

Abstract
We observed a remarkable reduction in the frequency and immunosuppressive activity of splenic CD4+CD25+ T cells in C57BL/6 mice with MOG33–55-induced experimental autoimmune encephalomyelitis (EAE). Our study revealed that pertussis toxin (PTx), one component of the immunogen used to induce murine EAE, was responsible for down-regulating splenic CD4+CD25+ cells. Treatment of normal BALB/c mice with PTx in vivo reduced the frequency, suppressive activity and FoxP3 expression by splenic CD4+CD25+ T cells. However, PTx treatment did not alter the expression of characteristic phenotypic markers (CD45RB, CD103, GITR and CTLA-4) and did not increase the expression of CD44 and CD69 by the residual splenic and lymph node CD4+CD25+ T cells. This property of PTx was attributable to its ADP-ribosyltransferase activity. PTx did not inhibit suppressive activity of purified CD4+CD25+ T regulatory (Treg) cells in vitro, but did so in vivo, presumably due to an indirect effect. Although the exact molecular target of PTx that reduces Treg activity remains to be defined, our data suggests that alteration of both distribution and function of splenic immunocytes should play a role. This study concludes that an underlying cause for the immunological adjuvanticity of PTx is down-regulation of Treg cell number and function.
Keywords: CD4+CD25+ T regulatory cells, Experimental autoimmune encephalomyelitis, Pertussis toxin
Introduction
Recently, the crucial role of FoxP3+CD4+CD25+ regulatory T cells (Treg cells) in suppression of responses to self antigens, and the maintenance of anergy has been demonstrated in both mice and humans [1]. Clinically, defects in Treg cells have been reported in various human autoimmune diseases, including psoriasis [2], myasthenia [3], rheumatoid arthritis [4], autoimmune liver disease [5] and hepatitis C-mixed cryoglobulinemia vasculitis [6], autoimmune polyglandular syndrome [7] and multiple sclerosis [8]. Deficiency of Treg cells was also found in murine autoimmune disease models such as collagen-induced arthritis [9] and in autoimmune prone strains of mice such as NOD/SCID mice [10].
Experimental autoimmune encephalomyelitis (EAE) is a mouse model of inflammatory demyelinating disease that shares many clinical and histological features with multiple sclerosis. In mice susceptible to developing EAE, immunization with myelin protein antigens such as proteolipid protein (PLP), myelin basic protein (MBP) or myelin oligodendrocyte glycoprotein (MOG) in complete Freund’s adjuvant (CFA) along with pertussis toxin (PTx) overcomes tolerant/anergic state, and induces a CD4+ Th1 cell-mediated inflammatory response in the central nervous system (CNS) [11]. The development of EAE is countered by Treg cells, since depletion of Treg cells increased the severity and mortality [12], while adoptive transfer of Treg cells inhibited the onset/progression of the disease [13]. Our previous work revealed that a mouse strain susceptible to EAE had fewer Treg cells than an EAE-resistant mouse strain [14]. Therefore, EAE mice may serve as a model to investigate the factors responsible for the reduction in Treg activity.
In this report, we found that the proportion of splenic CD4+CD25+ cells was significantly reduced in mice with induced EAE. Furthermore, the residual splenic CD4+CD25+ Tcells from EAE mice robustly proliferated, and produced high levels of IFN-γ in response to TCR stimulation. More importantly, they failed to suppress the proliferation of splenic CD4+CD25− T cells. We further found that PTx, one component in the EAE immunogen, was responsible for the reduction in the number of CD4+CD25+ T cells in the mouse spleen. PTx is one of the major virulence factors of Bordetella pertussis, which is composed of an active subunit (S1), that ADP-ribosylates the alpha subunit of several mammalian G proteins and the B oligomer (S2–S5), that binds glycoconjugate receptors on cells [15]. Historically, the means by which PTx was thought to act on EAE was by making the blood-brain barrier permeable. Recent work has shown that PTx actually increases adhesion molecule expression that initiates leukocyte infiltration into the brain [16]. Further, PTx appears to induce the maturation of dendritic cells [17–21], which results in the expansion of Teffector cells and secretion of IFN–γ [22].
We observed that, in vivo, PTx treatment resulted in a reduction in the expression of FoxP3 by purified splenic CD4+ cells and CD4+CD25+ cells. The immunosuppressive activity of splenic CD4+CD25+ cells was also reduced after PTx treatment. This property of PTx was dependent on its ADP-ribosyltransferase activity. Our data suggest that reduction in Treg activity is an important mechanism by which PTx acts as an immunological adjuvant.
Results
Splenic CD4+CD25+ T cells are reduced in number and immunosuppressive activity in EAE mice
To determine the effect of induction of EAE on the frequency and activity of Treg cells, C57BL/6 (B6) mice were killed and analyzed on day 10 after immunization with MOG33–55 peptide and CFA/PTx. After immunization, lymph nodes (LN; axillary, inguinal and mesenteric regions) of mice were dramatically withered and the cellularity was reduced to less than one tenth that of normal mice (data not shown). In contrast, the splenic cellularity of EAE mice was increased almost twofold over that of normal mice (data not shown). Thus, we focused on analyzing B6 splenic cells in subsequent experiments, since the paucity of LN cells made it difficult to determine the activity of Treg cells. Although the pathology of EAE is well known to be mediated by CD4 Tcells [23], the proportion of CD4+CD25+ Tcells in the spleen of B6 mice induced to develop EAE was significantly lower than that of control mice (p<0.001, Fig. 1A). The reduced frequency of CD4+CD25+ cells in the spleen persisted for at least 70 days after EAE induction, and the lower proportion of splenic CD4+CD25+ cells correlated with the higher clinical score (data not shown).
Figure 1.
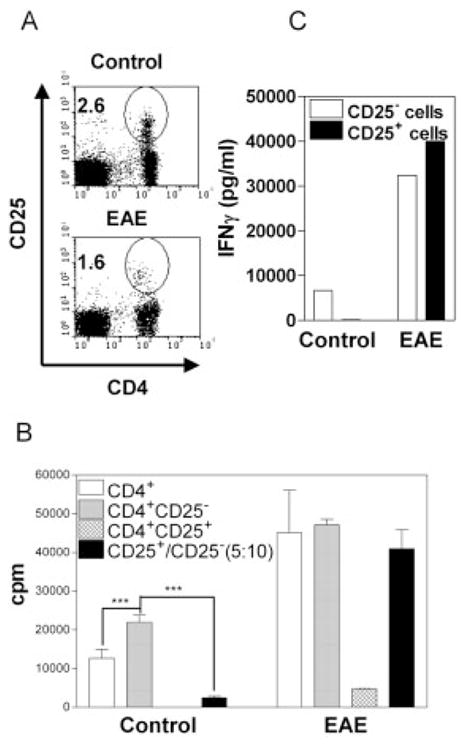
EAE induction reduces frequency and immunosuppressive function of CD4+CD25+ splenocytes. Splenocytes were isolated from B6 mice on day 10 post immunization with EAE immunogen. (A) The cells were analyzed for expression of CD4 and CD25 by FACS. Numbers in the dot plot indicate the percentage of CD4+CD25+ cells. The data are representative of five separate experiments. (B) MACS-purified CD4 (5 × 104 cells/well, open bar) cells and FACS-purified CD4+CD25− (5 × 104 cells/well, gray bar) and CD4+CD25+ (2.5 × 104 cells/ well, hatched bar) splenocytes were cultured separately or co-cultured (black bar, ratio of CD25− versus CD25+ cells was 10:5, from the same group). The cells were stimulated with normal B6 APC (2 × 105 cells/well) and an anti-CD3 antibody (0.5 μg/ mL) for 72 h. Proliferation was measured by [3H]thymidine incorporation. ***p<0.001 compared with proliferation of CD4+CD25− T cells. (C) FACS-purified CD4+CD25+ or CD4+CD25−T cells were stimulated with 10 μg/mL of a plate-bound anti-CD3 antibody and 2 μg/mL of a soluble anti-CD28 antibody. After 72 h, the supernatants were collected and IFN-γ levels were determine. Data (B, C) are representative of three separate experiments with the similar results.
We next examined the capacity of splenic CD4+CD25+ T cells to suppresses the proliferation of CD4+CD25− T cells from the same group, utilizing a standard in vitro functional assay for Treg cells. As shown in Fig. 1B, in the control group, the proliferation of splenic CD4+CD25− T cells was markedly higher than that of unfractionated splenic CD4 T cells (p<0.05), presumably due to the depletion of Treg cells. Control splenic CD4+CD25+ cells were hyporesponsive to TCR stimulation (cpm: 30) and potently inhibited proliferation of splenic CD4+CD25− cells (p<0.01), as expected. In the EAE group, the proliferation of splenic CD4 cells was much more vigorous than their normal counterparts (3.5-fold higher in cpm). Furthermore, splenic CD4+CD25+ cells from EAE mice exhibited a low level of proliferation and failed to suppress splenic CD4+CD25− T cells from the same group.
Once activated by TCR stimulation, normal splenic CD4+CD25− T cells, but not CD4+CD25+ T cells, produced considerable amounts of IFN-γ. In sharp contrast, splenic CD4+CD25+ T cells from EAE mice produced an even higher level of IFN-γ than CD4+CD25−T cells (Fig. 1C). Thus, EAE mice were deficient in Treg activity, displaying a decrease in the proportion of splenic CD4+CD25+ T cells and an increase in IFN-γ production by CD4+CD25+ cells.
PTx treatment reduced the frequency and number of splenic CD4+CD25+ T cells
To identify the immunogen component that reduced the number and activity of Treg cells, we injected MOG/incomplete Freund’s adjuvant (IFA), CFA, MOG/CFA and PTx, separately, into normal B6 mice. One week after injection, the proportion of splenic CD4+CD25+ cells was analyzed. As shown in Fig. 2A, only PTx markedly decreased the proportion of CD4+CD25+ cells in the spleen (<50% of normal mice, p<0.01).
Figure 2.
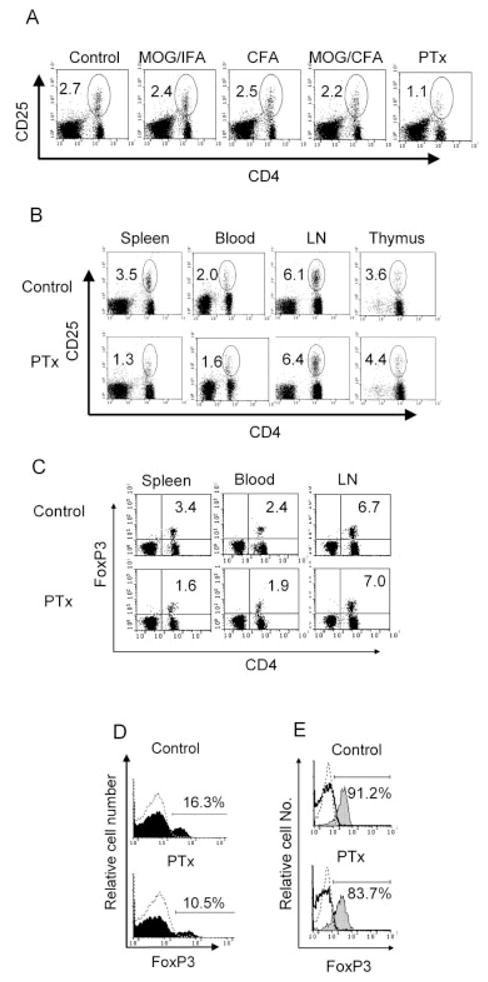
In vivo PTx treatment reduces the proportion of CD4+CD25+ T cells and CD4+FoxP3+ T cells. (A) Female B6 mice were treated with MOG peptide in IFA, or CFA, or MOG peptide in CFA or PTx for 2 days (once a day) as described in the Materials and methods. One week after the last treatment, spleens were harvested and the proportion of CD4+CD25+ T cells was analyzed by FACS. The data are representative of two separate experiments with similar results. (B–E) BALB/c mice were treated with PTx (400 ng/mouse/day, i.p.) for 2 days. One week after the last injection, the mice were killed and the percentage of CD4+CD25+ T cells (B) and CD4+FoxP3+ T cells (C) in the lymphoid tissues and peripheral blood was analyzed by FACS. (D) FoxP3 expression was analyzed by gating on CD3+CD4+ splenic cells. Dashed histogram indicates isotype control; black histogram indicates FoxP3 staining. (E) FoxP3 expression was analyzed by gating on CD4+CD25+ (gray histogram) and CD4+CD25− (solid line histogram) splenic cells. Dashed histogram indicates isotype control. The data are representative of at least three separate experiments with similar results. The number in the dot plot indicates the percentage of CD4+CD25+ T cells or CD4+FoxP3+ T cells.
PTx treatment of BALB/c mice also resulted in a reduction in splenic CD4+CD25+ cells. However, the proportion of CD8−CD4+CD25+ T cells in the thymus was not significantly decreased (Fig. 2B). Furthermore, the percentage of CD25+ T cells in the splenic CD3+CD4+ population was also reduced by 40% after PTx treatment (data not shown). In BALB/c mice, treatment with PTx resulted in a 30-fold decrease in the cellularity of the LN. The average pooled cellularity from axillary, inguinal and mesenteric LN was 24 × 106 in untreated mice and 0.8 × 106 in PTx-treated mice. The average cellularity of the spleen increased from 63.3 × 106 in a normal mouse to 102.4 × 106 in a PTx-treated mouse. The number of splenic CD4+CD25+ T cells in PTx-treated mice was 1.32 × 106 ± 0.08 × 106, which was less than the 2.21 × 106 ± 0.06 × 106 in the control mice (p<0.05). The total number of CD4+CD25+ T cells in the spleen and LN of PTx-treated mice (1.37 × 106 ± 0.08 × 106) was also less than that of normal mice (3.68 × 106 ± 0.07 × 106, p<0.001). Thus, PTx treatment not only reduced the proportion of splenic CD4+CD25+ cells, but also markedly diminished the absolute numbers of CD4+CD25+ cells in the peripheral lymphoid tissues.
We also observed a reduction in of CD4+FoxP3+ T cells in the spleen and peripheral blood. As shown in Fig. 2C, the proportions of CD4+FoxP3+ T cells in the spleen and peripheral blood were reduced from 3.4% and 2.4% to 1.6% and 1.9%, respectively, while there was no decrease in the LN. Furthermore, the proportion of FoxP3+ cells in the splenic CD4 population was reduced from 16.3% to 10.5% (Fig. 2D). The FoxP3+ cells in the splenic CD4+CD25+ population was also reduced from 91.2% to 83.7% (Fig. 2E). The FoxP3 mRNA expression was decreased in CD4 and CD4+CD25+ population, as measured by real-time PCR (data not shown). Thus, the decrease in CD4+CD25+ splenic cells and peripheral blood cells was reflected by the reduction of CD4+FoxP3+ Treg cells after PTx treatment.
PTx treatment impairs the suppressive function of splenic CD4+CD25+ T cells
Upon TCR stimulation, splenic CD4+CD25+ cells from PTx-treated BALB/c mice showed a low proliferative response; nevertheless, they were slightly more proliferative than the splenic CD4+CD25+ cells from untreated BALB/c mice (p<0.05, data not shown). However, the suppressive activity of PTx-treated splenic CD4+CD25+ cells on responder CD4+CD25− cells from either normal BALB/c mice or PTx-treated BALB/c mice was significantly reduced, and was observed across a spectrum of CD25+ versus CD25− ratios (Fig. 3A–C, p<0.05–0.01). Thus, PTx treatment not only reduced the number of splenic CD4+CD25+ cells, but also blunted the suppressive effect of these splenic CD4+CD25+ cells.
Figure 3.
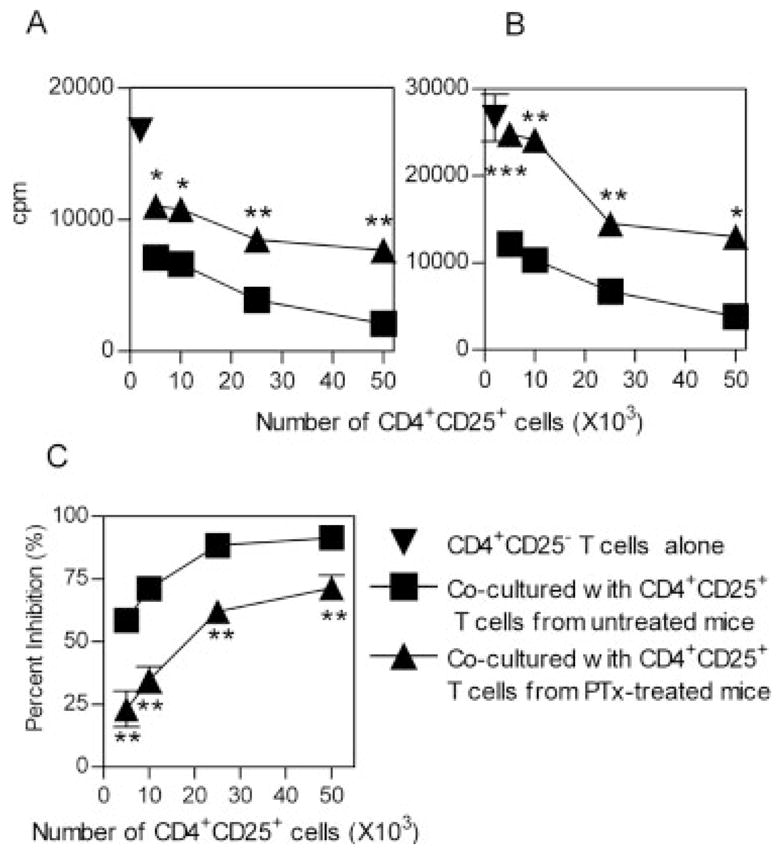
In vivo PTx treatment reduces the suppressor effect of splenic CD4+CD25+ T cells. CD4+CD25+ and CD4+CD25− T cells were purified by flow cytometry, from MACS-purified CD4 cells derived from spleen. CD4+CD25− T cells (5 × 104 cells/well) were mixed with increasing numbers of CD4+CD25+ T cells (5 × 103–5 × 104 cells/well). The cells were stimulated with APC (from normal control mice, 2 × 105 cells/well) plus a soluble anti-CD3 antibody (0.5 μg/mL) and cultured for 72 h. Proliferation was measured by [3H]thymidine incorporation. The responder CD4+CD25− T cells were either from normal control mice (A) or from PTx-treated mice (B). The inverted triangle indicates CD4+CD25− T cells alone; the square indicates untreated mice derived-CD4+CD25+ T cells mixed with CD4+CD25− T cells; the triangle indicates CD4+CD25− T cells mixed with CD4+CD25+ T cells derived from PTx-treated mice. The data shown are representative of at least five separate experiments with similar results. (C) Percent inhibition of CD4+CD25+ T cells from normal control (squares) or PTx-treated (triangles) mouse spleen to the proliferation of normal control mouse CD4+CD25− T cells. The data shown are summarized from five separate experiments (with three to five mice per group). *p<0.05, **p<0.01, and ***p<0.001, compared with inhibition elicited by normal mouse CD4+CD25+ T cells.
Effects of PTx treatment on the phenotype and production of cytokine by CD4+CD25+ T cells
Natural Treg cells express a spectrum of characteristic immunological markers such as a lower level of CD45RB, and a higher level of CD103 (integrin αEβ7), CTLA-4 (CD152) and GITR, compared with CD4+CD25− T cells [24]. We therefore compared expression of these markers by splenic and LN CD4+CD25+ cells and CD4+CD25− cells from normal and PTx-treated BALB/c mice. As shown in Fig. 4A, the expression of these surface markers by residual splenic and LN CD4+CD25+ cells was not altered after PTx treatment. Furthermore, the expression of CD44 and CD69 by splenic and LN CD4+CD25+ cells from PTx-treated mice was not significantly increased, suggesting PTx treatment did not activate CD4 cells.
Figure 4.
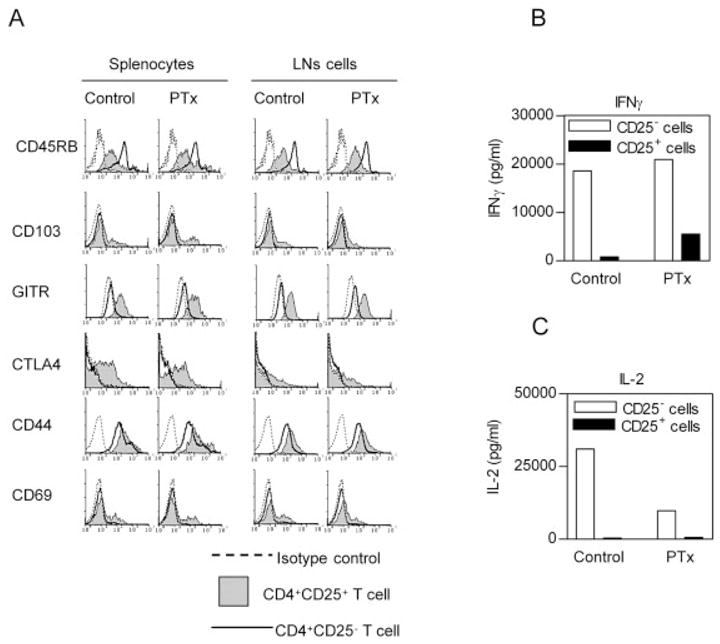
In vivo PTx treatment does not change phenotypic characteristics of CD4+CD25+ T cells (A) Female BALB/c mice were treated with PTx (400 ng/mouse) for 2 days. At 1 week after the last injection, the spleen and LN were harvested. After lysing erythrocytes, the splenic cell or LN cells were stained with Cy-chrome-CD4, PE- or FITC-conjugated CD25, FITC- or PE-conjugated third antibodies. Analyses were gated on CD4+CD25+ or CD4+CD25− T cells. Dashed line indicates isotype control, gray histogram indicates CD4+CD25+ T cells and solid line indicates CD4+CD25− T cells. Y-axis shows relative cell number. X-axis shows fluorescence intensity. The data are representative of three separate experiments with similar results. (B, C) Purified splenic CD4+CD25+ or CD4+CD25− cells (5 × 104 cells) were seeded in round-bottom 96-well plates. The cells were stimulated using plate-bound anti-CD3 (10 μg/mL) and soluble anti-CD28 (2 μg/mL) for 72 h. The production of IFN-γ (B) and IL-2 (C) in the supernatants was determined. Data shown are representatives of three separate experiments with similar results.
Upon stimulation by anti-CD3/CD28, splenic CD4+CD25+ cells from PTx-treated BALB/c mice produced lower levels of IFN-γ than their CD4+CD25−cells (while splenic CD4+CD25+ cells from EAE mice produced more IFN-γ, Fig. 1B). Splenic CD4+CD25+ cells from PTx-treated mice produced up to 6.8-fold higher levels of this Th1 cytokine than CD4+CD25+ cells from untreated mice (Fig. 4C).
Since IL-2 is a critical cytokine in the production of Treg cells, and maintenance of the Treg cell suppressive activity (reviewed in [25]), we also examined the production of IL-2 by cells from BALB/c mice. As shown in Fig. 4C, IL-2 was exclusively produced by normal splenic CD4+CD25− cells, but not by CD4+CD25+ cells. The capacity of splenic CD4+CD25− cells from PTx-treated mice to produce IL-2 was markedly suppressed (~30% of normal mice). The suppression of IL-2, which is essential for Treg cells, may account for the reduction of Treg activity.
Effect of APC from PTx-treated mice on Treg cells
To clarify whether in vivo effects of PTx were due to direct action of PTx on the Treg cells, we exposed purified Treg cells to PTx (1 μg/mL) for 24 h and measured their FoxP3 mRNA expression and suppressive activity. The results showed direct in vitro treatment with PTx neither down-regulated FoxP3 mRNA expression, nor reduced the suppressive activity of Treg cells (data not shown). We therefore evaluated the possibility that PTx affected other splenic cell types that contributed to the down-regulation of Treg activity.
Up-regulation of costimulatory molecules on activated APC has been reported to be responsible for breaking T cell anergy and reversing the suppressive activity of Treg cells in vitro [26]. In vitro studies have shown that PTx activated APC by up-regulating antigen-presenting molecules (MHC) and costimulatory molecules (CD40, CD80 and CD86) [17, 20, 21]. We therefore examined the effect of APC from PTx-treated mice on the activity of Treg cells. T-depleted/irradiated splenocytes from PTx-treated mice were prepared and used as APC in the standard in vitro functional assay for Treg cells. These APC did not interfere with the capacity of splenic CD4+CD25+ cells from normal control mice to markedly inhibit proliferation of responder CD4+CD25− cells. Additionally, the APC did not block the inhibition of splenic CD4+CD25+ cell from PTx-treated mice (Fig. 5A). Therefore, these APC did not interfere with the inhibitory activity of Treg cells.
Figure 5.
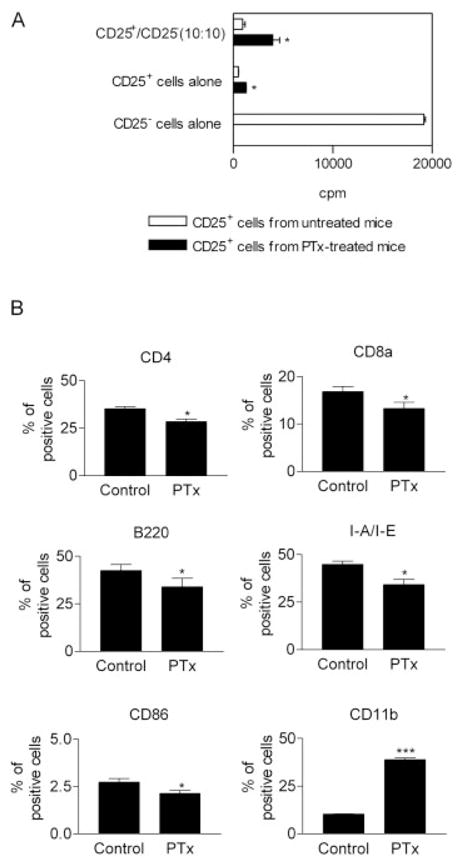
APC from PTx-treated mice support suppression by Treg cells. (A) The suppression by CD4+CD25+ splenocytes (5 × 104 cells/well) from normal control mice or PTx-treated mice (400 ng/mouse/day, 2 days) were assessed by culture with the same number of normal BALB/c CD4+CD25− splenocytes. The cells were stimulated with APC from PTx-treated mice (2 × 105 cells/well) and 0.5 μg/mL anti-CD3. The proliferation was measured by [3H]thymidine incorporation. Data shown are representative of three separate experiments with similar results. *p<0.05, compared with cells from control mice. (B) BALB/c mice were treated with PTx (400 ng/mouse/day, 2 days) and splenocytes were isolated and analyzed for expression of surface markers by FACS. The data shown are percent of positive cells (means ± SD), which is summarized from seven separate experiments (n=35). *p<0.05, ***p<0.001, compared with normal control mice.
We next analyzed the phenotype of unfractionated splenocytes after PTx treatment in vivo. Quite surprisingly, in vivo treatment with PTx did not enhance, but actually reduced, the expression of MHC class II and costimulatory molecules (CD86) by splenocytes (p<0.05). This is distinctly different from the effect of in vitro PTx treatment on APC. The proportion of splenic CD4+, CD8α+ and B220+ cells was also decreased (p<0.05). However, CD11b+ myeloid cells were dramatically increased by almost fourfold by PTx treatment (p<0.001) (Fig. 5B).
S1 mutant PT (mPTx) fails to reduce the frequency and suppression of splenic CD4+CD25+ cells
To determine whether the ADP-ribosylation of PTx is required to reduce the activity of Treg cells, we compared the effect of PTx with a S1 subunit mutant inactive PTx (mPTx) [27]. Treatment with mPTx failed to alter the distribution of lymphocytes in the secondary lymphoid tissues. Unmutated PTx treatment reduced the proportion of splenic CD4+CD25+ cells by 56%. In contrast, treatment with mPTx failed to reduce the proportion of splenic CD4+CD25+ cells (data not shown). Furthermore, suppression by splenic CD4+CD25+ cells of proliferation of CD4+CD25− cells was not significantly impaired by treatment with mPTx, while markedly attenuated by treatment with PTx (p<0.01–0.05, Fig. 6). Thus, the reduction in suppressive activity of Treg cells by PTx is based on its ADP-ribosylation activity.
Figure 6.
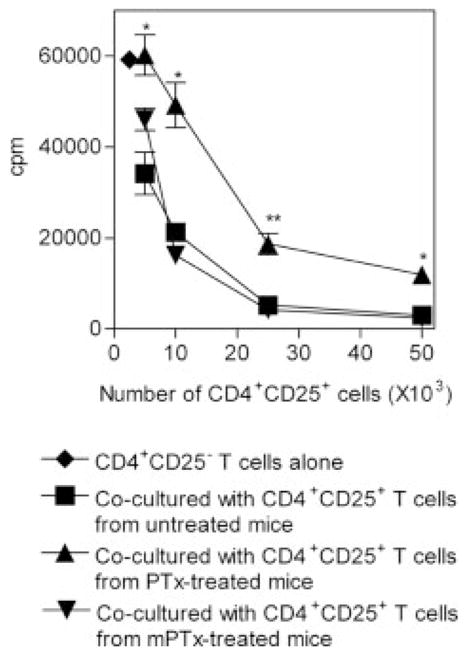
S1 mutant PTx fails to reduce the activity of CD4+CD25+ Treg cells. BALB/c mice were injected (i.p.) with 400 ng/mL of wild-type PTx or S1 mutant PTx (mPTx) for 2 days. Spleens were harvested 1 week after the last injection. The indicated number of FACS-sorted CD4+CD25+ splenocytes from either untreated mice (square), wild-type PTx-treated mice (triangle) or S1 mutant PTx (inverted triangle) were mixed with normal mouse CD4+CD25− splenocytes (5 × 104 cells/ well). The cells were stimulated with APC (from untreated mice) and 0.5 μg/mL soluble anti-CD3 and cultured for 72 h. Proliferation was measured by [3H]thymidine incorporation. *p<0.05, **p<0.01, compared with the inhibition elicited by normal BALB/c CD4+CD25+ splenocytes. The data shown are representative of three separate experiments with similar results.
Discussion
In this study, we observed that the murine EAE model was profoundly deficient in Treg activity. Further, we found that PTx, one of the components of the immunogen cocktail used for EAE induction, was largely responsible for the reduction in Treg activity. Thus, we hypothesize that suppression of Treg activity would account for the immunological adjuvant action of PTx. This hypothesis is supported by a recent report that administration of anti-CD25, an antibody that depletes Treg cells in vivo, abolishes the need for PTx to induce EAE [28].
PTx is a potent immunological adjuvant, and has been widely used to enhance Th1-mediated organ-specific autoimmune diseases, including EAE [29] and experimental autoimmune uveitis [30]. To date, the cellular target and action mechanism by which PTx mediates adjuvanticity remains to be defined [21]. Over the past few years, it has been reported that PTx promoted the activation and maturation of APC, especially dendritic cells [17–21], and a recent report suggests this may be based on the activation of TLR4 by PTx [16]. However, these findings were based on in vitro observations, and the mechanism of in vivo action of PTx remains largely obscure.
Although our data do not provide the precise molecular basis underlying the action of PTx in reducing Treg activity, there are a number of plausible contributory effects of PTx. First, PTx may suppress Treg cells due to PTx inhibition of G-protein-coupled chemokine receptors. PTx treatment inactivates chemokine receptors and results in the redistribution of lymphocytes [31], manifested by inhibition of entrance of lymphocytes into LN and Peyer’s patches and by accumulation of cells in the spleen [32]. Our data show that the ADP-ribosyltransferase activity of PTx is required for the reduction of Treg activity, whereas ADP-ribosyltransferase activity was reported to be unnecessary for PTx to stimulate immune responses (the activation of APC and production of cytokines) [18]. Therefore, inactivation of PTx-sensitive G-protein-coupled receptors and alteration of leukocyte trafficking could be critical for the reduction in Treg cells.
In addition, the suppression of Treg cells by PTx may involve the inhibition of IL-2 production in vivo by PTx. IL-2 has been firmly established as a vital cytokine for thymic development, peripheral expansion/ maintenance, and immunosuppressive activity of Treg cells (reviewed in [25]). Several reports suggest PTx blocks IL-2 production by T cells in response to diverse stimuli, including Con A [33, 34] and IL-1 [35, 36]. Indeed, once stimulated with anti-CD3 and CD28, splenic CD4+CD25− cells from PTx-treated mice produced a lower level of IL-2 than the cells from untreated mice (Fig. 4D). However, it has been reported that a high concentration of PTx itself stimulates IL-2 production [33], and we also observed that treatment with PTx (1 μg/mL) in vitro resulted in the release of IL-2 by mouse splenocytes (data not shown). This discrepancy may be the basis for the previously proposed dual effect of PTx: it activates mononuclear phagocytes and lymphocytes itself, while inhibiting the response of those immunocytes to other stimuli [37]. Although deprivation of IL-2 by PTx treatment is a very attractive explanation for the reduction of Treg activity, further research is needed to clarify exactly how PTx affects IL-2 production in vivo.
One possible explanation for the observed effect of PTx may be the expansion of effector T cells, rather than reducing the number of Treg cells. However, this possibility was not supported by results showing that the total number of CD4+FoxP3− T cells present in the spleen and LN (mesenteric, axillary and inguinal regions) was not increased by PTx treatment (data not shown). Furthermore, the cell cycle profile of CD4+ Tcells in the spleen and LN of PTx-treated and untreated mice was not different (data not shown). These observations are consistent with an earlier report that PTx-induced lymphocytosis was due to a redistribution of lymphocytes between the circulating and tissue pools, and not due to cellular proliferation [31]. The results observed in this study are unlikely to be due to potential LPS contamination in the PTx, since the in vitro action of PTx on the splenocytes was not blocked by the LPS neutralizer-polymycin B (data not shown).
Since CD25 is also a marker of activated effector CD4 cells, we wondered whether the residual splenic CD4+CD25+ cells after PTx treatment were Treg cells or activated CD4 cells. To answer this question, we examined the expression of FoxP3, a specific hallmark of Treg cells [38]. In agreement with the decrease of CD4+CD25+ cells, the FoxP3 mRNA expression by total splenocytes from PTx-treated mice was reduced (data not shown). The residual splenic CD4+CD25+ cells obtained from PTx-treated mice also exhibited a reduced level of FoxP3. It is reasonable to speculate that PTx treatment favors activation of effector CD4 cells, and consequently the residual splenic CD4+CD25+ cells from PTx-treated mice may consist of mixture of natural Treg cells and activated CD4 cells. However, this speculation is not supported by the observation that the phenotypic markers, especially CD44 and CD69, expressed by splenic and LN CD4+CD25+ cells from PTx-treated mice were not different from those of normal untreated mice. A previous study demonstrated that FoxP3 mRNA expression by Treg cells could be down-regulated, e.g., by lack of TGF-β signaling [39] or by activation of autoimmune pathogenesis [9]. It has also been reported that once anergy had been broken, CD4+CD25+ cells were able to proliferate and produce cytokine [26]. Since PTx treatment reduced the total number of CD4+CD25+ cells in the spleen and in the LN, we favor the idea that in vivo PTx treatment, by a mechanism to be further defined, reduces the functions of Treg cells by down-regulation of FoxP3 expression and immunosuppressive activity.
In summary, the results presented in this study demonstrate a profound defect in the immunosuppressive activity of splenic CD4+CD25+ cells in MOG33–55-specific EAE mice, which could be the underlying basis for the organ-specific immunopathology observed in this model. We found that PTx was largely responsible for the reduction in the frequency, number and suppressive activity of Treg cells in the peripheral lymphoid tissues. This property could be an underlying cause of the immunological adjuvancity of PTx.
Materials and methods
Mice and reagents
Female B6 mice and BALB/c mice, 8–12 weeks old, were purchased from Animal Production Area of the National Cancer Institute-Frederick (Frederick, MD). Animal care was provided in accordance with the procedures outlined in the “Guide for the Care and Use of Laboratory Animals” (NIH publication no. 86–23, 1985). Mice were maintained on standard laboratory food and water ad libitum. Paralyzed animals were afforded easier access to food and water. Anti-mouse antibodies [14] purchased from BD PharMingen (San Diego, CA) and R&D Systems (Minneapolis, MN). PE-conjugated anti-mouse/rat Foxp3 staining set was purchased from eBioscience (San Diego, CA). PTx was purchased from List Biological Laboratory, Inc. (Campbell, CA). S1 mutant PTx (PT-9 K129G) was purified in the laboratory of Dr. Nicholas H Carbonetti at University of Maryland, as previously described [27].
Induction and clinical evaluation of MOG35–55-induced EAE
Female B6 mice, 8–12 weeks old, were immunized s.c. with 200 μL of an emulsion containing CFA plus 300 μg Mycobacterium tuberculosis H37Ra (Difco Laboratories, Detroit, MI) and 200 μg MOG35–55 (Invitrogen Life Technologies, Carlsbad, CA). Additionally, 400 ng PTx in 200 μL PBS was administrated (i.p.) on days 0 and 2 post immunization. At day 7 the mice were immunized with MOG peptide once more.
MOG, CFA and PTx treatment
B6 mice were injected s.c. with 200 μL of an emulsion and 200 μg MOG35–55 in IFA or in CFA, or CFA alone, on the flank. Both B6 and BALB/c mice were used for the PTx treatment experiment in which the mice were injected (i.p.) with 400 ng (in 0.2 mL PBS) per day for 2 consecutive days. One week after the first treatment, the mice were killed and lymphoid organs were removed aseptically and a single-cell suspension was prepared for the assays.
Purification of cells
CD4+, CD4+CD25+ and CD4+CD25− T cells from spleen were separated using a MACS MS column (Miltenyi Biotech, Auburn, CA) as previously described [14]. The purity of CD4+CD25+ T cells was greater than 90% (after positive selection by two MS columns) and that of CD4+CD25− T cells was greater than 95%. In some experiments, the cells were sorted using a Cytomation MoFlo cytometer (Fort Collins, CO) yielding a purity for both subsets of nearly 98%. Purified cells were suspended and cultured in RPMI 10 [RPMI 1640 with 10% fetal bovine serum (FBS, Hyclone, Logan, UT) containing 2 mM glutamine, 100 IU/mL penicillin, and 100 μg/mL streptomycin, 10 mM HEPES, 1 mM sodium pyruvate and 50 μM 2-ME].
Flow cytometry
All incubation steps were conducted for 30 min at 4°C. After treatment with an anti-CD16/CD32 antibody, the cells were incubated with appropriately diluted antibodies for cell surface staining. For intracellular staining of anti-CTLA-4, cells were fixed and permeabilized using Cytofix/Cytoperm Kits (BD PharMingen) and then incubated with PE-conjugated anti-CTLA-4. Anti-FoxP3 Ab was stained using PE-conjugated anti-mouse/rat Foxp3 Staining Set (eBioscience). Flow cytometry analysis was performed on a FACScan (BD Biosciences, Mountain View, CA) using CellQuest software.
In vitro proliferation assay
CD4+CD25− T cells (5 × 104 cells/well) were seeded in a U-bottom 96-well plate in RPMI 10, with or without 2 × 105 cells/well of T cell-depleted, irradiated splenocytes (APC) plus 0.5 μg/mL of a soluble anti-CD3 antibody (145–2C11). CD4+CD25+ T cells were added to wells at ratios of 10:0, 10:1, 10:2, 10:5, 10:10 and 0:10 (CD4+CD25− T cell: CD4+CD25+ T cell). Cells were pulsed with 1 μCi [3H]thymidine (Amersham Pharmacia Biotech, Piscataway, NJ) per well for the last 15 h of the 72-h culture period. The amount of incorporated [3H]thymidine was measured with a Wallac Microbeta counter (Perkin-Elmer Life Sciences, Gaithersburg, MD).
Detection of cytokines
Purified splenic CD4+CD25+ or CD4+CD25− cells were seeded in U-bottom 96-well plates and were stimulated with plate-bound anti-CD3 (10 μg/mL) and soluble anti-CD28 (2 μg/ mL). After culturing for 72 h, the supernatants were collected and cytokine measurement was performed by analysis of supernatant with SearchLight Mouse Cytokine Array (Pierce Biotechnology, Woburn, MA).
Statistical analysis
Comparisons of data from experimental group and control group were analyzed by two-tailed Student’s t-test using Graphpad Prism 4.0. (GraphPad Software, Inc., San Diego, CA).
Acknowledgments
This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, and with federal funds from the National Cancer Institute, under contract no. N01-CO-012400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organization imply endorsement by the U.S. Government.
Abbreviations
- B6
C57BL/6
- MOG
myelin oligodendrocyte glycoprotein
- mPTx
S1 mutant pertussis toxin
- PTx
Pertussis toxin
- Treg cells
CD4+CD25+ T regulatory cells
References
- 1.Sakaguchi S. Regulatory T cells: key controllers of immunologic self-tolerance. Cell. 2000;101:455–458. doi: 10.1016/s0092-8674(00)80856-9. [DOI] [PubMed] [Google Scholar]
- 2.Sugiyama H, Gyulai R, Toichi E, Garaczi E, Shimada S, Stevens SR, McCormick TS, Cooper KD. Dysfunctional blood and target tissue CD4+CD25high regulatory T cells in psoriasis: mechanism underlying unrestrained pathogenic effector T cell proliferation. J Immunol. 2005;174:164–173. doi: 10.4049/jimmunol.174.1.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balandina A, Lecart S, Dartevelle P, Saoudi A, Berrih-Aknin S. Functional defect of regulatory CD4+CD25+ Tcells in the thymus of patients with autoimmune myasthenia gravis. Blood. 2005;105:735–741. doi: 10.1182/blood-2003-11-3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ehrenstein MR, Evans JG, Singh A, Moore S, Warnes G, Isenberg DA, Mauri C. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J Exp Med. 2004;200:277–285. doi: 10.1084/jem.20040165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Longhi MS, Ma Y, Bogdanos DP, Cheeseman P, Mieli-Vergani G, Vergani D. Impairment of CD4(+)CD25(+) regulatory T-cells in autoimmune liver disease. J Hepatol. 2004;41:31–37. doi: 10.1016/j.jhep.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 6.Boyer O, Saadoun D, Abriol J, Dodille M, Piette JC, Cacoub P, Klatzmann D. CD4+CD25+ regulatory T-cell deficiency in patients with hepatitis C-mixed cryoglobulinemia vasculitis. Blood. 2004;103:3428–3430. doi: 10.1182/blood-2003-07-2598. [DOI] [PubMed] [Google Scholar]
- 7.Kriegel MA, Lohmann T, Gabler C, Blank N, Kalden JR, Lorenz HM. Defective suppressor function of human CD4+ CD25+ regulatory T cells in autoimmune polyglandular syndrome type II. J Exp Med. 2004;199:1285–1291. doi: 10.1084/jem.20032158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med. 2004;199:971–979. doi: 10.1084/jem.20031579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kelchtermans H, De Klerck B, Mitera T, Van Balen M, Bullens D, Billiau A, Leclercq G, Matthys P. Defective CD4+CD25+ regulatory T cell functioning in collagen-induced arthritis: an important factor in pathogenesis, counter-regulated by endogenous IFN-gamma. Arthritis Res Ther. 2005;7:R402–415. doi: 10.1186/ar1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–440. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- 11.Zamvil SS, Steinman L. The T lymphocyte in experimental allergic encephalomyelitis. Annu Rev Immunol. 1990;8:579–621. doi: 10.1146/annurev.iy.08.040190.003051. [DOI] [PubMed] [Google Scholar]
- 12.Zhang X, Koldzic DN, Izikson L, Reddy J, Nazareno RF, Sakaguchi S, Kuchroo VK, Weiner HL. IL-10 is involved in the suppression of experimental autoimmune encephalomyelitis by CD25+CD4+ regulatory T cells. Int Immunol. 2004;16:249–256. doi: 10.1093/intimm/dxh029. [DOI] [PubMed] [Google Scholar]
- 13.Kohm AP, Carpentier PA, Anger HA, Miller SD. Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J Immunol. 2002;169:4712–4716. doi: 10.4049/jimmunol.169.9.4712. [DOI] [PubMed] [Google Scholar]
- 14.Chen X, Oppenheim JJ, Howard OM. BALB/c mice have more CD4+CD25+ T regulatory cells and show greater susceptibility to suppression of their CD4+CD25− responder T cells than C57BL/6 mice. J Leukoc Biol. 2005;78:114–121. doi: 10.1189/jlb.0604341. [DOI] [PubMed] [Google Scholar]
- 15.Moss J, Stanley SJ, Burns DL, Hsia JA, Yost DA, Myers GA, Hewlett EL. Activation by thiol of the latent NAD glycohydrolase and ADP-ribosyltransferase activities of Bordetella pertussis toxin (islet-activating protein) J Biol Chem. 1983;258:11879–11882. [PubMed] [Google Scholar]
- 16.Kerfoot SM, Long EM, Hickey MJ, Andonegui G, Lapointe BM, Zanardo RC, Bonder C, et al. TLR4 contributes to disease-inducing mechanisms resulting in central nervous system autoimmune disease. J Immunol. 2004;173:7070–7077. doi: 10.4049/jimmunol.173.11.7070. [DOI] [PubMed] [Google Scholar]
- 17.Hou W, Wu Y, Sun S, Shi M, Sun Y, Yang C, Pei G, et al. Pertussis toxin enhances Th1 responses by stimulation of dendritic cells. J Immunol. 2003;170:1728–1736. doi: 10.4049/jimmunol.170.4.1728. [DOI] [PubMed] [Google Scholar]
- 18.Tonon S, Goriely S, Aksoy E, Pradier O, Del Giudice G, Trannoy E, Willems F, et al. Bordetella pertussis toxin induces the release of inflammatory cytokines and dendritic cell activation in whole blood: impaired responses in human newborns. Eur J Immunol. 2002;32:3118–3125. doi: 10.1002/1521-4141(200211)32:11<3118::AID-IMMU3118>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 19.Shive CL, Hofstetter H, Arredondo L, Shaw C, Forsthuber TG. The enhanced antigen-specific production of cytokines induced by pertussis toxin is due to clonal expansion of Tcells and not to altered effector functions of long-term memory cells. Eur J Immunol. 2000;30:2422–2431. doi: 10.1002/1521-4141(2000)30:8<2422::AID-IMMU2422>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 20.Ryan M, McCarthy L, Rappuoli R, Mahon BP, Mills KH. Pertussis toxin potentiates Th1 and Th2 responses to co-injected antigen: adjuvant action is associated with enhanced regulatory cytokine production and expression of the co-stimulatory molecules B7–1, B7–2 and CD28. Int Immunol. 1998;10:651–662. doi: 10.1093/intimm/10.5.651. [DOI] [PubMed] [Google Scholar]
- 21.Bagley KC, Abdelwahab SF, Tuskan RG, Fouts TR, Lewis GK. Pertussis toxin and the adenylate cyclase toxin from Bordetella pertussis activate human monocyte-derived dendritic cells and dominantly inhibit cytokine production through a cAMP-dependent pathway. J Leukoc Biol. 2002;72:962–969. [PubMed] [Google Scholar]
- 22.Wakatsuki A, Borrow P, Rigley K, Beverley PC. Cell-surface bound pertussis toxin induces polyclonal T cell responses with high levels of interferon-gamma in the absence of interleukin-12. Eur J Immunol. 2003;33:1859–1868. doi: 10.1002/eji.200323675. [DOI] [PubMed] [Google Scholar]
- 23.Margot CD, Ford ML, Evavold BD. Amelioration of established experimental autoimmune encephalomyelitis by an MHC anchor-substituted variant of proteolipid protein 139–151. J Immunol. 2005;174:3352–3358. doi: 10.4049/jimmunol.174.6.3352. [DOI] [PubMed] [Google Scholar]
- 24.Wood KJ, Sakaguchi S. Regulatory T cells in transplantation tolerance. Nat Rev Immunol. 2003;3:199–210. doi: 10.1038/nri1027. [DOI] [PubMed] [Google Scholar]
- 25.Malek TR, Bayer AL. Tolerance, not immunity, crucially depends on IL-2. Nat Rev Immunol. 2004;4:665–674. doi: 10.1038/nri1435. [DOI] [PubMed] [Google Scholar]
- 26.Fehervari Z, Sakaguchi S. Control of Foxp3+ CD25+CD4+ regulatory cell activation and function by dendritic cells. Int Immunol. 2004;16:1769–1780. doi: 10.1093/intimm/dxh178. [DOI] [PubMed] [Google Scholar]
- 27.Carbonetti NH, Mays RM, Artamonova GV, Plaut RD, Worthington ZE. Proteolytic cleavage of pertussis toxin S1 subunit is not essential for its activity in mammalian cells. BMC Microbiol. 2005;5:7–21. doi: 10.1186/1471-2180-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Montero E, Nussbaum G, Kaye JF, Perez R, Lage A, Ben-Nun A, Cohen IR. Regulation of experimental autoimmune encephalomyelitis by CD4+, CD25+ and CD8+ T cells: analysis using depleting antibodies. J Autoimmun. 2004;23:1–7. doi: 10.1016/j.jaut.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 29.Hofstetter HH, Shive CL, Forsthuber TG. Pertussis toxin modulates the immune response to neuroantigens injected in incomplete Freund’s adjuvant: induction of Th1 cells and experimental autoimmune encephalomyelitis in the presence of high frequencies of Th2 cells. J Immunol. 2002;169:117–125. doi: 10.4049/jimmunol.169.1.117. [DOI] [PubMed] [Google Scholar]
- 30.Caspi RR, Roberge FG, Chan CC, Wiggert B, Chader GJ, Rozenszajn LA, Lando Z, Nussenblatt RB. A new model of autoimmune disease. Experimental autoimmune uveoretinitis induced in mice with two different retinal antigens. J Immunol. 1988;140:1490–1495. [PubMed] [Google Scholar]
- 31.Morse SI, Barron BA. Studies on the leukocytosis and lymphocytosis induced by Bordetella pertussis. 3. The distribution of transfused lymphocytes in pertussis-treated and normal mice. J Exp Med. 1970;132:663–672. doi: 10.1084/jem.132.4.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang K, Im SY, Samlowski WE, Daynes RA. Molecular mechanisms of lymphocyte extravasation. III. The loss of lymphocyte extravasation potential induced by pertussis toxin is not mediated via the activation of protein kinase C. J Immunol. 1989;143:229–238. [PubMed] [Google Scholar]
- 33.Gilmore W, Weiner LP. The effects of pertussis toxin and cholera toxin on mitogen-induced interleukin-2 production: evidence for G protein involvement in signal transduction. Cell Immunol. 1988;113:235–250. doi: 10.1016/0008-8749(88)90023-8. [DOI] [PubMed] [Google Scholar]
- 34.Stanley JB, Gorczynski RM, Delovitch TL, Mills GB. IL-2 secretion is pertussis toxin sensitive in a T lymphocyte hybridoma. J Immunol. 1989;142:3546–3552. [PubMed] [Google Scholar]
- 35.Zumbihl R, Dornand J, Fischer T, Cabane S, Rappuoli R, Bouaboula M, Casellas P, Rouot B. IL-1 stimulates a diverging signaling pathway in EL4 6.1 thymoma cells. IL-2 release, but not IL-2 receptor expression, is sensitive to pertussis toxin. J Immunol. 1995;155:181–189. [PubMed] [Google Scholar]
- 36.O’Neill LA, Ikebe T, Sarsfield SJ, Saklatvala J. The binding subunit of pertussis toxin inhibits IL-1 induction of IL-2 and prostaglandin production. J Immunol. 1992;148:474–479. [PubMed] [Google Scholar]
- 37.Vistica BP, McAllister CG, Sekura RD, Ihle JN, Gery I. Dual effects of pertussis toxin on lymphoid cells in culture. Cell Immunol. 1986;101:232–241. doi: 10.1016/0008-8749(86)90200-5. [DOI] [PubMed] [Google Scholar]
- 38.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 39.Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF-β1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med. 2005;201:1061–1067. doi: 10.1084/jem.20042276. [DOI] [PMC free article] [PubMed] [Google Scholar]