

Abstract
In this paper, we describe a new method to generate a smooth algebraic spline (AS) approximation of the molecular surface (MS) based on an initial coarse triangulation derived from the atomic coordinate information of the biomolecule, resident in the PDB (Protein data bank). Our method first constructs a triangular prism scaffold covering the PDB structure, and then generates a piecewise polynomial F on the Bernstein-Bezier (BB) basis within the scaffold. An ASMS model of the molecular surface is extracted as the zero contours of F which is nearly C1 and has dual implicit and parametric representations. The dual representations allow us easily do the point sampling on the ASMS model and apply it to the accurate estimation of the integrals involved in the electrostatic solvation energy computations. Meanwhile comparing with the trivial piecewise linear surface model, fewer number of sampling points are needed for the ASMS, which effectively reduces the complexity of the energy estimation.
Index Terms: Polynomial splines, molecular surfaces, prismatic scaffolds, Bernstein-Bezier basis, solvation energetics, error bounds, rate of convergence
I. Introduction
The computation of electrostatic solvation energy (also known as polarization energy) for biomolecules plays an important role in the molecular dynamics simulation [1], the analysis of stability in protein structure prediction [2], and the protein-ligand binding energy calculation [3]. The explicit model of the solvent provides the most rigorous solvation energy calculation [4]. However, due to the large amount of solvent molecules, most of the computation time is spent on the trajectories of the solvent molecules, which severely increases the computation cost of this method [5]. An alternative method is to represent the solvent implicitly as a dielectric continuum [6], then the electrostatic potential is known by solving the Poisson-Boltzmann (PB) equations [7] [8]. A more efficient method is to approximate the PB electrostatic solvation energy by the generalized Born (GB) model [9] [10] [11], which computes the electrostatic solvation energy ΔGelec as
| (1) |
where , εp is the solute (low) dielectric constant, εw is the solvent (high) dielectric constant, qi is the atomic charge of atom i, rij is the distance between atom i and j, F is an empirical factor (could be 4 [9] or 8 [11]), and Ri is the effective Born radius of atom i. The effective Born radius reflects how deep an atom is buried in the molecule and consequently determines the importance to the polarization. The formulation of the effective Born radii is derived in [12]:
| (2) |
where Γ is the molecular surface of the solute, xi is the center of atom i, and n(r) is the unit normal of the surface at r. The details of the derivation of (2) and a fast evaluation algorithm based on the fast Fourier transform (FFT) for (2) is discussed in [13]. Since the numerical integrations are done on the molecular surface Γ, an accurate and analytic representation of Γ is needed.
Three well-known molecular surfaces are shown in Figure 1 in 2D. The van der Waals surface (VWS) is the union of a set of spheres with atomic van der Waals radii. The solvent accessible surface (SAS) is the union of augmented van der Waals spheres with each radius enlarged by the solvent probe radius (normally taken as 1.4 Å) [14]. The solvent excluded surface (SES, also called molecular surface or Connolly surface) is the boundary of the union of all possible solvent probes that do not intersect with the interior of the VWS [15] [16]. As described in [15], the SES consists of the convex spherical patches which are parts of the VWS as well, the toroidal patches and the concave spherical patches, which are generated by the probes rolling along the intersections of neighboring atoms. The VWS causes an overestimation of the electrostatic solvation energy, while the SAS leads to an underestimation [11]. The SES is the most accurate when it is applied in the energetic calculation and therefore it is most often used to model the molecular surface. However the SES still has one significant drawback: it contains cusps when the rolling probe self-intersects, which may cause singularity in the Born radii and the force calculations.
Fig. 1.

Three molecular surfaces are shown for two atoms in two dimension. The boundary of the union of balls (dotted red line) with the van der Waals radii is the VWS. The SAS (solid thin line in purple) is the union of augmented van der Waals spheres with each radius enlarged by the radius of a solvent probe (light blue sphere). The SES (the solid thick line in blue) is boundary of all possible solvent probes that do not intersect with the interior of the VWS.
In the energetic computation, knowing the patch complexes of the molecular surface is not enough. For convenience, an analytical representation of the molecular surface is needed and the singularity should also be avoided. One way to generate such a model is to define an analytical volumetric density function, for example, the summation of Gaussian functions [17], Fermi-Dirac switching function [18], or piecewise polynomials [11], and approximate the SES by an iso-contour of the density function. Techniques of fast extracting an iso-contour of smooth kernel functions are developed in [19] [20]. However the error of the generated isosurface could be large and result in inaccurate energy computation. A NURBS representation for the SES is presented in [21]. Although it provides a parametric approximation to the SES, it does not solve the singularity problem. Edelsbrunner [22] defines another paradigm of a smooth surface referred to as skin which is based on the Voronoi, Delaunay, and Alpha complexes of a finite set of weighed points. The skin model has good geometric properties such as it is free of singularity and it can be decomposed into a collection of quadratic patches. Triangulation schemes based on the skin model are provided in [23] [24]. However when applied to the energetic computation, the skin triangulation which in fact is a linear approximation to the SES has to be very dense to gain accuracy, which causes oversampling on the surface and hence makes the computation very slow. Therefore it still remains a challenge to generate a model for the molecular surface which is accurate, smooth, and computable.
The main contribution of this paper is to provide a method to model the SES as piecewise algebraic spline patches with certain continuity at the boundary of the patches. Each patch has dual implicit and parametric representations. Hence high order implicit surfaces can be parameterized onto a planer domain and therefore higher order quadrature rules of 2D such as the Gaussian quadrature rules can be easily applied to the energetic computation. Moreover, because higher order spline patches are used to approximate the SES, fewer number of triangles are needed to obtain the same accuracy in the energetic computation as the linear model. The algebraic spline patches are generated based on the prism scaffold built surrounding the original triangular mesh of the SES and are defined implicitly by simple BB spline functions. Previous work on constructing piecewise spline patches within a simplical hull over a triangular mesh includes generating quadric patches [25], cubic patches [26] [27], and nonsingular and single sheeted cubic patches [28] in a tetrahedra scaffold. In this paper, we also show that the so generated algebraic spline patches are error bounded and free of singularity under certain conditions.
The paper is organized as follows: Section II describes the details of the algebraic spline molecular surface (ASMS) generation; Section III discusses the error of ASMS and Section IV discusses the application to the energetic computation and provides some examples.
II. Algebraic spline model
A. Algorithm Sketch
There are four main steps in our ASMS construction algorithm: (1) construct an initial triangular mesh of the SES; (2) build a prism scaffold surrounding the triangulation; (3) define a piecewise polynomial with certain continuity; (4) extract the 0-contour of the piecewise polynomial. We are going the explain each step in detail in the following and discuss how to make use the parametrization of the ASMS in the numerical integration.
B. Initial triangulation of the MS
So far a lot of work has been done on the triangulation of the SES or its approximation [24] [29] [30] [31] [32]. The ASMS generation could be applied to any of these triangulations. In our current research we use the triangulation generated by a program in the software TexMol [32] [33] as the initial. In this program the SES is described as an iso-contour of a sign distance function (SDF) with the isovalue equal to the radius of the water probe. The SDF measures the distance of any point in ℝ3 to the SAS where the sign indicates which side the point locates of the SAS. Here we define the SDF to be positive if the point is inside the SAS and negative if it is outside the SAS. A dual contour method is used to extract the iso-contour. The cusps created by the self-intersecting patches are detected and removed. Features of the molecular surface are well preserved in this triangulation. We then decimate the mesh by removing some of vertices from the triangulation. These vertices have the smallest normal variation, so the detailed features of the surface can still be captured after the vertices are removed [34].
C. Implicit/parametric patches generation
Given the triangulation mesh
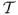 , let [vivjvk] be one of the triangles where vi, vj, vk are the vertices of the triangle. Suppose the unit normals of the surface at the vertices are also known, denoted as nl, (l = i, j, k). Let vl(λ) = vl + λnl. First we define a prism (Figure 2) Dijk:= {p: p = b1vi(λ) + b2vj(λ) + b3vk(λ), λ ∈ Iijk}, where (b1, b2, b3) are the barycentric coordinates of points in [vivjvk], and Iijk is a maximal open interval containing 0 and for any λ ∈ Iijk, vi(λ), vj(λ), vk(λ) are not collinear and ni, nj, nk point to the same side of the plane Pijk(λ):= {p: p = b1vi(λ) + b2vj(λ) + b3vk(λ)}. Next we define a function in the Bernstein-Bezier (BB) basis over the prism Dijk:
, let [vivjvk] be one of the triangles where vi, vj, vk are the vertices of the triangle. Suppose the unit normals of the surface at the vertices are also known, denoted as nl, (l = i, j, k). Let vl(λ) = vl + λnl. First we define a prism (Figure 2) Dijk:= {p: p = b1vi(λ) + b2vj(λ) + b3vk(λ), λ ∈ Iijk}, where (b1, b2, b3) are the barycentric coordinates of points in [vivjvk], and Iijk is a maximal open interval containing 0 and for any λ ∈ Iijk, vi(λ), vj(λ), vk(λ) are not collinear and ni, nj, nk point to the same side of the plane Pijk(λ):= {p: p = b1vi(λ) + b2vj(λ) + b3vk(λ)}. Next we define a function in the Bernstein-Bezier (BB) basis over the prism Dijk:
| (3) |
where is the Bezier basis
Fig. 2.
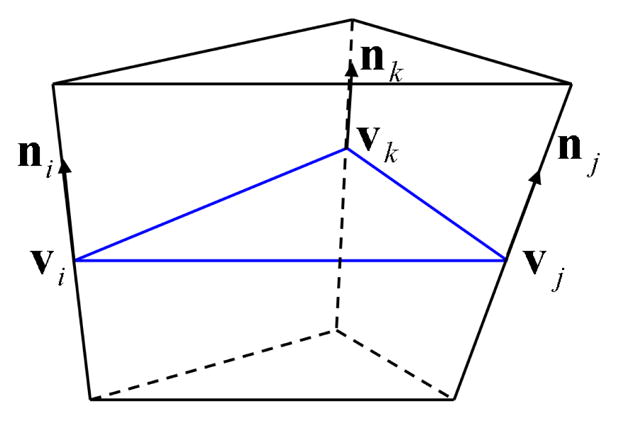
A prism Dijk constructed based on the triangle [vivjvk].
We approximate the molecular surface by the zero contour of F, denoted as S. In order to make S smooth, the degree of the Bezier basis n should be no less than 3. For simplicity, here we consider the case of n = 3. The control coefficients bijk(λ) should be properly defined such that S is continuous. In Figure 3 we show the relationship of the control coefficients and the points of the triangle when n = 3. Next we are going to discuss these coefficients are defined.
Fig. 3.
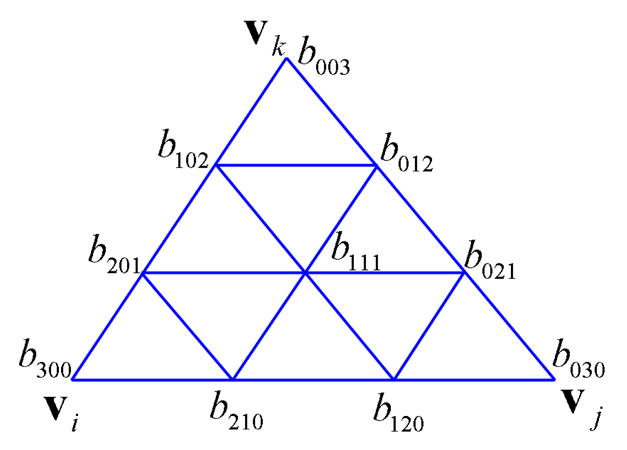
The control coefficients of the cubic Bezier basis of function F.
Since S passes through the vertices vi, vj, vk, we define
| (4) |
Next we are going to define the coefficients on the edges of the triangle in Figure 3. To obtain C1 continuity at vi, we require that the directional derivatives of F at vi in the direction of b2 and b3 are equal to ∇F · (vj − vi) and ∇F · (vk − vi), respectively. Noticing that F has the form of (3) and (b1, b2, b3) = (1, 0, 0) at vi, one can derive that , where ∇F(vi) = ni. Therefore
| (5) |
b120, b201, b102, b021, b012 are defined similarly.
To obtain the C1 continuity at the midpoints of the edges of
, we define b111 by using the side-vertex scheme [35]:
| (6) |
where
Next we are going to define and . In Appendix V-A we prove that our scheme of defining this three coefficients can guarantee the C1 continuity at the midpoints of the edges vjvk, vivk and vivj. Consider the edge vivj. Recall that any point p = (x, y, z) in Dijk can be represented by
| (7) |
Therefore differentiating both sides of (7) with respect to x, y and z, respectively, yields
| (8) |
where I3 is a 3 × 3 unit matrix. Denote
| (9) |
and let A = vi(λ) − vk(λ), B = vj(λ) − vk(λ) and C = b1ni +b2nj +b3nk, then M = (A B C)T. From (8) we have
| (10) |
According to (3), at the midpoint of vivj, , we have
By (6), at we have . Therefore the gradient at ( , 0) is
| (11) |
Define vectors
| (12) |
Let
| (13) |
| (14) |
Let . In order to have C1 continuity at ( , 0), we should have ∇F · d3(λ) = 0. Therefore, by (11) and (14), we have
| (15) |
Similarly, we may define and .
Now the function F(b1, b2, b3, λ) is well defined. The next step is to extract the zero level set S. Given the barycentric coordinates (b1, b2, b3) of a point in the triangle [vivjvk], we find the corresponding λ by solving the equation F(b1, b2, b3, λ) = 0 for λ and this could be done by the Newton’s method. Then we may get the corresponding point on S as
| (16) |
D. Smoothness
Theorem 2.1
The ASMS S is C1 at the vertices of
and the midpoints of the edges of
.
Theorem 2.2
S is C1 everywhere if every edge vivj of
satisfies ni·(vi − vj) = nj·(vj − vi).
Theorem 2.3
S is C1 everywhere if the unit normals at the vertices of
are the same.
Proofs of the theorems are shown in the Appendix.
E. Parametrization and quadrature
In this section, we would like to show how the ASMS is applied to the computation of (2). Since we use the ASMS to represent the molecular surface, now Γ = S. Let . We decompose the entire surface S into patches {Sj} with Sj being the AMSM generated over triangle j, then we have
| (17) |
For any point x = (x, y, z) on Sj, by the inverse map of (16), one can uniquely map x to a point in triangle j and get its baricentric coordinates (b1, b2, b3) with b3 = 1 − b1 − b2. Therefore, x, y, z can be represented in terms of (b1, b2):
Replacing (x, y, z) with (b1, b1, b3) in (17) and letting
we get
| (18) |
where
We then apply the Gaussian quadrature to (18):
| (19) |
where ( ) and Wk are the Gaussian integration nodes and weights on the triangles.
III. Error of the ASMS model
In order to show the error of S to the true surface S0, we do a test on some typical surfaces (Table I) S0:= {(x, y, z): z = f(x, y), (x, y) ∈ [0, 1]2} which are considered as the true surfaces. We generate a triangulation mesh over the true surface with the maximum edge length h being 0.1. Based on the mesh, we construct the ASMS model S. The error of S to S0 is defined as , where p ∈ S, q ∈ S0, and p and q have the same (b1, b2, b3) coordinates but different λ. We sample (p, q) on the surfaces and compute the maximum relative error. For the point pair p(b1, b2, b3, λp) and q(b1, b2, b3, λq) defined above, we prove that their Euclidean distance is bounded by the difference of their λ coordinates.
TABLE I.
Relative error and Convergence
| Function (x, y) ∈ [0, 1]2 | C | ||
|---|---|---|---|
| z = 0 | 0 | 0 | |
| z = x2 + y2 | 2.450030e-05 | 1.010636e-2 | |
| z = x3 + y3 | 1.063699e-04 | 2.610113e-2 | |
|
|
5.286856e-07 | 6.288604e-5 | |
|
|
2.555683e-04 | 4.58608e-2 | |
| z = tanh(9y − 9x) | 1.196519e-02 | 1.896754e-1 | |
|
|
8.614969e-05 | 1.744051e-1 (h4) | |
|
|
1.418242e-05 | 1.748754e-02 | |
Lemma 3.1
The error of the approximation point p to the true point q is bounded by |λp − λq|.
Proof
To study the rate of converges of S to S0, we gradually refine the initial mesh. Since the error is bounded by |λp − λq|, we compute the ratio of the maximum difference of λp and λq to h, h2, h3, and so forth. As h decreases, we check if the ratio converges or not, which allows us to know the highest rate of convergence of S to S0. For most of the test functions in Table I, we observe that S converges to S0 as fast as O(h3). We also observe that for the case , the rate of convergence reaches O(h4). We show the limit of the ratio as h ↓ 0, denoted as C, in Table I. Hence we draw the following claim:
Claim
Let h be the maximum side length of triangulation mesh
, p be the point on the ASMS, q be the corresponding point on the true surface, then p converges to q at the rate of O(h3). i.e. There exists a constant C such that ||p − q|| ≤ Ch3.
We generated the ASMS for the real proteins based on different size of meshes (Figure 4) and show the error of the ASMS to the SES of three proteins: 1GCQ (843 atoms), 1ML0 (1051 atoms), and 1KKL (1276 atoms) in Table II. Here the SES is modeled as a level set of the summation of fast decaying Gaussian functions. The ASMS is generated from the triangulation of the SES at different resolution. The number of triangles of the initial meshes are listed in Table II. The error εmax is defined as the one-way Hausdorff distance from the ASMS to the SES: . As we see in the table, the errors are small and decrease rapidly as the initial triangulation becomes dense.
Fig. 4.

The top row is the triangulation of the SES of protein 1ML0 with different number of triangles. The bottom row is the ASMS generated from the above corresponding triangulation.
TABLE II.
Error of ASMS to the SES
| 1GCQ | 1ML0 | 1KKL | |||
|---|---|---|---|---|---|
| No. of Δs | εmax | No. of Δs | εmax | No. of Δs | εmax |
| 16,312 | 0.266069 | 18,400 | 0.233949 | 19,968 | 0.260418 |
| 32,624 | 0.142149 | 36,864 | 0.142380 | 39,544 | 0.134689 |
| 65,456 | 0.082550 | 73,736 | 0.083895 | 79,096 | 0.085855 |
IV. Application to the biomolecular energetic computation
We apply the ASMS model to the GB electrostatic solvation energy computations of the example proteins 1PPE (436 atoms), 1HIA (693 atoms), 1CGI (852 atoms), 7CEI (1912 atoms), 1F15 (7704 atoms), and 1KXP (11859 atoms). The ASMS models S for the proteins are generated based on the initial mesh with different number of triangles (Table III). We show the ASMS of the example molecules generated from the decimated triangulations in Figure 5 and Figure 6. As a comparison, we compute the polarization energy Gpol for both the ASMS and the piecewise linear (PL) surfaces and show the energy results and the timing in Table III. For all the computations, a 4-point Gaussian quadrature rule over a triangle [36] is used for the numerical integration in (19) when computing the Born radii. The running time contains the time cost of computing the integration nodes over the surfaces, computing the Born radii, and evaluating Gpol. If we consider the energy computed from the dense mesh as accurate, as we see from the table, the Gpol computed from the coarse PL model has a large error, however for the coarse ASMS model, it is very close to the dense mesh result but with less time. On the other hand, to get a energy result of the same accuracy, fewer number of triangles are needed for the ASMS model than the PL model. For example, for the protein 1CGI, the Gpol computed from the ASMS with 3674 triangles is −1394.227 kcal/mol. However to get a similar result, 8712 triangles are needed for the piecewise linear model. Therefore the ASMS model is much more efficient in the energetic computation than trivial piecewise linear models.
TABLE III.
ELECTROSTATIC SOLVATION ENERGY AND TIMING
| Protein ID | No. of Triangles |
Gpol (kcal/mol)
|
Timing (s)
|
||
|---|---|---|---|---|---|
| PL | AS | PL | AS | ||
| 1PPE | 24244 | −835.5639 | −825.3252 | 17.27 | 18.26 |
| 6004 | −852.7130 | −828.2158 | 5.09 | 5.39 | |
| 2748 | −933.9562 | −845.5085 | 2.74 | 3.27 | |
|
| |||||
| 1HIA | 27480 | −1361.2266 | −1340.6384 | 30.23 | 31.18 |
| 7770 | −1389.0175 | −1347.8067 | 9.43 | 9.93 | |
| 3510 | −1571.8908 | −1388.4665 | 5.21 | 5.21 | |
|
| |||||
| 1CGI | 29108 | −1371.7419 | −1343.1496 | 39.64 | 40.31 |
| 8712 | −1399.1948 | −1346.2230 | 12.94 | 12.64 | |
| 3674 | −1678.4447 | −1394.2270 | 7.40 | 6.11 | |
|
| |||||
| 7CEI | 54544 | −3758.7928 | −3711.3626 | 29.04 | 29.96 |
| 17044 | −3771.7803 | −3753.3377 | 10.03 | 9.89 | |
| 5324 | −3876.7333 | −3826.1959 | 4.11 | 4.08 | |
|
| |||||
| 1F51 | 87516 | −11656.0327 | −11411.9689 | 123.55 | 121.52 |
| 33660 | −11691.4450 | −11622.8886 | 51.95 | 52.56 | |
| 8290 | −12527.8362 | −11721.4931 | 21.01 | 21.55 | |
|
| |||||
| 1KXP | 402812 | −13258.0206 | −13121.3053 | 975.08 | 977.96 |
| 134272 | −13325.0423 | −13264.7272 | 340.16 | 346.31 | |
| 94352 | −14669.1209 | −14071.9965 | 246.68 | 244.33 | |
Fig. 5.

Molecular models of a protein(1HIA). (a) is The atomic model. (b) is the initial dense mesh of the SES (27480 triangles). (c) is the decimated mesh of the SES model (7770 triangles). (d) is the ASMS (7770 patches) generated from (c).
Fig. 6.

The top row are the models of 1CGI and the bottom row are the models of 1PPE. (a) and (d) are the atomic structures of the proteins. (b) and (e) are the decimated triangular meshes of the proteins with 8712 triangles and 6004 triangles, respectively. (c) and (f) are the ASMS models generated from (b) and (e), respectively.
V. Conclusions
We have introduced a method to generate a model for the molecular surface. Like the other molecular surface models, this ASMS model is smooth and close to the SES as long as the initial triangulation is based on the SES. In addition, it has dual implicit and parametric representations. The implicit representation enables us to flexibly vary the surface by selecting different level sets, while the parametric representation allows us easily apply the ASMS to the numerical computations, such as the numerical integrations involved in the finite element method or the boundary element method. Moreover, unlike the other piecewise linear models, the ASMS surface is of higher degree, therefore, to get the same accuracy, fewer number of triangles (roughly one-third of the PL model) are needed for the ASMS when it is applied to the numerical integrations. For many large system problems, for example the atomistic molecular dynamics simulations, efficient computation is the most concerning issue, hence he ASMS is very suitable to be used in this kind of problems. We should mention that, while not detailed in this paper, the algorithm of Section II-C can, by repeated evocation, yield a hierarchical multiresolution spline model of the molecular surface. In the future research we could extend this algebraic patch model to the electrostatic solvation forces calculation which is crucial in the molecular dynamics simulations. Fast and accurate numerical integration is also one of the main tasks of the force calculation and is more challenging because the integration domain contains not only the surface but also a skin layer over each atom.
Acknowledgments
This research was supported in part by NSF grant CNS-0540033 and NIH contracts R01-EB00487, R01-GM074258, R01-GM07308. We wish to thank Vinay Siddavanahalli and other members of the CVC group for developing and maintaining TexMol, our molecular modeling and visualization software tool (http://cvcweb.ices.utexas.edu/software/). A substantial part of this work in this paper was done when Guoliang Xu was visiting Chandrajit Bajaj at UT-CVC. His visit was additionally supported by the J. T. Oden ICES visitor fellowship.
Appendix
A. Proof of Theorem 2.1
Proof
It is obvious that S is C1 at the vertices. For the continuity at the midpoints of edges, let us consider the edge vivj in triangle [vivjvk]. On the edge vivj, b3 = 0. So we may let b2 = t and b1 = 1 − t. Then matrix M can be written as
and
where A = vi(λ) − vk(λ), B = vj(λ) − vk(λ) and C(t) = ni + t(nj − ni). Therefore on the edge vivj,
The gradient of F on the edge vivj can be written as
| (20) |
When , therefore
Consider the function inside the square bracket of (20) and denote it as F1. Then
| (21) |
Since on the edge vivj, , substituting (15) into (21), we get F1 is 0. Therefore, at the midpoint
| (22) |
So S is C1 continuous at the midpoints of the edges.
B. Proof of Theorem 2.2
Proof
It is obvious that S is C1 within the triangles. By Theorem 2.1 we have already known that S is C1 at the vertices and the midpoints of the edges. Here we only need to show S is C1 at any points of the edges, let us consider the the edge vivj in the triangle [vivjvk].
Under the condition ni · (vi − vj) = nj · (vj − vi), we have b120 = b210, so (20) is written as
| (23) |
Similar as (12), we define
| (24) |
By (15) together with the facts that b120 = b210 and on edge vivj, we have
| (25) |
where d3(λ) is defined in (14). Plug (24) and (25) in (23), we get
| (26) |
Consider the function inside the square bracket of (26) and denote it as F2. Our goal is to show that F2 = 0. Since we have already known that when , F2 = 0, this prompts us to compute the derivative of F2 with respect to t and see if the derivative is 0. We observe that both the numerator of the denominator of F2 are linear in terms of t, so F2 is of the form with
In order to show , which is equivalent to show N:= ad − bc = 0, we compute
| (27) |
Under the condition ni · (vi − vj) = nj · (vj − vi), we have (B − A)Tc = (vj(λ) − vi(λ))Tc = 0, where . Therefore
| (28) |
and
| (29) |
Plug (28) and (29) into (27) and divide both sides by ||vj(λ) − vi(λ)||2, we get
| (30) |
If ni = nj, (30) is 0. Now let us assume ni ≠ nj. Recall that . we define another vector and let D = B − A. Then c is orthogonal to e and D:
| (31) |
Furthermore
| (32) |
By the definition of c and e,
| (33) |
Substitute (33) into (30) and replace A × B with A × D, we get
| (34) |
If e and D are linearly dependent, then e × D = 0, moreover e(A × D) = 0, which yields F3 = 0. Otherwise, we introduce a new matrix
Since c, e, and D are linearly independent, M is nonsingular. So F3 (a vector) is equal to
By the Lagrange’s formula:
| (35) |
and (32), (35) is zero and thus F3 = 0. So far we have proved that F2 is independent of t. Meanwhile in the proof of Theorem 2.1, we know that F2 = 0 at . Hence F2 = 0 for all t and therefore on the edge vivj, ∇F is
So S is C1 on the edges.
C. Proof of Theorem 2.3
Proof
As same as the proof of Theorem 2.2, we only need to show that S is C1 on the edge vivj. In the proof of Theorem 2.1, we have already derived the gradient function on the edge vivj (20):
Let
| (36) |
Following the same idea of the proof the Theorem 2.2, we compute . The numerator of is
| (37) |
Since
(37) is 0 when ni = nj. So F4 is independent of t. By the proof of Theorem 2.1, F4 = 0 at . So F4 = 0 for all t. So S is C1 continuous.
Contributor Information
Wenqi Zhao, Email: wzhao@ices.utexas.edu, The Institute for Computational Engineering and Science, University of Texas at Austin.
Chandrajit Bajaj, Email: bajaj@ices.utexas.edu, The Institute for Computational Engineering and Science, University of Texas at Austin.
Guoliang Xu, Email: xuguo@lsec.cc.ac.cn, The Institute of Computational Mathematics and Scientific/Engineering Computing, Chinese Academy of Sciences.
References
- 1.Karplus Martin, Andrew McCammon J. Molecular dynamics simulations of biomolecules. Nature Structural Biology. 2002;9:646–652. doi: 10.1038/nsb0902-646. [DOI] [PubMed] [Google Scholar]
- 2.Srinivasan J, Cheatham TE, Cieplak P, Kollman PA, Case DA. Continuum solvent studies of the stability of dna, rna, and phosphoramidate-dna helices. J Am Chem Soc. 1998;120:9401–9409. [Google Scholar]
- 3.Kuhn B, Kollman PA. A ligand that is predicted to bind better to avidin than biotin: insights from computational fluorine scanning. J Am Chem Soc. 2000;122:3909–3916. [Google Scholar]
- 4.Nina M, Beglov D, Roux B. Atomic radii for continuum electrostatics calculations based on molecular dynamics free energy simulations. J Phys Chem B. 1997;101:5239–5248. [Google Scholar]
- 5.Roux B, Simonson T. Implicit solvent models. Biophysical Chemistry. 1999;78:1–20. doi: 10.1016/s0301-4622(98)00226-9. [DOI] [PubMed] [Google Scholar]
- 6.Schaefer M, Karplus M. A comprehensive analytical treatment of continuum electrostatics. J Phys Chem. 1996;100:1578–1599. [Google Scholar]
- 7.Baker N, Holst M, Wang F. Adaptive multilevel finite element solution of the poisson-boltzmann equation ii. refinement at solvent-accessible surfaces in biomolecular systems. J Comput Chem. 2000;21:1343–1352. [Google Scholar]
- 8.Madura JD, Briggs JM, Wade RC, Davis ME, Luty BA, Ilin A, Antosiewicz J, Gilson MK, Bagheri B, Scott LR, McCammon JA. Electrostatics and diffusion of molecules in solution: simulations with the university of houston brownian dynamics program. Computer Physics Communications. 1995;91:57–95. [Google Scholar]
- 9.Still WC, Tempczyk A, Hawley RC, Hendrickson T. Semianalytical treatment of solvation for molecular mechanics and dynamics. J Am Chem Soc. 1990;112:6127–6129. [Google Scholar]
- 10.Bashford D, Case DA. Generalized born models of macromolecular solvation effects. Annu Rev Phys Chem. 2000;51:129–152. doi: 10.1146/annurev.physchem.51.1.129. [DOI] [PubMed] [Google Scholar]
- 11.Lee MS, Feig M, Salsbury FR, Brooks CL. New analytic approximation to the standard molecular volume definition and its application to generalized born calculations. J Comput Chem. 2003;24:1348–1356. doi: 10.1002/jcc.10272. [DOI] [PubMed] [Google Scholar]
- 12.Ghosh A, Rapp CS, Friesner RA. Generalized born model based on a surface integral formulation. J Phys Chem B. 1998;102:10983–10990. [Google Scholar]
- 13.Bajaj C, Siddavanahalli V, Zhao W. Fast algorithms for molecular interface triangulation and solvation energy computations. 2007 ICES Technical Report TR-07-06. [Google Scholar]
- 14.Lee B, Richards FM. The interpretation of protein structure: estimation of static accessiblilty. J Mol Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- 15.Connolly ML. Analytical molecular surface calculation. J Appl Cryst. 1983;16:548–558. [Google Scholar]
- 16.Richards FM. Areas, volumes, packing, and protein structure. Annu Rev Biophys Bioeng. 1977;6:151–176. doi: 10.1146/annurev.bb.06.060177.001055. [DOI] [PubMed] [Google Scholar]
- 17.Grant JA, Pickup BT. A gaussian description of molecular shape. J Phys Chem. 1995;99:3503–3510. [Google Scholar]
- 18.Lee MS, Salsbury FR, Brooks CL. Novel generalized born methods. J Chemical Physics. 2002;116:10606–10614. [Google Scholar]
- 19.Bajaj C, Castrillon-Candas J, Siddavanahalli V, Xu Z. Compressed representations of macromolecular structures and properties. Structure. 2005;13:463–471. doi: 10.1016/j.str.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 20.Bajaj C, Siddavanahalli V. Fast error-bounded surfaces and derivatives computation for volumetric particle data. 2006 ICES Technical Report TR-06-06. [Google Scholar]
- 21.Bajaj C, Lee H, Merkert R, Pascucci V. Nurbs based b-rep models from macromolecules and their properties. Proceedings of Fourth Symposium on Solid Modeling and Applications; 1997. pp. 217–228. [Google Scholar]
- 22.Edelsbrunner H. Deformable smooth surface design. Discrete Computational Geometry. 1999;21:87–115. [Google Scholar]
- 23.Cheng H, Shi X. Guaranteed quality triangulation of molecular skin surfaces. IEEE Visualization. 2004:481–488. [Google Scholar]
- 24.Cheng H, Shi X. Quality mesh generation for molecular skin surfaces using restricted union of balls. IEEE Visualization. 2005:51–58. [Google Scholar]
- 25.Dahmen W. Smooth piecewise quadratic surfaces. In: Lyche T, Schumaker L, editors. Mathematical methods in computer aided geometric design. Academic Press; Boston: 1989. pp. 181–193. [Google Scholar]
- 26.Guo B. PhD thesis. Cornell University; 1991. Modeling arbitrary smooth objects with algebraic surfaces. [Google Scholar]
- 27.Dahmen W, Thamm-Schaar TM. Cubicoids: modeling and visualization. Computer Aided Geometric Design. 1993;10:89–108. [Google Scholar]
- 28.Bajaj C, Chen J, Xu G. Modeling with cubic A-patches. ACM Transactions on Graphics. 1995;14:103–133. [Google Scholar]
- 29.Akkiraju N, Edelsbrunner H. Triangulating the surface of a molecule. Discrete Applied Mathematics. 1996;71:5–22. [Google Scholar]
- 30.Laug P, Borouchaki H. Molecular surface modeling and meshing. Engineering with Computers. 2002;18:199–210. [Google Scholar]
- 31.Zhang Y, Xu G, Bajaj C. Quality meshing of implicit solvation models of biomolecular structures. Computer Aided Geometric Design. 2006;23:510–530. doi: 10.1016/j.cagd.2006.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bajaj C, Siddavanahalli V. An adaptive grid based method for computing molecular surfaces and properties. 2006 ICES Technical Report TR-06-57. [Google Scholar]
- 33.Bajaj C, Djeu P, Siddavanahalli V, Thane A. Texmol: Interactive visual exploration of large flexible multi-component molecular complexes. Proc. of the Annual IEEE Visualization Conference; 2004. pp. 243–250. [Google Scholar]
- 34.Bajaj C, Xu G, Holt R, Netravali A. Hierarchical multiresolution reconstruction of shell surfaces. Computer Aided Geometric Design. 2002;19:89–112. [Google Scholar]
- 35.Nielson G. The side-vertex method for interpolation in triangles. J Apprrox Theory. 1979;25:318–336. [Google Scholar]
- 36.Dunavant D. High degree efficient symmetrical gaussian quadrature rules for the triangle. International Journal for Numerical Methods in Engineering. 1985;21:1129–1148. [Google Scholar]