

Abstract
We have developed an experimental model of spontaneous intracranial hemorrhage (ICH) in transgenic mice expressing human renin and human angiotensinogen (R + /A +) treated with high-salt diet and Nω-nitro-l-arginine methyl ester (l-NAME). We investigated whether oxidative stress is associated with spontaneous ICH in R + /A + mice. R + /A + mice on high-salt diet and l-NAME presented neurologic signs 57±13 (mean±s.e.m.) days after the start of treatment. Intracranial hemorrhage was shown with histologic examination. Levels of superoxide in brain homogenate were significantly increased in R + /A + mice with ICH (118±10RLU per sec per mg; RLU, relative light unit) compared with age-matched control mice (19±1) and R + /A + mice without ICH (53±3). NAD(P)H oxidase activity was significantly higher in R + /A + mice with ICH (34,933±2,420 RLU per sec per mg) than in control mice (4,984±248) and R + /A + mice without ICH (15,069±917). These results suggest that increased levels of superoxide are due, at least in part, to increased NAD(P)H oxidase activity. Increased NAD(P)H oxidase activity preceded signs of ICH, and increased further when R+ /A + mice developed ICH. These findings suggest that oxidative stress may contribute to spontaneous ICH in chronic hypertension.
Keywords: hypertension, intracranial hemorrhage, oxidative stress
Introduction
Spontaneous intracranial hemorrhage (ICH) is a devastating type of stroke that accounts for 10% to 15% of all strokes, and is associated with high mortality and morbidity (Xi et al, 2006). Hypertension is an important risk factor for ICH. Mechanisms that lead to ICH during hypertension and the pathogenesis of brain injury after ICH, however, remain poorly understood.
Recently, we have developed the first experimental model of spontaneous ICH in hypertensive mice: double transgenic mice with overexpression of human renin and human angiotensinogen (R + /A +) are treated with Nω-nitro-l-arginine methyl ester (l-NAME; an inhibitor of nitric oxide synthase (NOS)) and high-salt diet (Iida et al, 2005). These mice developed accelerated hypertension with ICH in basal ganglia, brainstem, and cerebellum, as well as in cerebral cortex. These stroke-prone hypertensive mice are useful to study fundamental mechanisms that lead to ICH.
An important mechanism of vascular injury in hypertension is oxidative stress (Heistad, 2006). Oxidative stress, produced in part by NAD(P)H oxidase, induces dysfunction and cell death of endothelium and vascular muscle (Bhunia et al, 2002; Burlacu et al, 2001; Didion and Faraci, 2003). Angiotensin II activates NAD(P)H oxidase and thereby generates high levels of superoxide in cerebral vessels as well as in the central nervous system (Hanna et al, 2002; Didion and Faraci, 2003; Zimmerman et al, 2004). In addition, several studies suggest that activation of matrix metalloproteinases (MMPs) is redox-sensitive (Gu et al, 2002; Rajagopalan et al, 1996b). Matrix metalloproteinases are a family of zinc endopeptidases involved in the degradation of basal lamina and extracellular matrix (Woessner, 1991). Expression and activity of MMP-9 are increased in cerebral microvessels in rats with chronic hypertension (Liebetrau et al, 2005). Thus, in our model of ICH in double transgenic R + /A + mice, we hypothesize that oxidative stress in intracranial vessels may predispose to ICH.
Oxidative stress may also contribute to brain injury after ICH. Superoxide, visualized by in situ detection of oxyethidium, is increased in the perihematomal region after ICH (Wang and Tsirka, 2005). Activation of NAD(P)H oxidase contributes to brain injury after ICH (Tang et al, 2005). Previous studies were performed after injection of bacterial collagenase into the brain to produce ICH (Tang et al, 2005; Wang and Tsirka, 2005). However, no studies have examined the role of oxidative stress in spontaneous ICH.
The major goal of this study was to determine whether generation of superoxide and activation of NAD(P)H oxidase are associated with spontaneous ICH during chronic hypertension in R + /A + mice.
Materials and methods
Experimental Animals
Studies were conducted in female double transgenic mice (R + /A +), which were generated by cross-breeding human renin (R + ) transgenic mice with human angiotensinogen (A + ) transgenic mice (Merrill et al, 1996). Nontransgenic (R−/A−) and single transgenic littermate mice (R + /A− or R−/A + ) were used as controls for R + /A + mice, because strict specificity in the enzymatic reaction exists between the mouse and human renin–angiotensinogen system (Merrill et al, 1996). Breeding and genotyping of R + /A + mice and their control mice were performed in the transgenic animal facility located in a virus- and pathogen-free animal-care facility.
We also studied male wild-type (C57BL/6) mice and gp91phox-deficient mice (C57BL/6 background) that lack the membrane gp91phox (Nox2) subunit of NAD(P)H oxidase. The gp91phox-deficient and C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA) and were housed in the Animal Care Facility at the University of Iowa.
All mice were housed in individual cages and kept on a 12-h light/dark cycle (light from 0600 to 1800 hours). All experimental protocols were approved by the Institutional Animal Care and Use Committee at the University of Iowa.
Spontaneous Intracranial Hemorrhage
Mice developing spontaneous ICH were generated as described previously (Iida et al, 2005). Briefly, female R + /A + and control mice (4 months old) were given an 8% high-salt diet (TD-92012; Harlan Teklad, Madison, WI, USA) and 100 to 120 mg/kg l-NAME per day in drinking water to increase blood pressure (Iida et al, 2005).
Systolic blood pressure (SBP) was measured using a tail cuff (Vistech System BP-2000, Apex, NC, USA). R + /A + and control mice on treatment were trained for 3 consecutive days in the prewarmed tail-cuff device, and then SBP was measured for 2 days. On each day, 30 measurements were obtained and averaged for each mouse. Averaged values of SBP were measured at baseline and once every month for 24 weeks.
Clinical signs of stroke were assessed by daily neurologic examinations, including contralateral forelimb extension, circling behavior, or other motor dysfunction (Garcia et al, 1995). When an R + /A + mouse developed these signs, the mouse was euthanized with Nembutal (150 mg/kg intraperitoneally), and an age-matched R + /A + mouse without signs of stroke and an age-matched control mouse were euthanized. The mice were perfused in situ for 10 mins through a cannula in the left ventricle with phosphate-buffered 0.9% saline (PBS) at about 70 mm Hg. Brains were cut sagittally into half and analyses were conducted according to several procedures described below.
Half of the brain of each mouse was immersion-fixed in 7% formaldehyde for histometric analysis. Paraffin-embedded tissue was serially sectioned at 5 μm and stained with hematoxylin and eosin (H&E). To facilitate precise identification of ICH, one section of every five serial sections was stained with DAB (diaminobenzidine), which reacts with peroxidases in red blood cells. Diaminobenzidine highlights hemorrhages, leaving non-hemorrhagic areas unstained. We have found that this method is much more sensitive in detecting ICH, and thus allows far more accurate quantification of ICH, than examining sections stained with H&E. All H&E- and DAB-stained sections were screened using a light microscope, and each section was scanned in toto using an Aperio Scanscope System (Model T3; Aperio, Vista, CA, USA) to create a digital image with a final resolution of 0.5 μm/pixel (equivalent to a final magnification × 200) of the entire section. Digital images were analyzed with Image J software (NIH) to identify the location and quantify the number of hemorrhages.
Fresh, unfixed half brains were used to evaluate oxidative stress by lucigenin-enhanced chemiluminescence and to estimate mRNA levels by real-time reverse-transcription PCR. These unfixed half brains were cut and divided into four parts at 3 mm intervals from anterior pole to cerebellum. The most anterior quarter of brain contained cerebral cortex and anterior part of striatum. The second anterior quarter of brain contained cerebral cortex, posterior part of striatum, and anterior part of thalamus and hippocampus. The third anterior quarter of brain contained posterior part of thalamus and hippocampus, and upper part of brainstem. The most posterior quarter of brain contained cerebellum and middle and lower part of brainstem (Franklin and Paxinos, 1997).
Striatal Injection of Blood
To determine whether ICH itself produces oxidative stress in the brain, we examined the levels of superoxide and the activity of NAD(P)H oxide after injection of blood in the striatum of male gp91phox-deficient mice and C57BL/6 mice (4 months old). The gp91phox-deficient mice were used to investigate whether Nox2 is associated with the oxidative stress after ICH. Mice were placed in a stereotactic frame, and a 27-gauge stainless-steel cannula was introduced through a burr hole into each striatum (2 mm lateral to midline, 1 mm anterior to bregma, and depth 4 mm below the surface of the skull). Each mouse received a 4-μL injection over 5 mins of autologous whole-blood into the left striatum and artificial cerebrospinal fluid (aCSF) (temperature 371C; ionic composition (in mmol/L): 132 NaCl, 2.95 KCl, 1.71 CaCl2, 0.65 MgCl2, 24.6 NaHCO3, 3.69 d-glucose) into the right striatum. Nonheparinized blood was taken from the tail vein of a mouse and injected immediately into the striatum. Rectal temperature was maintained at 36°C to 37°C with a heating pad. In some studies, C57BL/6 mice were treated immediately after the injection of blood with apocynin (3 mg/kg intraperitoneally), a drug that inhibits NAD(P)H oxidase activity by interfering with binding of the cytosolic subunit of p47phox to the membranous subunit of gp91phox (Williams and Griendling, 2007).
The injection cannula was slowly withdrawn 10 mins after the injection of blood or aCSF. The wound was sutured, and the mouse was returned to its cage with free access to normal food and water. Twenty-four hours later, mice were euthanized by an injection of overdose of Nembutal (150 mg/kg intraperitoneally) and perfused transcardially with PBS as described above. The brain was immediately removed and cut coronally into a section 2 mm anterior and posterior to the injection site. The brain sections with injected blood and aCSF were used for the measurements described below.
Evaluation of Oxidative Stress
Superoxide levels were quantified with lucigenin-enhanced chemiluminescence. Fresh, unfixed brains were homogenized and sonicated in PBS containing protease inhibitors (protease inhibitor cocktail, Complete Mini; Roche Diagnostic, Mannheim, Germany) at 0°C to 5°C. Brain homogenates were placed in 0.5 mL PBS and 5 μmol/L lucigenin, preincubated for 60 mins at room temperature with or without 1 mmol/L tiron (a superoxide dismutase mimetic) or 100 μmol/L diphenyliodinium (DPI; an inhibitor of flavoprotein-containing enzymes, including NAD(P)H oxidases). Relative light units (RLUs) were measured for 10 mins. Background counts were determined and subtracted, and values were normalized per 1 mg brain tissue. NAD(P)H oxidase activity was estimated by adding NADPH (1, 10, or 100 mmol/L) to brain homogenates with or without addition of tiron (1 mmol/L). Under all conditions, the tiron-inhibitable chemiluminescence value (RLU per sec per mg brain tissue) was used as a measure of superoxide levels.
Dihydroethidium (DHE), an oxidative fluorescent dye, was used to localize superoxide in brain, in situ, after intrastitial injection of blood. Some mice were treated with DHE (300 μL; stock solution of DHE: 20 mmol/L in dimethylsulfoxide, diluted to 3.3 mmol/L in PBS just before use). Dihydroethidium solution was injected intraperitoneally 3 h before the mice were euthanized. In these mice treated with DHE, we did not use brain tissue for estimating superoxide levels using lucigenin-enhanced chemiluminescence because DHE was reported to be neuroprotective and reduce superoxide in mice after stroke (Yu et al, 2003). Fresh, unfixed brains were frozen in TBS compound (Triangle Biomedical Sciences, Durham, NC, USA). Coronal sections (20-μm thick) were cut in a cryostat and placed on glass slides, and images were obtained using the Bio-Rad MRC-1024 laser (krypton/argon)-scanning confocal microscope. The fluorescence excitation/emission spectra for ethidium bromide used during the imaging process were 488 and 610 nm, respectively. Fluorescence was detected with a 585-nm long-pass filter.
Statistics
Results are expressed as mean±s.e.m. Repeated-measures ANOVA followed by Scheffé test was used for comparison of multiple groups. Mann–Whitney’s U test was used for comparison of two groups. A probability value of P < 0.05 was considered significant.
Results
Spontaneous Intracranial Hemorrhage
R + /A + mice presented neurologic signs 57±13 (mean±s.e.m.) days after the start of feeding high-salt diet and l-NAME. None of the control mice developed neurologic signs.
Systolic blood pressure
In R + /A + mice with ICH, SBP was higher at baseline (4 months of age) than in control mice. In R + /A + mice without ICH, SBP also tended to be higher at baseline than in control mice (P=0.07) (Table 1). R + /A + mice developed a progressive increase in SBP compared with control mice within 4 to 8 weeks of treatment with high-salt diet and l-NAME. In addition, SBP was much higher in R + /A + mice with ICH than in R + /A + mice without ICH (Table 1). Seven R + /A + mice with ICH developed marked increases in SBP (204±7 mm Hg, mean±s.e.m.) at 6±1 days (mean±s.e.m.) before they showed signs of stroke. One mouse developed ICH only 9 days after high-salt diet and l-NAME were started, before we measured SBP during this treatment.
Table 1.
Systolic blood pressure in control mice, R+/A+ mice without ICH, and R+/A+ mice with ICH
| Baseline | 4 weeks | 8 weeks | 12 weeks | |
|---|---|---|---|---|
| Control mice | 108±4 (8) | 117±3 (5) | 123±4 (5) | 126±2 (3) |
| R+/A+ ICH(−) | 120±6 (8) | 146±6 (5)** | 160±5 (5)** | 173±7 (3)** |
| R+/A+ ICH(+) | 124±4 (8)* | 179±7 (5)**,† | 208±5 (5)**,† | 216±4 (3)**,† |
ICH, intracranial hemorrhage; R+/A+, human renin and human angiotensinogen expressing mice.
Values are mean ± s.e.m.; number of mice in parentheses.
R+/A+ ICH(−), R+/A+ mice without ICH; R+/A+ IVH(+), R+/A+ mice with ICH.
P < 0.05
P < 0.01 compared with control mice at each time point.
P < 0.01 compared with R+/A+ ICH(−) at each time point.
Histometric analysis of the brain
All R + /A + mice with neurologic signs had multiple ICH, shown in H&E- and DAB-stained sections (Figures 1A to 1H). Hemorrhagic legions were mainly located in the brain stem, although ICHs were distributed widely in the brain including the cerebral cortex, basal ganglia, and cerebellum (Figure 1K). R + /A + mice with neurologic signs also had a small number of ischemic infarcts in the brain (Figure 1L). Most ischemic infarcts were separated from hemorrhagic lesions (Figure 1I), both longitudinally and horizontally, but a few ischemic infarcts colocalized with hemorrhagic lesions (Figure 1J). None of the control mice or R + /A + mice without signs of stroke had ICH or ischemic infarcts.
Figure 1.

Histologic changes in brains of R + /A + mice with signs of stroke. (A, E) Cerebral cortex, (B, F) basal ganglia, (C, G) brain stem, (D, H) cerebellum. (A to D) Hematoxylin and eosin staining, (E to H) DAB staining. (I) Ischemic lesion in brain stem. (J) Colocalization of ICH (arrowhead) and ischemic lesions (arrows) in brain stem. (K) Total number of hemorrhagic lesions (n = 5 mice). (L) Total number of ischemic lesions (n = 5 mice). Scale bars = 100 μm.
Superoxide levels
In R + /A + mice with ICH, lucigenin-enhanced chemiluminescence signals in each quarter of brain were significantly attenuated by preincubation with tiron (Figure 2). In R + /A + mice without ICH, tiron reduced chemiluminescence signals in three of four quarter-parts of the brain (Figure 2). In control mice, chemiluminescence signals were reduced only in the posterior quarter of the brain (Figure 2). Diphenyliodinium significantly reduced chemiluminescence signals in each quarter of the brain in R + /A + mice with and without ICH. Chemiluminescence signals were decreased in posterior half of the brain in control mice (Figure 2).
Figure 2.

Effects of tiron and DPI on lucigenin-enhanced chemiluminescence signals in brain tissue of control mice, R + /A + mice without ICH (R + /A + ICH(−)), and R + /A + mice with ICH (R + /A + ICH(+)). (A) Anterior quarter of brain, (B) second quarter of brain, (C) third quarter of brain, and (D) posterior quarter of brain. Values are means±s.e.m. (n = 8 in each group); RLU, relative light units. *P < 0.05, **P < 0.01 versus respective brain tissue.
Levels of superoxide under basal conditions, as measured using tiron-inhibitable chemiluminescence, were significantly higher in R + /A + mice with ICH than in control mice and R + /A + mice without ICH (Figure 3). The increase in superoxide was observed in each quarter of the brain in R + /A + mice with ICH. In the most posterior quarter of the brain of R + /A + mice with ICH, where spontaneous ICH was frequently observed, superoxide levels were greater than in the other parts of the brain (P < 0.01). In R + /A + mice without ICH, superoxide levels were also significantly higher than in control mice in each quarter of the brain (Figure 3).
Figure 3.
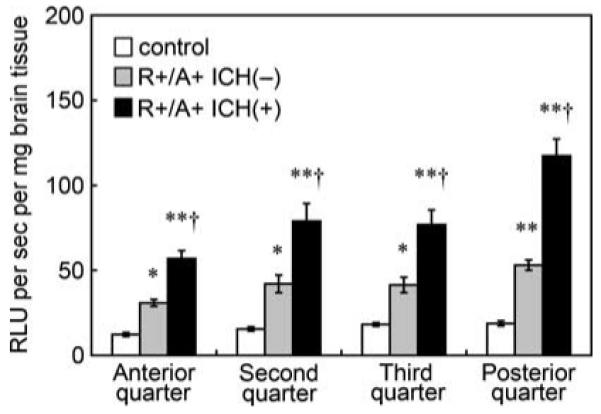
Basal levels of superoxide, calculated from Figure 2 as tiron-inhibitable signal, in brain tissue of control mice, R + /A + mice without ICH (R + /A + ICH(−)), and R + /A + mice with ICH (R + /A + ICH(+)). Values are means±s.e.m. (n =8 in each group); RLU, relative light units. *P < 0.05, **P < 0.01 versus respective control mice. †P < 0.01 versus respective R + /A + mice without ICH.
NAD(P)H oxidase activity
NAD(P)H oxidase activity, estimated as levels of superoxide using tiron-inhibitable chemiluminescence after adding NADPH, was higher in R + /A + mice with ICH than in control mice and in R + /A + mice without ICH (Figure 4). In R + /A + mice with ICH, NAD(P)H oxidase activity of the most posterior quarter of brain was significantly higher than in the anterior and second quarter-parts of the brain (P < 0.05). NAD(P)H oxidase activity was also higher in R + /A + mice without ICH than in control mice in all the quarters of the brain.
Figure 4.

NAD(P)H oxidase activity in brain tissue of control mice, R + /A + mice without ICH (R + /A + ICH(−)), and R + /A + mice with ICH (R + /A + ICH(+)). (A) Anterior, (B) second, (C) third and (D) posterior quarters of brain. Values are means±s.e.m. (n = 8 in each group); RLU, relative light units. *P < 0.05, **P < 0.01.
To examine the mechanisms that contributed to increased basal levels of superoxide in R + /A + mice with and without ICH, we measured the expression of the catalytic subunits of NAD(P)H oxidase (Nox2 and Nox4). Measurements were made in the posterior quarter of the brain in which basal levels of superoxide were greater than in other parts of the brain in R + /A + mice with ICH. In contrast to evidence for increased activity of NAD(P)H oxidase in R + /A + mice with and without ICH, quantitative real-time reverse-transcription PCR showed no significant difference in the expression of Nox2 and Nox4 among the three groups (data not shown).
Striatal Blood Injection
Superoxide levels
We used two methods to detect superoxide after injection of blood in the striatum: tiron-inhibitable chemiluminescence and DHE staining. In wild-type mice and in wild-type mice treated with apocynin, lucigenin-enhanced chemiluminescence was greater in slices of brain after the injection of blood than in slices after the injection of aCSF (Figure 5A). In gp91phox-deficient mice, there was no significant difference in lucigenin-enhanced chemiluminescence between brain sections after injection of blood and after injection of aCSF (P = 0.09). The increase in chemiluminescence after injection of blood was, however, significantly reduced by tiron or DPI in all groups (Figure 5A).
Figure 5.

(A) Effects of tiron and DPI on lucigenin-enhanced chemiluminescence signals in brain tissue of wild-type mice, wild-type mice treated with apocynin, and gp91phox-deficient mice after injection of blood or aCSF in striatum (n = 8 in each group). *P < 0.01 versus respective blood-injected brain tissue. †P < 0.01 versus respective aCSF-injected brain tissue. (B) Basal levels of superoxide, estimated as tiron-inhibitable signal, in brain tissue of wild-type mice, wild-type mice treated with apocynin, and gp91phox-deficient mice after injection of blood or aCSF in striatum (n = 7 to 10 in each group). †P < 0.05, ††P < 0.01 versus respective aCSF-injected brain tissue. *P < 0.05 versus blood-injected brain tissue of wild-type mice. (C) Superoxide (DHE fluorescence) in brain (striatum) of wild-type mice after injection of aCSF or blood. Hematoma is dark, and there was intense red fluorescence around the hematoma. Scale bars = 200 μm. (D) NAD(P)H oxidase activity in brain tissue of wild-type mice, wild-type mice treated with apocynin, and gp91phox-deficient mice after injection of blood or aCSF in striatum (n = 8 in each group). *P < 0.05, **P < 0.01. Values are means±s.e.m. RLU, relative light units.
In wild-type mice, basal levels of superoxide, as measured using tiron-inhibitable chemiluminescence, were significantly higher in the blood-injected slices of brain than in aCSF-injected slices (Figure 5B). There was no significant difference in basal levels of superoxide in blood-injected brain between male and female wild-type mice in a study of eight male and four female mice (data not shown). In wild-type mice treated with apocynin, basal superoxide levels were also higher in blood-injected brain than in aCSF-injected brain, but the increase in superoxide was significantly attenuated compared with the wild-type mice that did not receive apocynin (Figure 5B). The increase in levels of superoxide after injection of blood was less in gp91phox-deficient mice than in wild-type mice, and there was no significant difference in levels of superoxide between brain sections after injection of blood and after injection of aCSF (P = 0.20) (Figure 5B).
Consistent with results obtained with lucigenin-enhanced chemiluminescence, DHE staining showed higher levels of superoxide in blood-injected brain than in aCSF-injected brain in wild-type mice. Increases in DHE fluorescence were restricted to the area of needle insertion in aCSF-injected brain, whereas enhanced DHE fluorescence was widely distributed around the intraparenchymal hematoma after injection of blood (Figure 5C).
NAD(P)H oxidase activity
NAD(P)H oxidase activity was higher after injection of blood than after aCSF in both apocynin-treated wild-type mice and in mice that did not receive apocynin (Figure 5D). The increase in NAD(P)H oxidase activity after injection of blood was largely prevented by apocynin (Figure 5D). In gp91phox-deficient mice, the increase in NAD(P)H oxidase activity after injection of blood was less than in wild-type mice, and there was no significant difference in NAD(P)H oxidase activity between brain sections after injection of blood and after injection of aCSF (P = 0.11) (Figure 5D).
In contrast to the evidence for increased activity of NAD(P)H oxidase in brain sections after injection of blood, expressions of Nox2 mRNA and Nox4 mRNA were not different between brain sections after injection of blood and after injection of aCSF in wild-type mice (data not shown).
Discussion
There are several major findings in this study. First, basal superoxide levels were significantly higher in brains from R + /A + mice treated with high-salt diet and l-NAME than in control mice, even preceding ICH. This finding is compatible with the hypothesis that enhanced superoxide may play a role in the pathogenesis of hypertensive ICH. Second, preincubation with DPI markedly reduced the lucigenin-enhanced chemiluminescence signals in brain tissue from R + /A + mice with and without ICH. The effect of DPI, as well as NADPH-stimulated increases in superoxide levels, suggests that an increase in NAD(P)H oxidase activity may account for increases in basal levels of superoxide in R + /A + mice without ICH, and greater increase in basal levels of superoxide in R + /A + mice with ICH. Third, basal levels of superoxide were higher in brains from R + /A + mice with ICH than without ICH. Moreover, injection of blood in the striatum increased levels of superoxide. These findings suggest that extravascular blood is a source of oxidative stress after ICH.
Experimental Model of Spontaneous Intracranial Hemorrhage
We have previously observed spontaneous ICH in R + /A + mice treated with high-salt diet and l-NAME (Iida et al, 2005). R + /A + mice are transgenic mice that express human renin and angiotensinogen genes in many tissues, including brain, kidney, and liver. The mice have fourfold higher plasma levels of angiotensin II than normal mice and are chronically hypertensive, with mean arterial pressure of about 150 to 160 mm Hg (Merrill et al, 1996). We found that R + /A + mice treated with high-salt diet and l-NAME have augmented hypertension and spontaneously developed ICH. In R + /A + mice on high-salt diet and l-NAME, a small number of ischemic infarcts developed in the brain, and many ICHs were observed in the brainstem, cerebellum, and basal ganglia, as well as in the cerebral cortex. Thus, R + /A + mice are an excellent genetic model to study mechanisms that lead to ICH associated with hypertension.
Superoxide and Hypertension
Treatment of R + /A + mice with high-salt diet and l-NAME not only augmented hypertension, but also increased basal levels of superoxide, compared with control mice. We did not measure levels of superoxide in R + /A + mice on normal diet in this study, but a previous study from our lab showed that R + /A + mice on normal diet have an increased level of superoxide in cerebral vessels (Faraci et al, 2006). For two reasons it is likely that the levels of superoxide in R + /A + mice on normal diet are lower than in R + /A + mice on high-salt diet and l-NAME. First, the increase in blood pressure in R + /A + mice on normal diet is lower than in R + /A + mice on high-salt diet and l-NAME (Table 1). Because hypertension produced by the renin–angiotensin system is associated with increases in superoxide in blood vessels (Didion et al, 2002; Faraci et al, 2006), we expect that levels of superoxide may be lower in R + /A + mice on normal diet with lower arterial blood pressure. Second, despite lack of direct evidence for a relationship between levels of superoxide and severity of hypertension, severe hypertension may be associated with greater increases in oxidative stress than mild hypertension (Simic et al, 2006). Therefore, it seems reasonable to suggest that basal superoxide levels of R + /A + mice without ICH were increased even preceding ICH. These increases in superoxide appear to be produced in part by activation of NAD(P)H oxidase.
NAD(P)H oxidases are a major enzymatic source of superoxide in the systemic vasculature, and activity of NAD(P)H oxidase is increased in several pathophysiological states, including hypertension (Didion et al, 2002; Griendling et al, 2000; Rajagopalan et al, 1996a). NAD(P)H oxidase is also expressed and activated in cerebral blood vessels during chronic hypertension (Didion and Faraci, 2003; Girouard et al, 2007; Kazama et al, 2004; Paravicini et al, 2004).
We observed in this study that superoxide is markedly reduced by preincubation of brain tissue with DPI, an inhibitor of NAD(P)H oxidase. We are aware of limitations of use of DPI as an NAD(P)H oxidase inhibitor, and the suggestion that DPI also inhibits electron flow through other enzymes, including mitochondrial respiratory chain complex I, NADH reductase, and NOS (Li et al, 2003). We cannot exclude the possibility that mitochondrial enzyme is a major source of increased oxidative stress observed in our study. The possibility that NOS is the source of increased oxidative stress is unlikely in our study, because R + /A + mice received l-NAME, which is an NOS inhibitor. In addition, in R + /A + mice treated with high-salt diet and l-NAME, NADPH-stimulated superoxide levels were higher than in control mice. We expected, for three reasons, that expression of the catalytic subunits of NAD(P)H (Nox2, Nox4) would be increased in R + /A + mice treated with high-salt diet and l-NAME. First, Nox2 and Nox4 are the predominant isoforms that are expressed in the brain of mice (Infanger et al, 2006). Second, angiotensin II produces endothelial dysfunction in the cerebral microcirculation of mice through superoxide derived from Nox2-containing NAD(P)H oxidase (Girouard et al, 2007; Kazama et al, 2004). Third, Nox4-containing NAD(P)H oxidase is upregulated in the basilar artery of rats with chronic hypertension (Paravicini et al, 2004). We did not detect, however, any statistically significant differences among groups in the expression of Nox2 or Nox4 in this study. Nevertheless, our results suggest that increased basal levels of superoxide in R + /A + mice treated with high-salt diet and l-NAME were due, at least in part, to increased activity of NAD(P)H oxidase.
Role of Superoxide in Spontaneous Intracranial Hemorrhage
A major finding in this study was that basal levels of superoxide and NAD(P)H oxidase activity in brain tissue were greater in R + /A + with ICH than in R + /A + mice without ICH. Although there is no direct evidence that superoxide contributes to spontaneous ICH, accumulating evidence suggests that oxidative stress may be involved in spontaneous ICH.
Chronic oxidative stress, especially from activation of NAD(P)H oxidase, produces dysfunction of endothelium and smooth muscle in blood vessels and induces cell death (Bhunia et al, 2002; Burlacu et al, 2001; Didion and Faraci, 2003). Furthermore, activity of MMPs is upregulated by oxidative stress, including superoxide (Gu et al, 2002; Rajagopalan et al, 1996b). The MMPs are a family of zinc endopeptidases involved in the degradation of basal lamina and extracellular matrix (Woessner, 1991). They are also reported to disrupt the blood–brain barrier in experimental ischemia–reperfusion model with transient middle cerebral artery occlusion (Yang et al, 2006). Expression and activity of MMP-9 are increased in cerebral microvessels in rats with chronic hypertension (Liebetrau et al, 2005). In addition, permeability of the blood–brain barrier was increased when blood pressure was greatly, but not moderately, elevated (Tuor et al, 1986; Nukhet Turkel and Ziya Ziylan, 2004). In this study, we observed that ICH occurred in R + /A + mice with signs of stroke, in which there were greater increases in blood pressure in most mice (Table 1) and in basal levels of superoxide in all cases (Figure 3), than in other R + /A + mice without hemorrhages, in which there were only moderate increases in blood pressure and basal level of superoxide. Thus, it seems reasonable to suggest that increased MMP activity, through oxidative stress associated with severe hypertension perhaps beyond a threshold level of blood pressure, may facilitate proteolysis of extracellular matrix components of the basement membrane, contributing to the pathogenesis of ICH. Therefore, we speculate that increases in superoxide in R + /A + mice treated with high-salt diet and l-NAME may contribute to the development of spontaneous ICH.
Oxidative Stress after Intracranial Hemorrhage
The increase in superoxide levels in R + /A + mice with ICH may also reflect brain injury after ICH. Enhanced superoxide levels resulting from activation of NAD(P)H oxidase have been reported to be associated with brain injury after ICH in experimental studies (Tang et al, 2005; Wang and Tsirka, 2005). Increased superoxide levels were shown in the perihematoma region after injection of bacterial collagenase, and the authors suggested that superoxide may mediate ICH injury (Wang and Tsirka, 2005). Expression of the gp91phox subunit of NAD(P)H oxidase was upregulated in brain after injection of collagenase in wild-type mice (Tang et al, 2005). In addition, brain edema, neurologic deficit, and mortality rate were reduced after injection of collagenase in gp91phox-deficient mice, which suggested that oxidative stress from activation of NAD(P)H oxidase contributed to brain injury (Tang et al, 2005). In those studies, bacterial collagenase was injected in the brain. Collagenase dissolves extracellular matrix and blood vessel lamina, and produces ICH (Brown et al, 1995). Collagenase also produces inflammation, which may contribute to oxidative stress, because inflammatory cells can generate superoxide (Green et al, 2001; Wang and Tsirka, 2005).
To determine whether ICH itself, in the absence of bacterial collagenase, produces oxidative stress in the brain, we examined superoxide and NAD(P)H oxidase activity after injection of blood in the striatum. Basal superoxide levels and NAD(P)H oxidase activity were increased after injection of blood in wild-type mice. The increase in superoxide and NAD(P)H oxidase activity after injection of blood was significantly, but not completely, reduced in wild-type mice treated with apocynin and in gp91phox-deficient mice. For several reasons, we are aware of the possibility that an NAD(P)H oxidase containing a Nox subunit other than Nox2, as well as other sources beside NAD(P)H oxidase, may contribute to increases in chemiluminescence signals in brain sections after injection of blood. First, DPI significantly reduced lucigenin-enhanced chemiluminescence signals in blood-injected brain tissue of wild-type mice treated with apocynin and in brain tissue from gp91phox-deficient mice, as well as in blood-injected brain tissue of wild-type mice. Second, apocynin does not inhibit activation of Nox4, as Nox4 does not require the cytosolic subunit p47phox for its activation (Infanger et al, 2006). Third, endothelial NOS (eNOS) could also produce superoxide (Vásquez-Vivar et al, 1998), and DPI also inhibits eNOS-mediated generation of superoxide (Li et al, 2003). Nevertheless, these findings suggest that intraparenchymal blood per se produces an increase in superoxide due, at least in part, to activation of the gp91phox subunit of NAD(P)H oxidase.
We cannot exclude the possibility that brain infarction may affect basal levels of superoxide in R + /A + mice with ICH. The levels of superoxide were reported to increase in cerebral ischemic lesions produced by permanent as well as transient middle cerebral artery occlusion (Fujimura et al, 1999; Noshita et al, 2003). The ischemic lesions in R + /A + mice with ICH were, however, far smaller than the ischemic lesions produced by middle cerebral artery occlusion. In addition, the number of ischemic lesions was much less than hemorrhagic lesions in R + /A + mice with ICH. Thus, it is likely that ischemic lesions have much less effect than hemorrhagic lesions on basal levels of superoxide in R + /A + mice with ICH.
Limitations of Study
There are methodological limitations of the present study. First, angiotensin II without hypertension generates superoxide in vascular cells in vitro (Berry et al, 2000; Zhang et al, 1999). In addition, because long-term administration of l-NAME stimulates angiotensin-converting enzyme, l-NAME increases angiotensin II. Thus, it is not clear whether increases in basal levels of superoxide in R + /A + mice treated with high-salt diet and l-NAME are because of increases in blood pressure per se or mediated by angiotensin II. Second, we did not identify the specific blood vessel that produced spontaneous ICH. A variety of vascular changes may contribute to ICH in our model. It will be of great interest in future studies to examine precise mechanisms, including, perhaps, the activation of MMPs that account for spontaneous ICH during hypertension. Third, hemoglobin and its metabolites, as well as inflammation, can produce oxidative stress after ICH (Wagner et al, 2003). Thus, we cannot exclude the possibility that increased oxidative stress in R + /A + mice with ICH reflects at least in oxidative stress secondary to ICH.
Acknowledgements
We thank the University of Iowa Transgenic Facility for genotyping R + /A + mice used in this study. We also thank Arlinda A LaRose for typing the manuscript and Robert M Brooks II for technical assistance.
This study was supported by National Institutes of Health Grant nos. NS24621, HL55006, HL62984; funds provided by the VA Medical Service; and the Carver Research Program of Excellence.
References
- Berry C, Hamilton CA, Brosnan J, Magill FG, Berg GA, McMurray JJV, Domininiczak AF. Investigation into sources of superoxide in human blood vessels. Angiotensin II increases superoxide production in human internal mammary arteries. Circulation. 2000;101:2206–12. doi: 10.1161/01.cir.101.18.2206. [DOI] [PubMed] [Google Scholar]
- Bhunia AK, Schwarzmann G, Chatterjee S. GD3 recruits reactive oxygen species to induce cell proliferation and apoptosis in human aortic smooth muscle cells. J Biol Chem. 2002;277:16396–402. doi: 10.1074/jbc.M200877200. [DOI] [PubMed] [Google Scholar]
- Brown MS, Kornfeld M, Mun-Bryce S, Sibbitt RR, Rosenberg GA. Comparison of magnetic resonance imaging and histology in collagenase-induced hemorrhage in the rat. J Neuroimaging. 1995;5:23–33. doi: 10.1111/jon19955123. [DOI] [PubMed] [Google Scholar]
- Burlacu A, Jinga V, Gafencu AV, Simionescu M. Severity of oxidative stress generates different mechanisms of endothelial cell death. Cell Tissue Res. 2001;306:409–416. doi: 10.1007/s004410100424. [DOI] [PubMed] [Google Scholar]
- Didion SP, Faraci FM. Angiotensin II produces superoxide-mediated impairment of endothelial function in cerebral arterioles. Stroke. 2003;34:2038–42. doi: 10.1161/01.STR.0000081225.46324.AA. [DOI] [PubMed] [Google Scholar]
- Didion SP, Ryan MJ, Baumbach GL, Sigmund CD, Faraci FM. Superoxide contributes to vascular dysfunction in mice that express human renin and angiotensinogen. Am J Physiol Heart Circ Physiol. 2002;283:H1569–76. doi: 10.1152/ajpheart.00079.2002. [DOI] [PubMed] [Google Scholar]
- Faraci FM, Lamping KG, Modrick ML, Ryan MJ, Sigmund CD, Didion SP. Cerebral vascular effects of angiotensin II: new insights from genetic models. J Cereb Blood Flow Metab. 2006;26:449–55. doi: 10.1038/sj.jcbfm.9600204. [DOI] [PubMed] [Google Scholar]
- Franklin K, Paxinos G, editors. The mouse brain in stereotaxic coordinates. Academic Press; San Diego, CA: 1997. [Google Scholar]
- Fujimura M, Morita-Fujimura Y, Kawase M, Copin JC, Calagui B, Epstein CJ, Chan PH. Manganese superoxide dismutase mediates the early release of mitochondrial cytochrome c and subsequent DNA fragmentation after permanent focal cerebral ischemia in mice. J Neurosci. 1999;19:3414–22. doi: 10.1523/JNEUROSCI.19-09-03414.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia JH, Wagner S, Liu KF, Hu XJ. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Stroke. 1995;26:627–635. doi: 10.1161/01.str.26.4.627. [DOI] [PubMed] [Google Scholar]
- Girouard H, Park L, Anrather J, Zhou P, Iadecola C. Cerebrovascular nitrosative stress mediates neurovascular and endothelial dysfunction induced by angiotensin II. Arterioscler Thromb Vasc Biol. 2007;27:303–9. doi: 10.1161/01.ATV.0000253885.41509.25. [DOI] [PubMed] [Google Scholar]
- Green SP, Cairns B, Rae J, Errett-Baroncini C, Hongo JA, Erickson RW, Curnutte JT. Induction of gp91-phox, a component of the phagocyte NADPH oxidase, in microglial cells during central nervous system inflammation. J Cereb Blood Flow Metab. 2001;21:374–84. doi: 10.1097/00004647-200104000-00006. [DOI] [PubMed] [Google Scholar]
- Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- Gu Z, Kaul M, Yan B, Kridel SJ, Cui J, Strongin A, Smith JW, Liddington RC, Lipton SA. S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science. 2002;297:1186–90. doi: 10.1126/science.1073634. [DOI] [PubMed] [Google Scholar]
- Hanna IR, Taniyama Y, Szocs K, Rocic P, Griendling KK. NAD(P)H oxidase-derived reactive oxygen species as mediators of angiotensin II signaling. Antioxid Redox Signal. 2002;4:899–914. doi: 10.1089/152308602762197443. [DOI] [PubMed] [Google Scholar]
- Heistad DD. Oxidative stress and vascular disease. Arterioscler Thromb Vasc Biol. 2006;26:689–95. doi: 10.1161/01.ATV.0000203525.62147.28. [DOI] [PubMed] [Google Scholar]
- Iida S, Baumbach GL, Lavoie JL, Faraci FM, Sigmund CD, Heistad DD. Spontaneous stroke in a genetic model of hypertension in mice. Stroke. 2005;36:1253–8. doi: 10.1161/01.str.0000167694.58419.a2. [DOI] [PubMed] [Google Scholar]
- Infanger DW, Sharma RV, Davisson R. NADPH oxidase of the brain: distribution, regulation, and function. Antioxid Redox Signal. 2006;8:1583–96. doi: 10.1089/ars.2006.8.1583. [DOI] [PubMed] [Google Scholar]
- Kazama K, Anrather J, Zhou P, Girouard H, Frys K, Milner TA, Iadecola C. Angiotensin II impairs neurovascular coupling in neocortex through NADPH oxidase-derived radicals. Circ Res. 2004;95:1019–26. doi: 10.1161/01.RES.0000148637.85595.c5. [DOI] [PubMed] [Google Scholar]
- Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, Melendez JA, Robinson JP. DPI induces mitochondrial superoxide-mediated apotosis. Free Radic Biol Med. 2003;34:465–477. doi: 10.1016/s0891-5849(02)01325-4. [DOI] [PubMed] [Google Scholar]
- Liebetrau M, Burggraf D, Wunderlich N, Jager G, Linz W, Hamann GF. ACE inhibition reduces activity of the plasminogen/plasmin and MMP systems in the brain of spontaneous hypertensive stroke-prone rats. Neurosci Lett. 2005;376:205–9. doi: 10.1016/j.neulet.2004.11.061. [DOI] [PubMed] [Google Scholar]
- Merrill DC, Thompson MW, Carney CL, Granwehr BP, Schlager G, Robillard JE, Sigmund CD. Chronic hypertension and altered baroreflex responses in transgenic mice containing the human renin and human angiotensinogen genes. J Clin Invest. 1996;97:1047–55. doi: 10.1172/JCI118497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noshita N, Sugawara T, Lewen A, Hayashi T, Chan PH. Copper–zinc superoxide dismutase affects Akt activation after transient focal cerebral ischemia in mice. Stroke. 2003;34:1513–8. doi: 10.1161/01.STR.0000072986.46924.F4. [DOI] [PubMed] [Google Scholar]
- Nukhet Turkel A, Ziya Ziylan Y. Protection of blood–brain barrier breakdown by nifedipine in adrenaline-induced acute hypertension. Intern J Neurosci. 2004;114:517–28. doi: 10.1080/00207450490422821. [DOI] [PubMed] [Google Scholar]
- Paravicini TM, Chrissobolis S, Drummond GR, Sobey CG. Increased NADPH-oxidase activity and Nox4 expression during chronic hypertension is associated with enhanced cerebral vasodilatation to NADPH in vivo. Stroke. 2004;35:584–9. doi: 10.1161/01.STR.0000112974.37028.58. [DOI] [PubMed] [Google Scholar]
- Rajagopalan S, Kurz S, Munzel T, Tarpey M, Freeman BA, Griendling KK, Harrison DG. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest. 1996a;97:1916–23. doi: 10.1172/JCI118623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan S, Meng XP, Ramasamy S, Harrison DG, Galis ZS. Reactive oxygen species produced by macrophage-derived foam cells regulate the activity of vascular matrix metalloproteinases in vitro. Implications for atherosclerotic plaque stability. J Clin Invest. 1996b;98:2572–9. doi: 10.1172/JCI119076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simic DV, Mimic-Oka J, Pljesa-Ercegovac M, Savic-Radojevic A, Opacic M, Matic D, Ivanovic B, Simic T. Byproducts of oxidative protein damage and antioxidant enzyme activities in plasma of patients with different degrees of essential hypertension. J Hum Hypertens. 2006;20:149–55. doi: 10.1038/sj.jhh.1001945. [DOI] [PubMed] [Google Scholar]
- Tang J, Liu J, Zhou C, Ostanin D, Grisham MB, Neil Granger D, Zhang JH. Role of NADPH oxidase in the brain injury of intracerebral hemorrhage. J Neurochem. 2005;94:1342–50. doi: 10.1111/j.1471-4159.2005.03292.x. [DOI] [PubMed] [Google Scholar]
- Tuor UI, Edvinsson L, McCulloch J. Catecholamines and the relationship between cerebral blood flow and glucose use. Am J Physiol. 1986;251:H824–33. doi: 10.1152/ajpheart.1986.251.4.H824. [DOI] [PubMed] [Google Scholar]
- Vásquez-Vivar J, Kalyanaraman B, Martásek P, Hogg N, Masters BS, Karoui H, Tordo P, Pritchard KA., Jr Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci USA. 1998;95:9220–5. doi: 10.1073/pnas.95.16.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner KR, Sharp FR, Ardizzone TD, Lu A, Clark JF. Heme and ion metabolism: role in cerebral hemorrhage. J Cereb Blood Flow Metab. 2003;23:629–52. doi: 10.1097/01.WCB.0000073905.87928.6D. [DOI] [PubMed] [Google Scholar]
- Wang J, Tsirka SE. Tuftsin fragment 1 to 3 is beneficial when delivered after the induction of intracerebral hemorrhage. Stroke. 2005;36:613–8. doi: 10.1161/01.STR.0000155729.12931.8f. [DOI] [PubMed] [Google Scholar]
- Williams HC, Griendling KK. NADPH oxidase inhibitors: new antihypertensive agents. J Cardiovasc Pharmacol. 2007;50:9–16. doi: 10.1097/FJC.0b013e318063e820. [DOI] [PubMed] [Google Scholar]
- Woessner JF., Jr Matrix metalloproteinases and their inhibitors in connective tissue remodeling. FASEB J. 1991;5:2145–54. [PubMed] [Google Scholar]
- Xi G, Keep RF, Hoff JT. Mechanisms of brain injury after intracerebral hemorrhage. Lancet Neurol. 2006;5:53–63. doi: 10.1016/S1474-4422(05)70283-0. [DOI] [PubMed] [Google Scholar]
- Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab. 2006;27:697–709. doi: 10.1038/sj.jcbfm.9600375. [DOI] [PubMed] [Google Scholar]
- Yu F, Sugawara T, Chan PH. Treatment with dihydroethidium reduces infarct size after transient focal cerebral ischemia in mice. Brain Res. 2003;978:223–7. doi: 10.1016/s0006-8993(03)02775-6. [DOI] [PubMed] [Google Scholar]
- Zhang H, Schmieber A, Garlichs CD, Plotze K, Damme U, Mugge A, Daniel WG. Angiotensin II-induced superoxide anion generation in human vascular endothelial cells: role of membrane-bound NADH-/NADPH-oxidases. Cardiovasc Res. 1999;44:215–22. doi: 10.1016/s0008-6363(99)00183-2. [DOI] [PubMed] [Google Scholar]
- Zimmerman MC, Lazartigues E, Sharma RV, Davisson RL. Hypertension caused by angiotensin II infusion involves increased superoxide production in the central nervous system. Circ Res. 2004;95:210–6. doi: 10.1161/01.RES.0000135483.12297.e4. [DOI] [PubMed] [Google Scholar]