

Abstract
Ribonuclease P (RNase P) is an essential endoribonuclease for which the best-characterized function is processing the 5′ leader of pre-tRNAs. Compared to bacterial RNase P, which contains a single small protein subunit and a large catalytic RNA subunit, eukaryotic nuclear RNase P is more complex, containing nine proteins and an RNA subunit in Saccharomyces cerevisiae. Consistent with this, nuclear RNase P has been shown to possess unique RNA binding capabilities. To understand the unique molecular recognition of nuclear RNase P, the interaction of S. cerevisiae RNase P with single-stranded RNA was characterized. Unstructured, single-stranded RNA inhibits RNase P in a size-dependent manner, suggesting that multiple interactions are required for high affinity binding. Mixed-sequence RNAs from protein-coding regions also bind strongly to the RNase P holoenzyme. However, in contrast to poly(U) homopolymer RNA that is not cleaved, a variety of mixed-sequence RNAs have multiple preferential cleavage sites that do not correspond to identifiable consensus structures or sequences. In addition, pre-tRNATyr, poly(U)50 RNA, and mixed-sequence RNA cross-link with purified RNase P in the RNA subunit Rpr1 near the active site in “Conserved Region I,” although the exact positions vary. Additional contacts between poly(U)50 and the RNase P proteins Rpr2p and Pop4p were identified. We conclude that unstructured RNAs interact with multiple protein and RNA contacts near the RNase P RNA active site, but that cleavage depends on the nature of interaction with the active site.
Keywords: Ribonuclease P, single-stranded RNA, antisense RNA, non-tRNA substrate
INTRODUCTION
Ribonuclease P (RNase P) is a highly conserved complex of RNA and protein subunits with the well-defined function of processing precursor transfer RNAs (pre-tRNAs) via an endonucleolytic cleavage to create their mature 5′ termini (Frank and Pace 1998; Walker and Engelke 2006). In almost all reported examples, RNase P has a similar catalytic RNA subunit that is responsible for pre-tRNA cleavage (Guerrier-Takada et al. 1983; Holzmann et al. 2008; Gobert et al. 2010). In vivo, protein subunit(s) are also required for proper function, and they are present in varying numbers, generally one in Bacteria, four to five in some Archaea, and nine to 10 in Eukarya (Hall and Brown 2002; Jarrous 2002; Evans et al. 2006; Smith et al. 2007). All but one of the eukaryotic nuclear RNase P proteins are also present in a separate enzyme, RNase MRP, which processes a number of RNAs that are not affected by RNase P including pre-rRNA and a cell cycle–linked mRNA (Schmitt and Clayton 1993; Lee and Clayton 1998; Gill et al. 2004). In addition, it was recently shown that early observations of RNase MRP cleavage of mitochondrial RNA primers were actually due to a distinct mitochondrial RNase MRP, which has a unique composition (Chang and Clayton 1987; Lu et al. 2010). In addition, RNase P and RNase MRP ribonucleoprotein complexes possess distinct, although related RNA subunits (Walker et al. 2010).
One function of the additional protein subunits in the nuclear enzyme may be to expand the RNA binding and cleavage potential beyond that of pre-tRNAs (Marvin and Engelke 2009; Marvin et al. 2011). Many additional substrates have been identified for the bacterial enzyme, which uses only a single small protein in vivo to expand its range of substrates (Bothwell et al. 1976; Peck-Miller and Altman 1991; Giegé et al. 1993; Alifano et al. 1994; Komine et al. 1994; Liu and Altman 1994; Hartmann et al. 1995; Jung and Lee 1995; Gimple and Schön 2001; Hansen et al. 2001; Li and Altman 2003; Altman et al. 2005; Wilusz et al. 2008). Adding a larger number of protein subunits to RNase P RNA in eukaryotes appears to further broaden the recognition potential of the complex. Eukaryotic RNase P binds a diverse set of RNAs in vivo, and mutations in RNase P subunits affect processing and turnover of other RNAs, including antisense RNAs, certain snoRNAs, and ribosomal RNAs (Chamberlain et al. 1996; Ziehler et al. 2000; Yang and Altman 2007; Coughlin et al. 2008; Marvin et al. 2011). Furthermore, the yeast nuclear enzyme has previously been shown to bind homopolymer RNAs with a marked sequence preference for poly(U) and poly(G), which are both potent inhibitors of pre-tRNA cleavage (Ziehler et al. 2000). However, the bacterial holoenzyme is not affected by homopolymer RNAs. It is likely that the bacterial and eukaryotic enzymes use alternative strategies to interact with non-tRNA substrates as a result of their differing subunit compositions. These observations suggest that one function of the additional protein complexity in nuclear RNase P might be to allow broader substrate recognition.
To further explore the interaction of eukaryotic RNase P with potential alternative substrates and inhibitors, we investigated the binding and cleavage of a variety of single-stranded RNA sequences by nuclear RNase P. Given the diversity of non-tRNA substrates, we chose to examine how single-stranded RNA interacts with yeast nuclear RNase P in vitro. We found that the unstructured homopolymer RNA, poly(U), binds strongly and in a length-dependent manner to RNase P. Mixed-sequence RNAs also bind tightly, but in contrast to poly(U), are cleaved by RNase P with no obvious sequence dependence or structural consensus. Cross-linking experiments show multiple sites of interaction for poly(U) with several RNase P protein subunits (Rpr2p, Pop4p) and, most prominently, with the RNA subunit (Rpr1r). Pre-tRNATyr, mixed-sequence RNA, and poly(U)50 RNA form cross-links with the same conserved region of Rpr1r, which is thought to constitute the catalytic center of RNase P, although not at the exact same positions. These results indicate that single-stranded RNA binds in a relatively sequence-independent fashion near the active site, and that this tight binding is independent of efficient cleavage of the bound RNA.
RESULTS
Binding of single-stranded RNA to RNase P
Earlier results demonstrated that nuclear RNase P, but not bacterial RNase P, was strongly inhibited by RNA homopolymers. Although some homopolymers inhibited pre-tRNA cleavage more readily, there was no obvious relationship to structural potential [poly(U) ∼ poly(G) ≫ poly(A) ⋙poly(C)] (Ziehler et al. 2000). Poly(U) RNA was chosen for further study of the length requirements for inhibition of RNase P-catalyzed pre-tRNA cleavage due to the predicted lack of secondary and tertiary structure (Kankia 2003; Davis 2004).
A range of poly(U) sizes from 25 to 60 nt (±2 nt) was isolated by alkaline hydrolysis of poly(U) RNA and denaturing electrophoretic separation as shown in Supplemental Figure S1. The poly(U) sizes were used to examine the size dependence of inhibition of pre-tRNATyr cleavage catalyzed by purified yeast nuclear RNase P (Fig. 1A). Measurement of the cleavage activity at a single concentration of poly(U) RNA demonstrates that poly(U) RNA must be >40 nt to observe significant inhibition of pre-tRNA cleavage, and inhibition increases as the length of the RNA increases (up to 63 nt). While inhibition demonstrates both binding of the inhibitor to RNase P and interference with activity, lack of inhibition does not distinguish between the smaller poly(U) RNAs binding more weakly to RNase P or not affecting activity once bound. Therefore, a larger poly(U) inhibitor was used to further analyze binding to RNase P, as estimated by inhibition of pre-tRNA cleavage.
FIGURE 1.

Poly(U) RNA inhibition of RNase P–catalyzed pre-tRNATyr cleavage is length-dependent. (A) Denaturing polyacrylamide gel electrophoresis of radiolabeled pre-tRNATyr cleavage products was used to monitor RNase P activity in the presence of 100 nM poly(U) RNA of increasing size (25–63 nt) (Supplemental Fig. S1). All samples are from the same gel with the control sample indicating migration of pre-tRNATyr without RNase P and poly(U) repositioned graphically, which is indicated by a small space between the samples. (B,C) Poly(U)50 RNA inhibition of RNase P–catalyzed cleavage of radiolabeled pre-tRNATyr when [pre-tRNATyr] = 4 nM (<Km) (B) or [pre-tRNATyr] = 107 nM (>Km) (C) was measured in triplicate. The data are curve-fit using a binding isotherm with a nonzero activity at saturating inhibitor; the value of the apparent IC50, the endpoint, and the percent inhibition at the endpoint are indicated with the error from curve fitting shown as standard error (SEM). The Km for this pre-tRNA was previously determined to be 55 nM (Ziehler et al. 2000).
For further analysis of inhibition by poly(U) RNA, a chemically synthesized 50-nt poly(U), poly(U)50, was used. Relative IC50 values for poly(U)50 were obtained for conditions where the concentration of pre-tRNATyr was both below (Fig. 1B) and above (Fig. 1C) the Km value (Ziehler et al. 2000). The relative IC50 values for poly(U)50 are in the nanomolar range, indicative of potent inhibition; furthermore, the IC50 value increases significantly at the higher substrate concentration, indicating that the inhibitor and substrate are at least partially competitive for binding to RNase P. However, even at high concentrations, poly(U)50 does not completely inhibit RNase P–catalyzed cleavage; at saturating inhibitor 20% and 29% of the starting activity remain at high and low substrate concentrations, respectively. This result demonstrates that RNase P is capable of binding and cleaving substrate in the presence of bound inhibitor. These data are consistent with poly(U)50 binding to one or more sites in RNase P that directly or indirectly interfere with pre-tRNA binding or cleavage, but are not consistent with the inhibitor binding at a position that completely overlaps the pre-tRNA binding site (see Discussion for model). Our cross-linking studies, outlined below, characterize the sites of interaction between poly(U)50 and purified RNase P.
Mixed-sequence RNA binding can lead to RNase P cleavage
Given the previously observed sequence preference for homopolymer binding by RNase P (Ziehler et al. 2000), we wanted to determine if mixed-sequence RNA also inhibits RNase P. With the diversity of RNA that has been previously identified to both copurify with RNase P and change in abundance in strains with RNase P temperature-sensitive mutations, we chose multiple in vitro transcripts from both strands of the PHO84 locus as a representative region for potential RNase P non-tRNA substrates (Coughlin et al. 2008). Both strands were tested because this locus has been shown to have physiologically relevant antisense RNA (Camblong et al. 2007). Both sense and antisense RNA transcripts (250 nt each) comprising the entire locus were tested for inhibition of RNase P–catalyzed pre-tRNATyr cleavage. At a concentration of 100 nM inhibitor using a pre-tRNA substrate concentration <Km, all 24 sense and antisense RNAs inhibited RNase P activity, although there was variability in the amount of inhibition (Fig. 2A; Supplemental Fig. S2). This variability in inhibition could be dependent on both the binding affinity and the amount of residual activity at saturating inhibitor.
FIGURE 2.

Inhibition of pre-tRNATyr cleavage by mixed-sequence RNAs. (A) Partially overlapping transcripts of 250 nt from both the top strand (1S–12S) and the bottom strand (1AS–12AS) of the S. cerevisiae PHO84 locus and neighboring YML122C were tested for inhibition of RNase P–catalyzed pre-tRNATyr cleavage. Inhibition of radiolabeled pre-tRNATyr cleavage by (B) RNA 3S titrated in triplicate or (C) RNA 3AS titrated in duplicate. The endpoint indicates the catalytic activity at saturating inhibitor and the apparent IC50 value is indicated with error from curve fitting (SEM). The percent inhibition at the endpoint is also indicated.
To further assess inhibition by the mixed-sequence RNAs, relative IC50 values at a pre-tRNA concentration <Km were obtained for one pair of sense and antisense RNAs, RNA 3S and RNA 3AS (Fig. 2B,C). Relative IC50 values for these RNAs are in the same range as that observed for poly(U)50 RNA. In addition, as was the case with poly(U)50, these RNAs did not completely inhibit RNase P; 6%–20% of the starting activity remains even at concentrations of mixed-sequence RNA that are 15–100 times higher than the IC50 values. Therefore, nuclear RNase P has a broad ability to bind mixed-sequence RNA in a way that partially conflicts with pre-tRNA cleavage, as estimated from the capacity of these RNAs to inhibit pre-tRNA cleavage. These data, combined with the diversity of RNAs both bound to and affected by RNase P, suggest that in vivo RNA interactions with RNase P are determined primarily by factors other than the sequence of the RNA ligands.
Next, we determined if RNase P could cleave any of these RNA inhibitors in vitro. At all levels of RNase P tested, no detectable cleavage products were observed after incubation of poly(U)50 RNA with RNase P (Fig. 3A), consistent with the results observed using longer poly(U) homopolymers (Ziehler et al. 2000; data not shown). However, both of the mixed-sequence RNAs (RNA 3S and RNA 3AS) used in the IC50 experiments were cleaved at multiple sites (Fig. 3A), as well as most other mixed-sequence RNAs tested (data not shown). Pre-treatment of highly purified RNase P with micrococcal nuclease (MNase) prior to adding mixed-sequence RNA resulted in the loss of cleaved product (Supplemental Fig. S3). This is consistent with the requirement for the nucleic acid subunit of RNase P, Rpr1r, for cleavage. We conclude that nuclear RNase P, and not a minor nuclease contaminant, is directly responsible for the cleavage of this mixed-sequence RNA.
FIGURE 3.

Cleavage of single-stranded RNA by RNase P. (A) Twofold increasing concentrations of RNase P (3.8 pM–60 pM for pre-tRNATyr; 10 pM–85 pM for all other RNA) were used for testing cleavage of radiolabeled RNAs (pre-tRNATyr, RNA 3S, and RNA 3AS) and 5′ radiolabeled poly(U)50 after incubation for 15 min. (B) The cleavage sites in 5′ radiolabeled RNA 3S at increasing amounts of RNase P were identified by comparison to cleavage of mixed sequence RNA by RNases with known specificity. The nucleotide specificity of RNases used for mapping is indicated. Five major sites of RNase P cleavage are indicated.
To determine if there is a sequence preference for cleavage of RNA by RNase P, we mapped the multiple cleavage sites obtained from RNase P–catalyzed cleavage of the mixed-sequence RNA 3S (Fig. 3B). As is shown in Figure 4, no strong sequence specificity for the cleavage sites was identified, nor is there evidence of structural consensus in the area surrounding the position of cleavage (Fig. 4A; Zuker 2003). In addition, upon comparison of regions surrounding the cleavage site with other previously identified RNAs cleaved by nuclear RNase P, there was no major sequence specificity for cleavage except a possible preference (12/17) for adenosine at position +3 relative to the cleavage site (Fig. 4B; Chamberlain et al. 1996; Coughlin et al. 2008). This is in contrast to the cleavage preference recently determined for RNase MRP, which also does not show extensive requirements for cleaving naked RNA, but has a strong consensus for cytosine at position +4 relative to the cleavage site (Esakova et al. 2011).
FIGURE 4.

RNA 3S does not fold into predicted tRNA-like structures, and RNase P cleavage sites do not show strong consensus sequences. (A) mFold (http://www.bioinfo.rpi.edu/applications/mfold) was used to predict RNA secondary structures at cleavage sites and the surrounding RNA 3S sequence. Sixty-one-nucleotide RNA fragments were used for folding to examine regions in close proximity to the cleavage sites. Arrows indicate sites of RNase P cleavage. (B) Cleavage site alignment of RNA 3S (cleavage sites 1–5) (Fig. 3B) is shown with previously identified preferential cleavage sites for yeast nuclear RNase P in snoRNAs and pre-ribosomal RNA ITS1 (Chamberlain et al. 1996; Coughlin et al. 2008). Multiple cleavages from the same RNA are indicated by numerals after the name. Cleavage sites are centered and indicated by an arrow and a bold line. Only a very weak consensus was obtained using the EDNAFULL alignment matrix (XNNANAUN5UN16UU) with X indicating the cleavage site.
Even though our data do not indicate strong sequence or structural consensus for RNase P cleavage, it is possible that local RNA structure might play a role in cleavage that we do not currently understand given that poly(U) RNA, which is predicted to lack stable secondary and tertiary structures, is not cleaved (Fig. 3A). Furthermore, only a small fraction of the mixed-sequence RNA is cleaved by RNase P (Fig. 3A). Increasing the RNase P concentration by as much as eightfold only enhances cleavage by a small fraction, perhaps suggesting the existence of multiple, slowly interconverting conformers of the mixed-sequence RNAs. Also, RNase P cleaves the mixed-sequence RNA at multiple positions, which suggests that slow cleavage could be due to suboptimal positioning of RNA in the active site. These data suggest that RNase P binds RNA in multiple positions or in multiple conformers with some of these bound complexes, leading to low levels of cleavage.
Identification of RNA contact sites in RNase P
We used cross-linking to investigate contacts between bound RNA and highly purified RNase P, comparing uncleavable poly(U)50 RNA with cleavable RNAs (pre-tRNATyr and RNA 3S). Covalently linked complexes were separated by denaturing polyacrylamide gel electrophoresis (Fig. 5A). We tested multiple cross-linking reagents, but only ultraviolet light irradiation (UV, 254 nm) provided discrete, reproducible cross-linked complexes between RNA and RNase P. Treatment with either formaldehyde or glutaraldehyde resulted in extremely heterogeneous or high-molecular-weight migration on gels, consistent with multiple cross-linking events per complex (data not shown). Using poly(U)50 RNA the major UV cross-links formed are with the RNA subunit, Rpr1r, as initially judged by insensitivity of the major shifted band to proteinase K on denaturing gels containing 7 M urea (Fig. 5A). Unlabeled pre-tRNATyr competes with poly(U)50 for all RNase P–dependent shifts, indicating that substrate pre-tRNAs compete for binding to a majority of the cross-linking contacts.
FIGURE 5.

Single-stranded RNAs and pre-tRNA cross-link to RNase P RNA. (A) 5′ radiolabeled poly(U)50 is shown on a denaturing polyacrylamide gel. A cross-link-dependent shift is observed with UV light and RNase P, which is also shown to be resistant to deproteinization by proteinase K. Unlabeled pre-tRNATyr is shown to compete for the RNase P–poly(U)50 cross-links with cross-linking also observed to poly(U)50 in the absence of RNase P regardless of deproteinization. (B) RNA ligands [pre-tRNATyr, RNA 3S, poly(U)50] cross-link to the CR-I region of Rpr1 RNA. After deproteinization, primer extension stops were compared from uncross-linked and cross-linked RNase P to RNase P cross-linked in the presence of various RNA ligands. Unique extension stops in the presence of RNA ligands represent sites of cross-linking between RNase P RNA Rpr1 and the RNA ligand. The primer extensions are mapped using the indicated dideoxy sequencing ladder lanes. Only the Rpr1r sequence where ligands cross-link is shown with ligand type indicated. Rpr1r sequence where the cross-links occur is shown next to the primer extension data. (C) Secondary structure of Rpr1 RNA showing positions of cross-linking by poly(U), RNA 3S, and pre-tRNATyr, as identified in B. Conserved stems are indicated (P1, etc.) along with eukaryotic specific helixes (eP8, etc.). Also, universally conserved sequence regions are shown (CRI-CRV).
To clarify if poly(U)50, RNA 3S, and pre-tRNATyr bind to similar site(s) in Rpr1r, we identified cross-linking positions for each of the RNA ligands with purified RNase P. After UV cross-linking and deproteinization, cross-linked sites were identified by primer extension analysis (Fig. 5B). Primer extensions were done to examine the entire sequence of Rpr1r, minus only the extreme 3′ end for technical reasons (primer hybridization), and the only significant cross-links to all three RNA ligands were found in a single region. Each of the identified cross-links to poly(U)50, pre-tRNATyr, or RNA 3S were found within “Conserved Region I” (CR-I) (Fig. 5B,C). This region is an absolutely conserved feature of all known RNase P RNAs and is thought to comprise part of the catalytic core of the ribozyme (Chen and Pace 1997; Reiter et al. 2010).
Although the major UV-induced cross-links to the RNA ligands occur with the Rpr1 RNA subunit, we also investigated whether cross-links could be detected with protein subunits. Multiple cross-linked RNase P proteins were identified, although possible approaches were limited by inefficiency of cross-linking and the small amounts of the low-copy and unstable holoenzyme that could be purified to homogeneity (Hsieh et al. 2009). When separated on SDS-polyacrylamide gels to observe denatured protein migration (Fig. 6), poly(U)50–RNase P cross-links were visible as multiple discrete shifted bands. The smaller of these bands was not identified previously using 7 M urea gels (Fig. 5A), possibly because these protein-containing complexes were part of insoluble aggregates that routinely failed to enter the urea gels. The two upper-shifted bands are consistent with cross-linking of poly(U)50 to the Rpr1 RNA subunit since they were proteinase K–resistant (data not shown). Furthermore, this large doublet is consistent with internally cross-linked Rpr1r, which gives two or more distinct bands on urea gels (Supplemental Fig. S4). However, the two lower-shifted bands were sensitive to proteinase K treatment, indicating probable protein subunit cross-links (data not shown).
FIGURE 6.

Poly(U)50 RNA cross-links to RNase P proteins. 5′ radiolabeled poly(U)50 is shown with RNase P separated on a SDS polyacrylamide gel. A cross-link-dependent shift is observed with UV light and RNase P. Indicated regions (gel slices 1, 2, 3) were analyzed by mass spectrometry in cross-linked and uncross-linked lanes (Supplemental Table S1). Proteins that are interpreted to cross-link to poly(U)50 (found only in cross-linked gel slices) are in bold type, while ambiguous proteins (found in both cross-linked and uncross-linked gel slices) are in parenthesis. Relative migration of a protein ladder (SeeBlue Plus2) is shown.
LC-MS/MS analysis of gel slices from equivalent positions containing the poly(U)50 RNA with or without cross-linking showed the cross-linking-dependent association of several proteins with poly(U) (Fig. 6; Supplemental Table S1). Pop4p and Rpr2p associated with shifted poly(U)50 only in the cross-linked lanes, and we interpret this as indicating that these proteins are in close contact with the bound single-stranded RNA. Although tested extensively, we did not detect UV cross-links between pre-tRNA substrate and RNase P protein subunits, perhaps due to rapid cleavage of the substrate. However, given that pre-tRNA competes for cross-link shifts with radiolabeled poly(U)50 (data not shown), the identified RNase P proteins (Pop4, Rpr2) that cross-link to poly(U)50 RNA could also be near the pre-tRNA substrate.
It is possible that the largest protein, Pop1p, is also bound to the poly(U)50, although the data are ambiguous. Peptides from the C terminus of Pop1p are found migrating in the smallest shifted band in a UV-dependent fashion, as well as throughout the gel at larger (slower migrating) positions in a UV-independent fashion (Fig. 6; Supplemental Table S1). Full-length Pop1 would be expected to substantially retard the migration of the cross-linked poly(U)50. Since Pop1 is known to be particularly susceptible to proteolysis, the data would be consistent with proteolysis either before or after UV-induced cross-linking (Lygerou et al. 1994; Chamberlain et al. 1998). The only other RNase P protein identified by mass spectrometry was Rpp1p, which migrates at analyzed gel positions whether or not UV cross-linking takes place, so if there was an Rpp1p-dependent shift, it would be masked (Supplemental Table S1). In addition, given the strict spatial limitations for UV-light-induced protein–RNA cross-linking combined with the low efficiency of obtained RNase P cross-links, it is entirely possible that other RNase P protein subunits make physiologically important contacts with substrates.
DISCUSSION
Broad RNA recognition potential for yeast nuclear RNase P
Previous work indicated a strong sequence preference for inhibition of yeast nuclear RNase P by polynucleotide homopolymers (Ziehler et al. 2000). These RNAs did not similarly inhibit the simpler bacterial enzyme, and it is proposed that the additional proteins found in the eukaryotic enzyme may have a direct influence on ligand binding. Here we have observed that many mixed-sequence RNAs bind to yeast nuclear RNase P and inhibit cleavage of pre-tRNAs. Although there is some variability in the apparent affinity of different RNAs, we find no obvious sequence preferences, suggesting that yeast nuclear RNase P can bind a broad set of mixed-sequence RNAs.
We also observe that RNase P cleaves mixed-sequence RNAs at multiple sites, whereas poly(U) RNA is not cleaved despite being a potent inhibitor (Figs. 3, 4). Our cross-linking data indicate that poly(U) RNA binds close to the active site of RNase P and is positioned similarly to the substrates that can be cleaved, pre-tRNA and mixed-sequence RNA (Fig. 3A). Given that poly(U) RNA is not predicted to form stable secondary structure, these observations are consistent with a model in which RNA sequence or structure is a determinant for RNA cleavage catalyzed by RNase P, even though inhibition of RNase P is a relatively sequence-independent event (Figs. 1B,C, 2A; Supplemental Fig. S2).
It is also noted that only a small fraction of a particular mixed-sequence RNA was cleaved, despite increasing levels of RNase P. This would be consistent with only a fraction of RNA existing in the correct conformation for cleavage at any given moment. Not surprisingly, multiple secondary structure variations of similar stability can be predicted for RNA3S fragments surrounding the site of RNase P cleavage (Supplemental Fig. S5). This does not mean that this cleavage might not be important, however, as in vivo non-tRNA substrates might be correctly positioned for cleavage by protein cofactors that assist the folding of the RNA into the correct, cleavable conformation (Chamberlain et al. 1996; Coughlin et al. 2008). This could explain why multiple positions of the RNA are cleaved and why cleavage is slow (Fig. 3).
A recent study indicated that RNase MRP has a different and also limited sequence preference for substrate cleavage (Esakova et al. 2011). Even though most of the protein subunits are shared between RNase P and RNase MRP, it appears that RNase MRP possesses a strict requirement for a cytosine at position +4 relative to the cleavage site. This difference could be due to variances in the RNA subunits between the two complexes, protein subunits, or a combination of these possibilities. Future study is warranted to more fully characterize RNase P and RNase MRP cleavage products in vivo, which will yield physiologically relevant results that account for the RNP structure of these substrates.
Model of single-stranded RNA binding RNase P
As expected, poly(U) RNA and mixed-sequence RNAs inhibit pre-tRNA cleavage by RNase P (Ziehler et al. 2000). The measured IC50 value increases at higher concentrations of pre-tRNA, consistent with a competitive inhibition model in which the RNA inhibitors compete with pre-tRNA for binding to RNase P. However, even at concentrations significantly higher than the apparent IC50, these RNAs do not completely inhibit RNase P (Fig. 1B,C). This residual activity is not explained by a simple competitive inhibition model with the formation of an E•I complex that is in direct competition with E•S. One model that is consistent with our data is the formation of a ternary complex (E•S•I) that retains a small amount of cleavage activity (Scheme 1). However, our data do not preclude the formation of other ternary complexes, such as E•P•I, that are in equilibrium with E•S. Finally, an alternative model consistent with the data is the existence of multiple forms of RNase P, perhaps in equilibrium, where one or more forms are inhibited by the non-tRNA substrates and the others are not inhibited. One possibility is that some protein subunits are modified or even missing from a subpopulation of the isolated holoenzyme, altering the RNA binding properties. One specialized example of this is that Pop1 tends to be partially proteolyzed, which is consistent with our observation of poly(U) cross-linking to Pop1 fragments of multiple apparent sizes (Fig. 6; Supplemental Table S1). However, all of the analytical data indicate that our purified RNase P is homogeneous, and RNase P from multiple preparations showed consistent levels of activity and inhibition levels. All of these possibilities point to the existence of at least two RNA binding sites in RNase P, one site that is competitive with pre-tRNA and a second site that is not competitive with tRNA.
SCHEME 1.

Inhibition of RNase P by mixed-sequence RNA was analyzed using the same method as poly(U) even though at high levels of RNase P, some cleavage of mixed-sequence RNA was observed (Fig. 3). Given the slow rate of cleavage at the concentrations of RNase P used for the inhibition experiments, the data can be explained by the same inhibition mechanisms used for poly(U) (Scheme 1), except that the observed IC50 value may reflect the value of KM rather than an apparent dissociation constant.
The catalytic RNA core of RNase P can interact with a diverse set of RNAs
The catalytic core of RNase P (helix P4) is formed by sequences from CR-I and CR-V and makes only limited contacts with pre-tRNATyr substrates within the CR-I region (Fig. 5). Earlier cross-linking results with the deproteinized Schizosaccharomyces pombe RNase P RNA subunit and mature tRNA found multiple cross-links throughout the RNA subunit (Marquez et al. 2006). However, we find that all of the tested pre-tRNA and single-stranded RNA ligands cross-link with helix P4 in the active site of the S. cerevisiae holoenzyme, consistent with the position of tRNA in the recent bacterial crystal structure from Reiter et al. (2010). This could be due to more precise positioning of the RNAs in the holoenzyme, protection of inappropriate sites in the RNA subunit by protein coverage, or both.
The precise positioning of RNA ligands bound to the RNase P active site was shown with our cross-linking results, although the exact nucleotides within Rpr1r that were in contact with the pre-tRNATyr, poly(U)50, and RNA 3S ligands differ (Fig. 5B). This could be consistent with the observed differences in cleavage competency among the RNA ligands, especially if the cross-linked single-stranded ligand was not the same as the small population that was cleavable (Fig. 3). The lack of complete congruence of the cross-linking sites is also consistent with the inhibition curves for poly(U) and the mixed-sequence RNAs, which suggests that they do not completely block the active site (Figs. 1B,C, 2B,C).
RNase P protein subunits interact with single-stranded RNA
Although yeast nuclear RNase P and bacterial RNase P have been shown to have similar kinetic behavior with pre-tRNA substrates, the significantly increased content of basic proteins of the yeast enzyme and the inhibition by homopolymer RNA argued for a broadened ability to bind single-stranded RNAs (Hsieh et al. 2009). We strove to understand how single-stranded RNA bound to the complex to identify eukaryotic-specific modes of interaction. The size dependence of unstructured poly(U) binding suggests that multiple interactions with the holoenzyme are required for tight binding and inhibition of pre-tRNA cleavage (Fig. 1A). Many of the protein subunits are potential candidates for RNA binding, given that seven of the nine RNase P proteins are highly basic and several studies have shown that most of these proteins can bind RNA in vitro (Walker and Engelke 2006). In addition, structural studies have shown that Pop6p and Pop7p bind specifically to the P3 region of Rpr1r in S. cerevisiae (Perederina et al. 2010). Consistent with this, we do not find that Pop6p and Pop7p cross-link to bound RNA ligands, but rather at least two other proteins (Pop4p and Rpr2p), and possibly more (including Pop1p), are in close contact with poly(U)50 RNA (Fig. 6; Supplemental Table S1). These two proteins interacting with RNA ligands is consistent with the finding that archaeal homologs of yeast Rpr2p and Pop4p, RPP21 and RPP29, increase substrate binding affinity, but not pre-tRNA cleavage in reconstituted enzymes (Chen et al. 2010). Future studies of RNase P should examine the mechanism(s) of how the extensive protein complement of nuclear RNase P helps to capture and control the cleavage of physiological substrates.
In summary, our results show that yeast nuclear RNase P can bind and cleave a diverse set of RNAs in vitro and suggests that future studies of non-tRNA RNase P substrates will need to identify determinants other than intrinsic RNA sequence for investigating non-tRNA substrates in vivo. In addition, we have shown that both pre-tRNAs and diverse single-stranded RNAs bind to the active site of the Rpr1r RNA subunit. These data provide a model of nuclear RNase P in which the increased protein content allows binding of non-tRNA substrates in such a way as to allow positioning of RNA within the same catalytic site used by the ancient ribozyme for pre-tRNA 5′ end removal.
MATERIALS AND METHODS
Yeast strains
Yeast nuclear RNase P was isolated from the S. cerevisiae strain SCWY10 (Hsieh et al. 2009). For some experiments, this strain contained a C-terminal 6xHis tag on one RNase P protein subunit (pop6::6HIS-HYG or pop8::6HIS-HYG).
Yeast extract preparation
The yeast strain SCWY10, and its 6xHIS-tagged derivatives were prepared as in Hsieh et al. (2009) with the following exceptions: Yeast (9–36 L) were lysed in 10 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM Mg-acetate, 1 mM imidazole, 2 mM CaCl2, 0.1% NP-40, and EDTA free Complete protease inhibitors (Roche) by passing through 200-μm and 100-μm chambers 10 times each. Extract was centrifuged at 17,000g for 30 min and stored at −80°C.
RNase P purification
RNase P was purified using multiple affinity-based methods described below, with extract preparation derived from Hsieh et al. (2009). RNase P purifications did not contain RNase MRP RNA as determined by Northern blot, which was also used to determine the amount of RNase P relative to control RNA synthesized in vitro, and for RNase P cross-linking controls (Hsieh et al. 2009). The RNase P fraction used in cross-linking was highly purified, showing only the subunit pattern expected for RNase P in denaturing protein gels (Hsieh et al. 2009).
For isolation Method 1, yeast extract was bound in batch to 5 μL of packed calmodulin resin per 1 mL of extract with constant mixing for 2 h at 4°C. Calmodulin affinity resin (Stratagene) was washed three times with 40 resin volumes of lysis buffer without protease inhibitors in batch. Two consecutive elutions were carried out with five resin volumes of lysis buffer plus 20 mM EGTA (calmodulin elution buffer). Pooled elutions were diluted to a conductivity equivalent of 100 mM NaCl with calmodulin elution buffer lacking NaCl. Samples were then bound to 125 μL of packed DEAE cellulose resin (DE52, Whatman) per 1 mL of sample for 1 h at 4°C with constant mixing. Washes were carried out in batches with five resin volumes of calmodulin elution buffer, followed by two washes with a 50% mix of calmodulin elution buffer and DEAE wash buffer (10 mM HEPES at pH 7.5, 10 mM MgCl2, 0.1% NP-40), and finally two washes with DEAE wash buffer. Samples were then eluted using DEAE elution buffer (400 mM NaCl, 10% glycerol, 10 mM HEPES at pH 7.5, 10 mM MgCl2, 0.1% NP-40).
For isolation Method 2, samples from Method 1 were diluted to a monovalent salt equivalent of 150 mM with calmodulin elution buffer (above) and applied to a 1-mL mono-Q column (Amersham-Pharmacia). Bound sample was washed with 0.1 M NaCl buffer (10 mM HEPES at pH 7.5, 10 mM MgCl2, 100 mM NaCl, 10% glycerol). RNase P was eluted using a 15-mL linear gradient of 0.1–0.8 M NaCl. Samples reproducibly eluted between 200 mM and 230 mM equivalent salt.
RNA preparation
Pre-tRNATyr with a 12-nt leader was transcribed from a linearized plasmid using titrated amounts of T7 RNA polymerase (Milligan and Uhlenbeck 1989; Hsieh et al. 2009). Mixed-sequence RNA from the PHO84 locus was transcribed using templates generated by PCR products containing T7 promoters from S. cerevisiae genomic DNA (Supplemental Table S2; Ziehler et al. 2000). Radiolabeled RNA was transcribed with either [α-32P]UTP (3000 Ci/mmol) or [α-32P]GTP (3000 Ci/mmol) and 0.1 mM UTP or GTP in modified buffer (Milligan and Uhlenbeck 1989). HPLC-purified poly(U)50 RNA was purchased from Integrated DNA Technology, whereas poly(U) of various sizes was produced by alkaline hydrolysis and separated by size (see below). Transcribed RNA was treated with Antarctic Phosphatase (NEB), 5′ radiolabeled with [γ-32P]ATP (6000 Ci/mmol) using T4 polynucleotide kinase (NEB), and purified using 6%–8% denaturing polyacrylamide gel electrophoresis (PAGE). Poly(U)50 RNA was 5′ radiolabeled without Antarctic Phosphatase treatment.
Alkaline hydrolysis of Poly(U) RNA
Alkaline hydrolysis of a mix of poly(U) RNA (Sigma-Aldrich) was done by incubating 10 mg of poly(U) RNA with 20 mM NaOH for 8 min at 65°C. The reaction was quenched by adding 188 mM NaOAc (pH 5.2). Samples were separated on a 10% polyacrylamide gel, and regions every 10 cm were eluted out of the gel. Samples were 5′ radiolabeled with [γ-32P]ATP using T4 polynucleotide kinase (NEB) and separated on a 10% denaturing polyacrylamide gel next to Decade Markers (Ambion).
Inhibition studies
RNase P (17.4 pM) was incubated for 15 min at 25°C with 4 nM both radiolabeled and unlabeled pre-tRNATyr in the presence of unlabeled inhibitor RNA [poly(U) and mixed-sequence RNA] in RNase P buffer (10 mM HEPES at pH 7.5, 10 mM MgCl2, 100 mM NaCl). Higher amounts of RNase P (152 pM) were needed to result in observable cleavage at 107 nM pre-tRNATyr. Reactions were stopped by adding an equal volume of 2× FEXBS (47.5% formamide, 7.5 mM EDTA, 0.0125% SDS, 0.01% xylene cyanol dye, 0.01% bromophenol blue dye). Reactions were separated using denaturing 8% PAGE and visualized with a Typhoon Trio+ imager.
Prism 5.0a (GraphPad Software) was used for nonlinear regression of the fraction of radiolabeled pre-tRNATyr produced under increasing concentrations of RNA inhibitors using Equation 1:
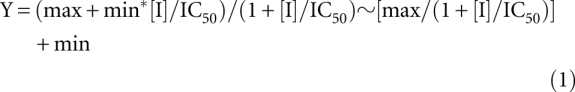 |
In Equation 1, max is the initial velocity in the absence of inhibitor and min is the velocity in the presence of saturating inhibitor. Initial velocity (per minute) was calculated using moles of substrate divided by moles of RNase P per time of the reaction.
Cleavage assays
In vitro cleavage of ∼1–2 ng of radiolabeled RNA by twofold dilutions of RNase P (0.038–0.6 fmol for pre-tRNATyr and poly(U)50; 0.1–0.85 fmol for RNA 3S/AS) was carried out for 15 min in RNase P buffer at 25°C. Cleavage of 1 ng of 5′ radiolabeled RNA 3S by RNase P (0.21–0.84 fmol) was carried out for 20 min at 25°C. Positions of cleavage were mapped using cleavage of RNA 3S using 0.2 ng of RNase A (Roche) and 0.2 units of RNase T1 (GIBCO BRL) as described (Ziehler and Engelke 2001). Reactions were stopped with 2× FEXBS and separated using 8% denaturing PAGE, then visualized.
Micrococcal nuclease digestion
One hundred or 200 units of micrococcal nuclease (MNase; Worthington Biochemical Corporation) was incubated in 10 mM HEPES (pH 7.5), 4 mM CaCl2, and 12 mM MgCl2 with RNase P (1 fmol) for 10 min at 37°C. Some reactions were pre-treated with 40 mM EGTA prior to the addition of RNase P, and all MNase reactions were terminated with 40 mM EGTA. RNase P cleavage was then started by the addition of radiolabeled RNA (pre-tRNATyr or RNA 3S) with RNase P buffer containing 12 mM MgCl2 for 20 min at 25°C. Reactions were stopped by EtOH precipitation, separated using 6% denaturing PAGE in 2× FEXBS, and then visualized.
Cross-linking
RNase P (100 fmol) was cross-linked in the presence of unlabeled RNA [100 nM poly(U)50, pre-tRNATyr, or RNA 3S] or 5′ radiolabeled RNA [1 ng of poly(U)50] using 254-nm UV light (Model UVG-11) at a distance of 20 mm for 2 min in RNase P buffer with 135 mM NaCl on ice. Cross-links were EtOH-precipitated and resuspended in either 2× FEXBS for separation using 6% denaturing PAGE or 2× Laemmli buffer (with 2-mercaptoethanol; Bio-Rad) for separation on a 4%–15% Tris-HCl acrylamide gel (Bio-Rad) with a SeeBlue Plus2 pre-stained protein ladder (Invitrogen). To determine the nature of the 5′ radiolabeled poly(U)50 cross-links, 120 fmol of pre-tRNATyr was added prior to cross-linking. In addition, some cross-linked samples were treated with 1/10 volume CP stop (2% SDS, 100 mM EDTA, 1 mg/mL proteinase K [Roche]) for 20 min at 42°C prior to denaturing PAGE and visualization.
Pre-tRNATyr, poly(U)50, and RNA 3S cross-link positions in Rpr1r were determined using a Sensiscript primer extension kit (QIAGEN) after CP stop treatment and acid phenol/chloroform treatment followed by EtoH precipitation. Primer extension for 50 min at 42°C was performed using oligonucleotides labeled with [γ-32P]ATP (6000 Ci/mmol) using PNK (NEB) (Supplemental Table S2). Dideoxy sequencing ladders were generated using Rpr1 DNA from a pUC19 plasmid (Hull et al. 1991). After this, EtOH precipitation samples were resuspended in 2× FEXBS and separated on a 6% denaturing polyacrylamide gel for visualization.
Mass spectroscopy
Uncross-linked and UV cross-linked RNase P (1 pmol) (Hsieh et al. 2009), both with 100 nM poly(U)50 RNA, was cut out of a 4%–15% Tris-HCl acrylamide gel (Bio-Rad). Gel slices were trypsin-digested, and peptide masses were identified using an LC-MS/MS (nano-UPLC coupled to a Q-Tof premier) at the UMMS Proteomics & Mass Spectrometry Facility (Rosenegger et al. 2010).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank May Tsoi for preparation of large volumes of yeast media. This work was supported by grant GM034869 (to D.R.E.), and UM Cellular Biotechnology Training Grant T32-GM08353 and a fellowship from Horace H. Rackham Graduate School (both to M.C.M.). In addition, funding was provided by grant GM 55387 (to C.A.F.) from the NIH.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.2633611.
REFERENCES
- Alifano P, Rivellini F, Piscitelli C, Arraiano CM, Bruni CB, Carlomagno MS 1994. Ribonuclease E provides substrates for ribonuclease P-dependent processing of a polycistronic mRNA. Genes Dev 8: 3021–3031 [DOI] [PubMed] [Google Scholar]
- Altman S, Wesolowski D, Guerrier-Takada C, Li Y 2005. RNase P cleaves transient structures in some riboswitches. Proc Natl Acad Sci 102: 11284–11289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothwell AL, Stark BC, Altman S 1976. Ribonuclease P substrate specificity: cleavage of a bacteriophage phi80-induced RNA. Proc Natl Acad Sci 73: 1912–1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camblong J, Iglesias N, Fickentscher C, Dieppois G, Stutz F 2007. Antisense RNA stabilization induces transcriptional gene silencing via histone deacetylation in S. cerevisiae. Cell 131: 706–717 [DOI] [PubMed] [Google Scholar]
- Chamberlain JR, Pagán-Ramos E, Kindelberger DW, Engelke DR 1996. An RNase P RNA subunit mutation affects ribosomal RNA processing. Nucleic Acids Res. 24: 3158–3166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain JR, Lee Y, Lane WS, Engelke DR 1998. Purification and characterization of the nuclear RNase P holoenzyme complex reveals extensive subunit overlap with RNase MRP. Genes Dev 12: 1678–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DD, Clayton DA 1987. A novel endoribonuclease cleaves at a priming site of mouse mitochondrial DNA replication. EMBO J 6: 409–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JL, Pace NR 1997. Identification of the universally conserved core of ribonuclease P RNA. RNA 3: 557–560 [PMC free article] [PubMed] [Google Scholar]
- Chen W-Y, Pulukkunat DK, Cho I-M, Tsai H-Y, Gopalan V 2010. Dissecting functional cooperation among protein subunits in archaeal RNase P, a catalytic ribonucleoprotein complex. Nucleic Acids Res 38: 8316–8327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlin DJ, Pleiss JA, Walker SC, Whitworth GB, Engelke DR 2008. Genome-wide search for yeast RNase P substrates reveals role in maturation of intron-encoded box C/D small nucleolar RNAs. Proc Natl Acad Sci 105: 12218–12223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JT 2004. G-quartets 40 years later: from 5′-GMP to molecular biology and supramolecular chemistry. Angew Chem Int Ed Engl 43: 668–698 [DOI] [PubMed] [Google Scholar]
- Esakova O, Perederina A, Quan C, Berezin I, Krasilnikov AS 2011. Substrate recognition by ribonucleoprotein ribonuclease MRP. RNA 17: 356–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans D, Marquez SM, Pace NR 2006. RNase P: interface of the RNA and protein worlds. Trends Biochem Sci 31: 333–341 [DOI] [PubMed] [Google Scholar]
- Frank DN, Pace NR 1998. Ribonuclease P: Unity and diversity in a tRNA processing ribozyme. Annu Rev Biochem 67: 153–180 [DOI] [PubMed] [Google Scholar]
- Giegé R, Florentz C, Dreher TW 1993. The TYMV tRNA-like structure. Biochimie 75: 569–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill T, Cai T, Aulds J, Wierzbicki S, Schmitt ME 2004. RNase MRP cleaves the CLB2 mRNA to promote cell cycle progression: Novel method of mRNA degradation. Mol Cell Biol 24: 945–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimple O, Schön A 2001. In vitro and in vivo processing of cyanelle tmRNA by RNase P. Biol Chem 382: 1421–1429 [DOI] [PubMed] [Google Scholar]
- Gobert A, Gutmann B, Taschner A, Gößringer M, Holzmann J, Hartmann RK, Rossmanith W, Giegé P 2010. A single Arabidopsis organellar protein has RNase P activity. Nat Struct Mol Biol 17: 740–744 [DOI] [PubMed] [Google Scholar]
- Guerrier-Takada C, Gardiner K, Marsh T, Pace N, Altman S 1983. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 35: 849–857 [DOI] [PubMed] [Google Scholar]
- Hall TA, Brown JW 2002. Archaeal RNase P has multiple protein subunits homologous to eukaryotic nuclear RNase P proteins. RNA 8: 296–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen A, Pfeiffer T, Zuleeg T, Limmer S, Ciesiolka J, Feltens R, Hartmann RK 2001. Exploring the minimal substrate requirements for trans-cleavage by RNase P holoenzymes from Escherichia coli and Bacillus subtilis. Mol Microbiol 41: 131–143 [DOI] [PubMed] [Google Scholar]
- Hartmann RK, Heinrich J, Schlegl J, Schuster H 1995. Precursor of C4 antisense RNA of bacteriophages P1 and P7 is a substrate for RNase P of Escherichia coli. Proc Natl Acad Sci 92: 5822–5826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzmann J, Frank P, Löffler E, Bennett KL, Gerner C, Rossmanith W 2008. RNase P without RNA: Identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell 135: 462–474 [DOI] [PubMed] [Google Scholar]
- Hsieh J, Walker SC, Fierke CA, Engelke DR 2009. Pre-tRNA turnover catalyzed by the yeast nuclear RNase P holoenzyme is limited by product release. RNA 15: 224–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull MW, Thomas G, Huibregtse JM, Engelke DR 1991. Protein–DNA interactions in vivo examining genes in Saccharomyces cerevisiae and Drosophila melanogaster by chromatin footprinting. Methods Cell Biol 35: 383–415 [PubMed] [Google Scholar]
- Jarrous N 2002. Human ribonuclease P: Subunits, function, and intranuclear localization. RNA 8: 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung YH, Lee Y 1995. RNases in ColE1 DNA metabolism. Mol Biol Rep 22: 195–200 [DOI] [PubMed] [Google Scholar]
- Kankia BI 2003. Mg2+-induced triplex formation of an equimolar mixture of poly(rA) and poly(rU). Nucleic Acids Res 31: 5101–5107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komine Y, Kitabatake M, Yokogawa T, Nishikawa K, Inokuchi H 1994. A tRNA-like structure is present in 10Sa RNA, a small stable RNA from Escherichia coli. Proc Natl Acad Sci 91: 9223–9227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DY, Clayton DA 1998. Initiation of mitochondrial DNA replication by transcription and R-loop processing. J Biol Chem 273: 30614–30621 [DOI] [PubMed] [Google Scholar]
- Li Y, Altman S 2003. A specific endoribonuclease, RNase P, affects gene expression of polycistronic operon mRNAs. Proc Natl Acad Sci 100: 13213–13218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Altman S 1994. Differential evolution of substrates for an RNA enzyme in the presence and absence of its protein cofactor. Cell 77: 1093–1100 [DOI] [PubMed] [Google Scholar]
- Lu Q, Wierzbicki S, Krasilnikov AS, Schmitt ME 2010. Comparison of mitochondrial and nucleolar RNase MRP reveals identical RNA components with distinct enzymatic activities and protein components. RNA 16: 529–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lygerou Z, Mitchell P, Petfalski E, Séraphin B, Tollervey D 1994. The POP1 gene encodes a protein component common to the RNase MRP and RNase P ribonucleoproteins. Genes Dev 8: 1423–1433 [DOI] [PubMed] [Google Scholar]
- Marquez SM, Chen JL, Evans D, Pace NR 2006. Structure and function of eukaryotic Ribonuclease P RNA. Mol Cell 24: 445–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvin MC, Engelke DR 2009. Broadening the mission of an RNA enzyme. J Cell Biochem 108: 1244–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvin MC, Clauder-Münster S, Walker SC, Sarkeshik A, Yates JR, III, Steinmetz LM, Engelke DR 2011. Accumulation of noncoding RNA due to an RNase P defect in Saccharomyces cerevisiae. RNA (this issue) doi: 10.1261/rna.2737511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan JF, Uhlenbeck OC 1989. Synthesis of small RNAs using T7 RNA polymerase. Methods Enzymol 180: 51–62 [DOI] [PubMed] [Google Scholar]
- Peck-Miller KA, Altman S 1991. Kinetics of the processing of the precursor to 4.5 S RNA, a naturally occurring substrate for RNase P from Escherichia coli. J Mol Biol 221: 1–5 [DOI] [PubMed] [Google Scholar]
- Perederina A, Esakova O, Quan C, Khanova E, Krasilnikov AS 2010. Eukaryotic ribonucleases P/MRP: the crystal structure of the P3 domain. EMBO J 29: 761–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter NJ, Osterman A, Torres-Larios A, Swinger KK, Pan T, Mondragón A 2010. Structure of a bacterial ribonuclease P holoenzyme in complex with tRNA. Nature 468: 784–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenegger D, Wright C, Lukowiak K 2010. A quantitative proteomic analysis of long-term memory. Mol Brain 3: 9 doi: 10.1186/1756-6606-3-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt ME, Clayton DA 1993. Nuclear RNase MRP is required for correct processing of pre-5.8S rRNA in Saccharomyces cerevisiae. Mol Cell Biol 13: 7935–7941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JK, Hsieh J, Fierke CA 2007. Importance of RNA–protein interactions in bacterial ribonuclease P structure and catalysis. Biopolymers 87: 329–338 [DOI] [PubMed] [Google Scholar]
- Walker SC, Engelke DR 2006. Ribonuclease P: The evolution of an ancient RNA enzyme. Crit Rev Biochem Mol Biol 41: 77–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker SC, Marvin MC, Engelke DR 2010. Eukaryote RNase P and RNase MRP. In Protein reviews: Ribonuclease P (ed. Liu F, Altman S), Vol. 10, pp. 173–202 Springer, New York [Google Scholar]
- Wilusz JE, Freier SM, Spector DL 2008. 3′ end processing of a long nuclear-retained noncoding RNA yields a tRNA-like cytoplasmic RNA. Cell 135: 919–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Altman S 2007. A noncoding RNA in Saccharomyces cerevisiae is an RNase P substrate. RNA 13: 682–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziehler WA, Engelke DR 2001. Probing RNA structure with chemical reagents and enzymes. Curr Protoc Nucleic Acid Chem 6: 6.1.1–6.1.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziehler WA, Day JJ, Fierke CA, Engelke DR 2000. Effects of 5′ leader and 3′ trailer structures on pre-tRNA processing by nuclear RNase P. Biochemistry 39: 9909–9916 [DOI] [PubMed] [Google Scholar]
- Zuker M 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31: 3406–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]