agonists can stimulate bone resorption in organ culture. Bone Miner. 1993;23:301–315. doi: 10.1016/s0169-6009(08)80105-5. [DOI] [PubMed] [Google Scholar]
- 30.Togari A, Arai M, Mizutani S, Mizutani S, Koshihara Y, Nagatsu T. Expression of mRNAs for neuropeptide receptors and beta-adrenergic receptors in human osteoblasts and human osteogenic sarcoma cells. Neurosci Lett. 1997;233:125–128. doi: 10.1016/s0304-3940(97)00649-6. [DOI] [PubMed] [Google Scholar]
- 31.Abe E, Marians RC, Yu W, et al. TSH is a negative regulator of skeletal remodeling. Cell. 2003;115:151–162. doi: 10.1016/s0092-8674(03)00771-2. [DOI] [PubMed] [Google Scholar]
- 32.Sakamoto A, Chen M, Nakamura T, Xie T, Karsenty G, Weinstein LS. Deficiency of the G protein alpha-subunit G(s)α in osteoblasts leads to differential effects on trabecular and cortical bone. J Biol Chem. 2005;280:21369–21375. doi: 10.1074/jbc.M500346200. [DOI] [PubMed] [Google Scholar]
- 33.Ogata N, Kawaguchi H, Chung UI, Roth SI, Segre GV. Continuous activation of GαQ in osteoblasts results in osteopenia through impaired osteoblast differentiation. J Biol Chem. 2007;282:35757–35764. doi: 10.1074/jbc.M611902200. [DOI] [PubMed] [Google Scholar]
- 34.Roy AA, Nunn C, Ming H, et al. Up-regulation of endogenous RGS2 mediates cross-desensitization between Gs and Gq signaling in osteoblasts. J Biol Chem. 2006;281:32684–32693. doi: 10.1074/jbc.M604416200. [DOI] [PubMed] [Google Scholar]
- 35.Riminucci M, Fisher LW, Shenker A, Spiegel AM, Bianco P, Gehron Robey P. Fibrous dysplasia of bone in the McCune-Albright syndrome: abnormalities in bone formation. Am J Pathol. 1997;151:1587–1600. [PMC free article] [PubMed] [Google Scholar]
- 36.Collins MT. Spectrum and natural history of fibrous dysplasia of bone. J Bone Miner Res. 2006;21:P99–104. doi: 10.1359/jbmr.06s219. [DOI] [PubMed] [Google Scholar]
- 37.Riminucci M, Saggio I, Robey PG, Bianco P. Fibrous dysplasia as a stem cell disease. J Bone Miner Res. 2006;21:P125–131. doi: 10.1359/jbmr.06s224. [DOI] [PubMed] [Google Scholar]
- 38.Bianco P, Kuznetsov SA, Riminucci M, Fisher LW, Spiegel AM, Robey PG. Reproduction of human fibrous dysplasia of bone in immunocompromised mice by transplanted mosaics of normal and Gsα-mutated skeletal progenitor cells. J Clin Invest. 1998;101:1737–1744. doi: 10.1172/JCI2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cao JJ, Wronski TJ, Iwaniec U, et al. Aging increases stromal/osteoblastic cell-induced osteoclastogenesis and alters the osteoclast precursor pool in the mouse. J Bone Miner Res. 2005;20:1659–1668. doi: 10.1359/JBMR.050503. [DOI] [PubMed] [Google Scholar]
- 40.Huang JC, Sakata T, Pfleger LL, et al. PTH differentially regulates expression of RANKL and OPG. J Bone Miner Res. 2004;19:235–244. doi: 10.1359/JBMR.0301226. [DOI] [PubMed] [Google Scholar]
- 41.Dacquin R, Starbuck M, Schinke T, Karsenty G. Mouse α1(I)-collagen promoter is the best known promoter to drive efficient Cre recombinase expression in osteoblast. Dev Dyn. 2002;224:245–251. doi: 10.1002/dvdy.10100. [DOI] [PubMed] [Google Scholar]
- 42.Dacic S, Kalajzic I, Visnjic D, Lichtler AC, Rowe DW. Col1a1-driven transgenic markers of osteoblast lineage progression. J Bone Miner Res. 2001;16:1228–1236. doi: 10.1359/jbmr.2001.16.7.1228. [DOI] [PubMed] [Google Scholar]
- 43.Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423:332–336. doi: 10.1038/nature01657. [DOI] [PubMed] [Google Scholar]
- 44.Robinson K. Evaluations of organ system development in juvenile toxicology testing. Reprod Toxicol. 2008;26:51–53. doi: 10.1016/j.reprotox.2008.05.056. [DOI] [PubMed] [Google Scholar]
- 45.Zoetis T, Tassinari MS, Bagi C, Walthall K, Hurtt ME. Species comparison of postnatal bone growth and development. Birth Defects Res B Dev Reprod Toxicol. 2003;68:86–110. doi: 10.1002/bdrb.10012. [DOI] [PubMed] [Google Scholar]
- 46.Warden SJ, Fuchs RK, Castillo AB, Nelson IR, Turner CH. Exercise when young provides lifelong benefits to bone structure and strength. J Bone Miner Res. 2007;22:251–259. doi: 10.1359/jbmr.061107. [DOI] [PubMed] [Google Scholar]
- 47.Weinstein LS, Shenker A. G protein mutations in human disease. Clin Biochem. 1993;26:333–338. doi: 10.1016/0009-9120(93)90109-j. [DOI] [PubMed] [Google Scholar]
- 48.Weinstein LS, Liu J, Sakamoto A, Xie T, Chen M. GNAS: normal and abnormal functions. Endocrinology. 2004;145:5459–5464. doi: 10.1210/en.2004-0865. [DOI] [PubMed] [Google Scholar]
- 49.Wilkes D, McDermott DA, Basson CT. Clinical phenotypes and molecular genetic mechanisms of Carney complex. Lancet Oncol. 2005;6:501–508. doi: 10.1016/S1470-2045(05)70244-8. [DOI] [PubMed] [Google Scholar]
- 50.Kurland ES, Heller SL, Diamond B, McMahon DJ, Cosman F, Bilezikian JP. The importance of bisphosphonate therapy in maintaining bone mass in men after therapy with teriparatide [human parathyroid hormone(1–34)] Osteoporos Int. 2004;15:992–997. doi: 10.1007/s00198-004-1636-z. [DOI] [PubMed] [Google Scholar]
- 51.Black DM, Bilezikian JP, et al. One year of alendronate after one year of parathyroid hormone (1–84) for osteoporosis. N Engl J Med. 2005;353:555–565. doi: 10.1056/NEJMoa050336. [DOI] [PubMed] [Google Scholar]