

Abstract
Inhibitory interneurons play a critical role in coordinating the activity of neural circuits. To explore the mechanisms that direct the organization of inhibitory circuits, we analyzed the involvement of tropomyosin-related kinase B (TrkB) in the assembly and maintenance of GABAergic inhibitory synapses between Golgi and granule cells in the mouse cerebellar cortex. We show that TrkB acts directly within each cell-type to regulate synaptic differentiation. TrkB is required not only for assembly, but also maintenance of these synapses and acts, primarily, by regulating the localization of synaptic constituents. Postsynaptically, TrkB controls the localization of a scaffolding protein, gephyrin, but acts at a step subsequent to the localization of a cell adhesion molecule, Neuroligin-2. Importantly, TrkB is required for the localization of an Ig superfamily cell adhesion molecule, Contactin-1, in Golgi and granule cells and the absence of Contactin-1 also results in deficits in inhibitory synaptic development. Thus, our findings demonstrate that TrkB controls the assembly and maintenance of GABAergic synapses and suggest that TrkB functions, in part, through promoting synaptic adhesion.
Introduction
Inhibitory interneurons are important for the processing of sensory information and generation of appropriate behaviors by modulating excitatory pathways in the nervous system. In the cerebellar cortex, different classes of inhibitory interneurons provide feedback and feedforward inhibition critical for cerebellar functions such as coordination, balance, gait and fine motor control (Ito, 1984). One class of inhibitory interneurons found in the internal granular layer (IGL), the Golgi cells, receives excitatory glutamatergic inputs from granule cells and in turn suppresses granule cell excitability via inhibitory GABAergic/glycinergic neurotransmission (Fig. 1A) (Ito, 1984). Thus, Golgi cells are the primary inhibitory neurons that regulate the activities of granule cells (Fig. 1A) (Ito, 1984). Consistent with this, elimination of Golgi cells results in overexcitation of granule cells and disruption in motor function (Watanabe et al., 1998). The synapses between Golgi cells and granule cells are localized within the cerebellar glomerulus, a highly organized structure, consisting of an excitatory mossy fiber ending, inhibitory Golgi axons and dendritic processes of granule cells (Fig. 1B) (Palay and Chan-Palay, 1974; Jakab and Hámori, 1988). The morphological and physiological properties of Golgi cells and granule cells have been characterized extensively (Eccles et al., 1967; Palay and Chan-Palay, 1974), but the mechanisms that control differentiation at their synapses are not well defined.
Figure 1.

Organization of GABAergic synaptic proteins in the developing and mature cerebellum. A, Major neuronal subtypes are organized in different lamina in the cerebellar cortex (+, excitatory input; −, inhibitory inputs). Golgi cells (G.c.) (green) provide feedback inhibition to granule cells (gr.c.) (red) in the IGL (Eccles et al., 1967). B, The glomerulus is a complex of synapses in which a mossy fiber ending (blue) forms glutamatergic synapses on the inner surface of the enveloping dendrites of granule cells (Berglund et al., 1999) and the axon termini of Golgi cells (green) form inhibitory synapses on the outer surface of the same dendrites (Jakab and Hámori, 1988). C–E, vGluT1 expression marks mossy fiber endings at P14 (C), P21 (D), and P90 (E). F, G, Quantification of the area of vGluT1 expression (F) expressed as sum of pixels per region analyzed, and the intensity of vGluT1 expression (G) expressed as fluorescent units (fl.u.) from P14–P90. H–J, Gephyrin expression (Berglund et al., 1999), found on granule cell dendrites, surrounds mossy fiber endings (blue) at P14 (H), P21 (I), and P90 (J). K, L, Quantification of the area (K) and intensity (L) of gephyrin expression in the IGL from P14–P90. M–O, GAD65 expression (green) marks Golgi axon varicosities which surrounds mossy fiber endings (blue) at P14 (M), P21 (N), and P90 (O). P, Q, Quantification of the area (P) and intensity (Q) of GAD65 expression. R–T, Golgi axon varicosities surrounding mossy fiber endings (blue) can also be marked by GAD67 expression (green) at P14 (R), P21 (S), and P90 (T). U, V, Quantification of the area (U) and intensity (V) of GAD67 expression. Scale bar, 10 μm. *Box plots contain lower and upper limits (“whiskers”), first and third quartile (edges of the box), median (white bar), mean (white dot), and outliers (black dots) of the dataset. From herein, refer to Tables S1–3 (available at www.jneurosci.org as supplemental material) for mean values and statistics (Mann–Whitney U test and Kruskal-Wallis test).
In addition to their involvement in neuronal survival and differentiation, neurotrophin-Trk receptor signaling also regulates synapse formation and plasticity (McAllister et al., 1999; Poo, 2001; Nagappan and Lu, 2005; Luikart and Parada, 2006). Several studies have implicated neurotrophin-Trk signaling in the development of cerebellar GABAergic synapses. In vivo and in vitro studies show that exogenous brain-derived neurotrophic factor (BDNF) promotes inhibitory synaptogenesis, whereas inhibition of BDNF binding results in a decrease in inhibitory synapses (Bao et al., 1999; Seil and Drake-Baumann, 2000). The primary BDNF receptor, tropomyosin-related kinase B (TrkB), is expressed in all neuronal subtypes in the developing and mature cerebellum (Klein et al., 1993; Yan et al., 1997). Loss of TrkB in all cerebellar cells leads to reduced number of GABAergic boutons in the molecular layer and IGL, and reduced number of Golgi-granule cell synapses (Rico et al., 2002). Moreover, the absence of TrkB in all cerebellar cells results in mice that are ataxic and have deficits in motor coordination (Rico et al., 2002). Thus, these studies show that TrkB controls important aspects of GABAergic synapse formation and cerebellar function, but raise several important questions. Does TrkB act within Golgi and/or granule cells to control GABAergic synaptic differentiation and ultrastructure? Is TrkB involved in the initial assembly and/or maintenance of these synapses? What downstream signaling pathways mediate the activities of TrkB? Does TrkB control the expression/localization of cell adhesion molecules known to control the formation of GABAergic inhibitory synapses?
Materials and Methods
Mouse strains
Mouse strains used in this study are as follows: Wnt1::Cre (Danielian et al., 1998), mGluR2::nlsCre-ires-eGFP, mα6::Cre (Funfschilling and Reichardt, 2002), TrkBfl (Grishanin et al., 2008), TrkBF616A (Chen et al., 2005), TrkBSHC/SHC and TrkBPLC/PLC (Minichiello et al., 1998, 2002), Contactin−/− (Berglund et al., 1999), ROSA::LacZ (Soriano, 1999), Thy1::YFP-H-line (Feng et al., 2000). For experiments using mice carrying a floxed TrkB allele (TrkBfl), both homozygous mice (TrkBfl/fl) without Cre and heterozygous mice (TrkBfl/+) with Cre were used as control.
To generate transgenic mice expressing Cre recombinase and eGFP under mGluR2 promoter elements, the Cre gene with a nuclear localization signal, followed by an internal ribosomal entry site (ires) to allow expression of enhanced green fluorescent protein (GFP), was cloned into vector containing the mouse 18.3 kb 5′-upstream genomic sequence including the first and second exons of the mGluR2 gene (vector containing mGluR2 promoter elements was a gift from S. Nakanishi, Osaka Bioscience Institute, Osaka, Japan) (Watanabe et al., 1998). For generation of mGluR2::nlsCre-ires-eGFP transgenic mice, the expression cassette was excised from the plasmid before pronuclear injection.
Generation of mice carrying a knock-in mutation with a substitution of phenylalanine (F) to alanine (A) at amino acid 616 of the TrkB gene (TrkBF616A) was described by others (Chen et al., 2005). Synthesis and preparation of [1-(1,1-dimethylethyl)-3-(1-naphthalenylmethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine] (1NMPP1) has been described by others (Chen et al., 2005). Homozygous TrkBF616A mice were treated with 50 mm 1NMPP1 or water for 28 d [postnatal day 0 (P0)–P28] and analyzed at P28, or for 20 d (P30–P50) and analyzed at P50.
Immunoblot and immunohistochemistry
Protein extracts were prepared from the cerebellum of control and Wnt1::Cre;TrkBfl/fl mice as previously described (Rico et al., 2002). We used 10 μg of protein per lane for SDS-PAGE and immunoblots. Antibodies used were: rabbit anti-TrkB extracellular domain (1:1000) (E. J. Huang et al., 1999), rabbit anti-GAD65 (glutamate decarboxylase 65) (1:640; Millipore Bioscience Research Reagents), mouse anti-GAD67 (1:7500; Millipore Bioscience Research Reagents), rabbit anti-gephyrin [1:1500; Synaptic Systems (SySy)], goat anti-Contactin-1 (1:1000; Neuromics), and mouse anti-β-tubulin (1:1000; Sigma). The level of each protein was normalized to the level of tubulin. Analysis was performed using chemifluorescence reagents (GE Healthcare) and a phosphor imager (Fujifilm FLA-2000).
Immunohistochemistry was performed on 15–20 μm cryosections using fluorophore-conjugated secondary antibodies (1:500–1:1000; Jackson Immunoresearch and Invitrogen). Slides containing cryosections were washed three times with PBS and then incubated in primary antibody [PBS, 0.1% Triton X-100 (TX100), 1% goat serum] overnight at 4°C. Slides were then washed three times with PBS (5 min each) and then incubated with secondary antibody (PBS, 0.1% TX100, 1% goat serum) for 1 h at room temperature. Slides were again washed three times with PBS (5 min each). Tissues from control and experimental conditions were placed on the same slide to ensure all tissues were processed the same.
Primary antibodies used in this study were as follows: guinea pig (gp) anti-vGluT1 (1:20,000; Millipore Bioscience Research Reagents); mouse anti-gephyrin (1:8000; SySy); rabbit anti-gephyrin (1:1000; SySy); rabbit anti-vGluT1 (1:10,000) and rabbit anti-GAD65 (1:8000) (gifts from T. Jessell, Columbia University, New York, NY) (Betley et al., 2009); mouse anti-GAD67 (1:10,000; Millipore Bioscience Research Reagents); rabbit vGAT (1:2000; SySy); goat anti-Contactin-1 (1:250; R&D Systems); goat anti-Contactin-1 (1:1000; Neuromics); rabbit anti-GFAP (1:1000; Dako); sheep anti-GFP (1:500; Biogenesis); rabbit anti-GFP (1:1500; Invitrogen); rabbit anti-Neurogranin (1:1000; Millipore Bioscience Research Reagents); mouse anti-mGluR2 (1:1500; Advanced Targeting Systems); gp anti-GlyT2 (1:5000; Millipore Bioscience Research Reagents); and goat anti-β-galactosidase (1:1000; AbD Serotec).
Quantification of synaptic protein expression
To determine the area and average pixel intensity of the expression of synaptic proteins surrounding individual mossy fiber endings within the IGL in lobules I-IV of the cerebellum (Rico et al., 2002), images of a fixed area in the IGL from different mice were collected on a Zeiss LSM 5 Pascal confocal microscope (63× and 100× objectives, numerical aperture 1.40, Plan-Apochromat), keeping individual pixel intensities in the linear range for control images. The mean area (sum of pixels above threshold) and fluorescent pixel intensity of the protein of interest surrounding an individual glomerulus within each analyzed region were determined on cryosections at subsaturating antibody concentrations and calculated using the histogram and color histogram function in NIH ImageJ (version 1.38x).
For analysis shown in Figure 1 and supplemental Figure S1 (available at www.jneurosci.org as supplemental material), the sum of pixels above threshold was measured in an area of 21,374 μm2 (63× objective) for each protein of interest and represented as an absolute number as previously described (Rico et al., 2002). For all other analysis, the sum of pixels above threshold was normalized to the sum of pixels of vGluT1 expression in an area of 8600 μm2 (100× objective) for each protein of interest and represented as an area ratio. The threshold was determined by measuring the average pixel intensity of an area of 225 μm2 between individual cerebellar glomerulus that does not express the protein of interest. At both 63× and 100×, it was possible to find areas not stained with individual markers between glomeruli. For analysis of the colocalized expression of Contactin-1 (see Fig. 8; supplemental Fig. S8, available at www.jneurosci.org as supplemental material), colocalization of Contactin-1 on individual cell-type is measured by using the RG2B Colocalization macros in ImageJ and Contactin-1 expression not localized on cell type-specific markers was omitted using the same macros (http://rsbweb.nih.gov/ij/plugins/rg2bcolocalization.html).
Figure 8.

Loss of Contactin-1 leads to a reduction of the localization of presynaptic and postsynaptic GABAergic proteins. A, B, The localization of GAD65 (green) in the IGL is reduced in P18 Contactin-1−/− mice (B) compared with control (A). C, Quantification of the area ratio of GAD65:vGluT1 expression in control and Contactin-1−/− mice. D, E, Similarly, the localization of GAD67 (green) in the IGL of is reduced in P18 Contactin-1−/− mice (E) compared with control (D). F, Quantification of the area ratio of GAD67:vGluT1 expression in control and Contactin-1−/− mice. G, H, The localization of gephyrin (Berglund et al., 1999) in the IGL is also reduced in P18 Contactin-1−/− mice (H) compared with control (G). I, Quantification of the area ratio of gephyrin:vGluT1 expression in control and Contactin-1−/− mice. Scale bar, 10 μm.
Quantification of synaptic junctions with electron microscopy
Mice were perfused and postfixed overnight using 4% paraformaldehyde. Cerebellar tissues were processed as previously described (Rico et al., 2002). We analyzed individual glomerulus from 20 electron micrographs (magnification of 11,500×, single 2 μm sections) for each experimental condition (n = 3). Only simple glomeruli were analyzed since large complex glomeruli containing more than one mossy fiber ending extended outside of the micrographs. Individual components of the glomerulus were identified by criteria described in the paragraph below using those published by others (Palay and Chan-Palay, 1974; Landis et al., 1983; Jakab and Hámori, 1988).
Mossy fiber endings.
Mossy fiber endings are interspersed between the cell bodies of granule cells (spherical and smooth surface with large blocks of condensed chromatin); they are filled with large, spheroid synaptic vesicles and contain numerous mitochondria typically concentrated in the center of the mossy fiber ending.
Granule cell dendrite.
Each finger-like dendritic branch contains a long mitochondria partially wrapped in endoplasmic reticulum; it does not contain synaptic vesicles and so appears much lighter in color compared with mossy fiber endings or Golgi axon varicosities.
Golgi axon varicosity.
The Golgi axon varicosity is located in the outskirts of the glomerulus. It is circular or elliptical-shaped compared with granule cell dendrites, contains ovoid or polygonal synaptic vesicles, and contains few mitochondria in the center of the varicosity. Synaptic junctions were counted when electron-dense material lining intracellular aspects of the presynaptic and postsynaptic membranes was observed between identified structures. Because only single-section electron micrographs were analyzed, not all synaptic junctions have clear membranes separated by electron-dense material. Plaques were counted whenever electron-dense material was observed between dendrites.
Results
Localization of GABAergic synaptic proteins within the glomeruli in the cerebellar cortex
Golgi cells and granule cells are major components of the cerebellar glomeruli located in the IGL (Fig. 1A). The glomerulus is composed of excitatory glutamatergic mossy fiber ending that synapses with the dendrites of granule cells (Fig. 1B). Additionally, the axon termini of Golgi cells form GABAergic/glycinergic synapses on the surface of granule cell dendrites opposite to the surface that is in contact with the mossy fiber ending (Fig. 1B). We analyzed the localization of proteins associated with both presynaptic and postsynaptic GABAergic synapses during their development at P14, P21, and in mature synapses at P90 (Altman and Bayer, 1997). Pre- and postsynaptic components were examined by monitoring the localization of presynaptic GABA synthetic enzymes (GAD65 and GAD67) and a postsynaptic scaffolding protein (gephyrin). The excitatory mossy fiber endings were marked by the localization of vesicular glutamate transporter-1 (vGluT1).
Focusing first on the development of the excitatory input provided by mossy fibers, the mean area of vGluT1 expression (expressed as an average of the sum of pixels) increased by 73% from P14 to P21 and did not significantly change from P21 to P90 [Fig. 1C–F; supplemental Table S1A, available at www.jneurosci.org as supplemental material; p < 0.001, Kruskal–Wallis (K.–W.)] (see Materials and Methods). The mean intensity of vGluT1 expression (fluorescence unit, fl.u.) remained unchanged from P14 to P21, but decreased by 6.5% from P21 to P90 (Fig. 1G; supplemental Table S1A, available at www.jneurosci.org as supplemental material; p < 0.05, K.–W.). The developmental profile of vGluT1 localization is consistent with an increased density and size of mossy fiber endings during P14–P90 (Mason and Gregory, 1984; Altman and Bayer, 1997). We next analyzed the development of inhibitory postsynaptic specializations formed by granule cell dendrites in contact with mossy fiber endings through examination of the localization of gephyrin, a postsynaptic scaffold protein associated with GABAergic and glycinergic synapses (Fritschy et al., 2008). The mean area covered by gephyrin expression increased by 79% from P14 to P21 and did not significantly change from P21 to P90 (Fig. 1H–K; supplemental Table S1A, available at www.jneurosci.org as supplemental material; p < 0.01, K.–W.). The mean intensity of gephyrin expression remained unchanged from P14 to P21, but increased by 10% between P21 and P90 (Fig. 1L; supplemental Table S1A, available at www.jneurosci.org as supplemental material; p < 0.001, K.–W.). Because these differences in intensity are small, we are unclear of their biological relevance.
The localization of GAD65 and GAD67 surrounding mossy fiber endings corresponds to the presynaptic axons/termini of Golgi cells (Fig. 1B,M–O,R–T). First, we observed an 18% reduction in the mean area of GAD65 expression between P14 and P21 (Fig. 1P; supplemental Table S1A, available at www.jneurosci.org as supplemental material). However, the mean area of GAD65 expression was only marginally different between P14, P21, and P90 (p = 0.07, K.–W.). The mean intensity of GAD65 expression remained unchanged from P14 to P21, but increased by 4% between P21 and P90 (Fig. 1Q; supplemental Table S1A, available at www.jneurosci.org as supplemental material; p < 0.05, K.–W.). Next, the mean area of GAD67 expression was reduced by 17% from P14 to P21 with a further reduction of 14% from P21 to P90 (Fig. 1U; supplemental Table S1A, available at www.jneurosci.org as supplemental material; p < 0.01, K.–W.). The mean intensity of GAD67 expression increased by 12% from P14 to P21 and then remained unchanged from P21 to P90 (Fig. 1V; supplemental Table S1A, available at www.jneurosci.org as supplemental material; p < 0.01, K.–W.). Our analysis suggests that the reduction in the area of the expression of GAD enzymes reflects an increased localization of these enzymes in regions juxtaposed to vGluT1+ mossy fiber ending. Studies in the cortex have reported a similar increase in localized GAD65 expression as inhibitory synapses mature (Z. J. Huang et al., 1999). More importantly, our analysis indicates that GAD65, GAD67 and gephyrin are present between P14 and P90 and can be used to monitor presynaptic and postsynaptic constituents of GABAergic synapses.
GABAergic synaptic differentiation requires TrkB
To gain further insight into the role of TrkB in the assembly and maintenance of GABAergic synapses, we used mice carrying a floxed TrkB allele (TrkBfl) in which loxP sites flank the first coding exon. Mice carrying the TrkBfl allele express full-length and truncated isoforms of TrkB at endogenous levels before Cre-mediated deletion (Grishanin et al., 2008). To determine the requirements of TrkB in GABAergic synaptic differentiation, we examined the consequences of TrkB deletion on the localization of presynaptic and postsynaptic proteins from P14 to P90.
Localized expression of presynaptic GABAergic proteins requires TrkB
We assessed the localization of presynaptic proteins in mice lacking TrkB in all cerebellar cell-types as a result of Wnt1::Cre-mediated deletion (Rico et al., 2002). Consistent with previous observation that the mean number of excitatory synapses between MFs and granule cells is not reduced in the absence of TrkB (Rico et al., 2002), we observed no reduction in vGluT1 localization from P14 to P90, in the IGL of mice lacking TrkB (supplemental Fig. S1, supplemental Table S1B, available at www.jneurosci.org as supplemental material).
Because vGluT1 localization is not perturbed in the cerebellum in mice lacking TrkB between P14 and P90, the area of the GABAergic synaptic protein of interest was normalized to the area of vGluT1 localization to generate an area ratio. At P14, the mean area ratio of GAD65:vGluT1 and GAD67:vGluT1 and the mean intensity of GAD65 and GAD67 expression were comparable between Wnt1::Cre;TrkBfl/fl mice and control (supplemental Fig. S2A–C,J–L; supplemental Table S1B, available at www.jneurosci.org as supplemental material). Strikingly, however, at P21, the mean area ratio of GAD65:vGluT1 and GAD67:vGluT was reduced by 71% and 64%, respectively, in Wnt1::Cre;TrkBfl/fl mice (Fig. 2A–C; supplemental Fig. S2D–F; supplemental Table S1B, available at www.jneurosci.org as supplemental material). In addition, we observed a 47% reduction in the localization of vesicular GABA transporter (vGAT) in P21 Wnt1::Cre;TrkBfl/fl mice (Fig. 2D–F). The localization of GAD65 and GAD67 remained reduced in the mature cerebellum at P90 in Wnt1::Cre;TrkBfl/fl mice (supplemental Fig. S2G–I,M–O; supplemental Table S1B, available at www.jneurosci.org as supplemental material). Together, our results indicate that TrkB is important for the maintenance of the localization of presynaptic proteins, GAD65 and GAD67, but not their initial expression and localization before P14.
Figure 2.

Loss of TrkB in the cerebellum disrupts the localization of GABAergic synaptic proteins. A–F, Loss of TrkB results in reduced localization of GAD67 (green) (B) and vGAT (green) (E) at GABAergic synapses in the IGL of P21 Wnt1::Cre;TrkBfl/fl mice compared with control (A, D). C, F, Quantification of the area ratio of GAD67:vGluT1 expression (C) and vGAT:vGluT1 expression (F) from control and Wnt1::Cre;TrkBfl/fl mice. G–L, Loss of TrkB results in reduced localization of gephyrin (red) (H), but not Neuroligin-2 (NL-2) (red) (K) in the IGL of Wnt1::Cre;TrkBfl/fl mice compared with control (G, J). I, L, Quantification of the area ratio of gephyrin:vGluT1 expression (I) and Neuroligin-2:vGluT1 expression (L) from control and Wnt1::Cre;TrkBfl/fl mice. The area and intensity of vGluT1 expression (blue) were not perturbed in mice lacking TrkB (supplemental Fig. S1, supplemental Table S1B, available at www.jneurosci.org as supplemental material), so vGluT1 expression was used to normalize the number and area of glomeruli per section. Scale bar, 10 μm. M, Immunoblot analysis of the expression level of synaptic proteins in cerebellar lysates from two P21 Wnt1::Cre;TrkBfl/fl mice compared with control. N–Q, Quantification of the intensity of individual bands for GAD65, GAD67 and two gephyrin isoforms (98 and 95 kDa) from control and Wnt1::Cre;TrkBfl/fl mice. N, The level of GAD65 was reduced in Wnt1::Cre;TrkBfl/fl mice (0.23 ± 0.02) compared with control (0.37 ± 0.02; p < 0.01). O, The level of GAD67, however, was not reduced in Wnt1::Cre;TrkBfl/fl mice (0.23 ± 0.02) compared with control (0.21 ± 0.01; p = 0.460). P, Q, Similarly, the level of both gephyrin isoforms was comparable between Wnt1::Cre;TrkBfl/fl mice (98 kDa: 0.56 ± 0.05; 95 kDa: 0.81 ± 0.07) and control (98 kDa: 0.48 ± 0.05, p = 0.222; 95 kDa: 0.76 ± 0.03, p = 0.841). Duplicates from 5 animals were analyzed for each condition.
In the IGL, a large subset of Golgi cells express the presynaptic plasmalemmal glycine transporter (GlyT2) together with GABAergic proteins (Simat et al., 2007). Interestingly, the localization of GlyT2 was not reduced in Wnt1::Cre;TrkBfl/fl mice (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). Since the localization of vGAT is impaired in mice lacking TrkB (Fig. 2D–F) and vGAT functions as the major vesicular transporter for glycine as well as GABA (Wojcik et al., 2006), our data imply that the absence of TrkB prevents normal glycinergic as well as GABAergic presynaptic differentiation.
Localized expression of postsynaptic GABAergic proteins requires TrkB
We next examined the localization of postsynaptic proteins at GABAergic synapses in Wnt1::Cre;TrkBfl/fl mice. The mean area ratio of gephyrin:vGluT1 was reduced by 77% at P14, 70% at P21, and 66% at P90 in Wnt1::Cre;TrkBfl/fl mice compared with control (Fig. 2G–I; supplemental Fig. S2P–U, supplemental Table S1B, available at www.jneurosci.org as supplemental material). At each stage, the reduction in gephyrin localization appears to reflect the presence of fewer, smaller gephyrin-expressing puncta within the granule cell dendrites adjacent to MF endings. Additionally, the loss of TrkB results in a reduction of gephyrin localization at P14, a stage where no deficits in the localization of presynaptic proteins were observed (supplemental Fig. S2B,K, available at www.jneurosci.org as supplemental material).
In addition, we examined the localization of Neuroligin-2, an adhesion molecule implicated in regulation of GABAergic synapses (Huang and Scheiffele, 2008) and found that loss of TrkB did not influence the localization of Neuroligin-2 (Fig. 2J–L). Our result is consistent with the finding that Neuroligin-2 localization is not impaired in mice lacking gephyrin (O'Sullivan et al., 2009). Together, these results indicate that TrkB is required for the localization of a major postsynaptic component of Golgi-granule cell synapses, but not required for the initial steps in differentiation of these synapses.
TrkB selectively regulates protein expression and localization of GABAergic synaptic proteins
To determine whether the reduced localization of GABAergic synaptic proteins in the IGL of cerebella lacking TrkB is caused by reduced synthesis or impaired localization of these proteins, we quantified the level of GABAergic proteins by immunoblot using cerebellar extracts from P21 Wnt1::Cre;TrkBfl/fl mice. Compared with control, the mean level of GAD65 was reduced by 38% in lysates obtained from Wnt1::Cre;TrkBfl/fl mice compared with control (Fig. 2M,N). In contrast, the mean level of GAD67 was not reduced in lysates obtained from Wnt1::Cre;TrkBfl/fl mice (Fig. 2M,O). The mean ratio of intensity of two distinct bands of gephyrin (98 kDa and 95 kDa) was also not reduced in Wnt1::Cre;TrkBfl/fl mice compared with control (Fig. 2M,P,Q). These results indicate that the reduced presence of GAD65 in Wnt1::Cre;TrkBfl/fl mice (supplemental Fig. S2E,H, available at www.jneurosci.org as supplemental material) can be explained, in part, by reduced expression of this protein. On the other hand, the reduced presence of GAD67 and gephyrin in Wnt1::Cre;TrkBfl/fl mice is likely due to mechanisms that influence transport or clustering. Our results are consistent with other studies that have demonstrated the expression or localization of GAD65 is dependent on BDNF-TrkB signaling (Z. J. Huang et al., 1999; Hong et al., 2008; Betley et al., 2009).
TrkB acts in both Golgi cells and granule cells to promote GABAergic synaptic differentiation
Our observation that TrkB deficiency results in impaired gephyrin localization before impairments in localization of GAD65 and GAD67 suggests that the presynaptic deficits observed in mice lacking TrkB in the cerebellum may be secondary consequences of a requirement for TrkB within postsynaptic granule cells. To explore this possibility, we analyzed the localization of GABAergic synaptic proteins in mice lacking TrkB specifically in either Golgi or granule cells.
To assess the role of TrkB in Golgi cells, we generated a transgenic allele that express Cre recombinase using the mGluR2 promoter-enhancer used by others (Watanabe et al., 1998). Within the cerebellum, the mGluR2::Cre allele promotes recombination specifically in Golgi cells and effectively mediates deletion of TrkB in mGluR2::Cre;TrkBfl/fl mice (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). Compared with control, we found that the mean area ratio of GAD67:vGluT1 and mean intensity of GAD67 expression was reduced by 66% and 7%, respectively, in mGluR2::Cre;TrkBfl/fl mice (Fig. 3A–D; supplemental Table S2A, available at www.jneurosci.org as supplemental material; data not shown). However, the localization and intensity of gephyrin were not reduced in mice lacking TrkB in Golgi cells (Fig. 3E–H; supplemental Table S2A, available at www.jneurosci.org as supplemental material). The number and morphology of Golgi cells were not noticeably different in mice lacking TrkB in Golgi cells (data not shown) consistent with observations from mice lacking TrkB from all cerebellar cells (Rico et al., 2002). Because mGluR2::Cre mediates recombination only shortly before P14 (Watanabe et al., 1998), we did not analyze mice lacking TrkB in Golgi cells at P14.
Figure 3.

Consequences of the specific deletion of TrkB in Golgi cells or granule cells. A–H, Loss of TrkB in presynaptic Golgi cells resulted in reduced GAD67 localization (green) (B), but did not alter gephyrin localization (red) (F) in the IGL of P21 mGluR2::Cre;TrkBfl/fl mice compared with control (A, E). C, D, G, H, Quantification of the area ratio of GAD67:vGluT1 (C), gephyrin:vGluT1 expression (G) and the intensity of GAD67 (D) and gephyrin expression (H) in control and mGluR2::Cre;TrkBfl/fl mice. I–P, Loss of TrkB in postsynaptic granule cells did not influence GAD67 localization (green) (J), but resulted in reduced gephyrin localization (red) (N) in the IGL of mα6::Cre;TrkBfl/fl mice compared with control (I, M). K, L, O, P, Quantification of the area ratio of GAD67:vGluT1 (K), gephyrin:vGluT1 expression (O) and the intensity of GAD67 (K), gephyrin expression (P) in control and mα6::Cre;TrkBfl/fl mice. Scale bar, 10 μm.
To examine the consequences of TrkB deletion in granule cells on the localization of GABAergic synaptic proteins, we used the enhancer of the mouse α6 GABAA subunit (mα6) gene to direct recombination specifically in granule cells (Funfschilling and Reichardt, 2002). In contrast to mice with Golgi cell-specific deletion of TrkB, the mean area ratio of GAD67:vGluT1 and mean intensity of GAD67 expression were comparable between mα6::Cre;TrkBfl/fl mice and control (Fig. 3I–L; supplemental Table S2A, available at www.jneurosci.org as supplemental material; data not shown). However, the mean area ratio of gephyrin:vGluT1 and mean intensity of gephyrin expression were reduced by 41% and 6%, respectively, in mice with granule cell-specific deletion of TrkB compared with controls (Fig. 3M–P; supplemental Table S2A, available at www.jneurosci.org as supplemental material). The number and morphology of granule cells were not noticeably different in mice lacking TrkB in granule cells (data not shown), consistent with observations from mice lacking TrkB in all cerebellar cells (Rico et al., 2002). Together, these results demonstrate that TrkB acts in both presynaptic Golgi cells and postsynaptic granule cells to regulate the localization of GABAergic synaptic proteins.
TrkB is required for formation of inhibitory synaptic junctions
To assess the consequences of perturbing the localization of presynaptic and postsynaptic proteins on the ultrastructure of GABAergic synapses, we analyzed synaptic junctions in the glomeruli in mice lacking TrkB. We focused on three main specialization: excitatory synaptic junctions between MF endings and granule cell dendrites (black arrows, asymmetric synapses), synaptic junctions between Golgi axon varicosities and granule cell dendrites (red arrows, symmetric synapses), and plaque-like junctions between granule cell dendrites (yellow arrows, plaques) (Landis et al., 1983; Jakab and Hámori, 1988) (Fig. 4A,B,G,H) (see Materials and Methods).
Figure 4.

Consequences of the deletion of TrkB on synaptic junctions in the cerebellar glomeruli. A, B, Loss of TrkB did not influence the number of excitatory synapses (black arrows) or granule cell plaques (yellow arrows) per glomerulus in P41 Wnt1::Cre;TrkBfl/fl mice (B) compared with control (A) (Ai/Aii and Bi/Bii correspond to dotted box in A and B). However, loss of TrkB resulted in a reduced number of inhibitory synapses per glomerulus and per Golgi cell ending in Wnt1::Cre;TrkBfl/fl mice (red arrows; B) compared with control (A). C–F, Quantification of the number of excitatory synapses (e.s.) (C; control, 6.37 ± 0.46; Wnt1::Cre;TrkBfl/fl, 6.66 ± 0.39; p = 0.863), granule cell plaques (D; control, 2.56 ± 0.15; Wnt1::Cre;TrkBfl/fl, 2.33 ± 0.17; p = 0.386), and inhibitory synapse per glomerulus (i.s.) (E; control, 4.22 ± 0.39; Wnt1::Cre;TrkBfl/fl, 2.33 ± 0.33, p < 0.01) and per Golgi cell ending (F; control, 2.10 ± 0.14; Wnt1::Cre;TrkBfl/fl, 1.20 ± 0.11, p < 0.001). G, H, Loss of TrkB in granule cells did not influence the number of excitatory synapses (black arrows) or granule cell plaques (yellow arrows) per glomerulus in mα6::Cre;TrkBfl/fl mice (H) compared with control (G) (Gi/Gii and Hi/Hii correspond to dotted box in G and H). However, loss of TrkB resulted in reduced number of inhibitory synapses per glomerulus and per Golgi cell ending in mα6::Cre;TrkBfl/fl mice (red arrows; H) compared with control (G). I–L, Quantification of the number of excitatory synapses (e.s.; I; control, 6.77 ± 0.51; Wnt1::Cre;TrkBfl/fl, 6.96 ± 0.49; p = 0.666), granule cell plaques (J; control, 2.37 ± 0.15; Wnt1::Cre;TrkBfl/fl, 2.59 ± 0.16; p = 0.436), and inhibitory synapses per glomerulus (i.e.; K; control, 4.48 ± 0.38; Wnt1::Cre;TrkBfl/fl, 2.56 ± 0.49; p < 0.01) and per Golgi cell ending (L; control, 1.83 ± 0.17, Wnt1::Cre;TrkBfl/fl, 0.93 ± 0.16; p < 0.001). Asterisk denotes Golgi cell ending. Scale bar, 2 μm.
We found that the mean number of excitatory synapses and plaques per glomerulus was comparable between Wnt1::Cre;TrkBfl/fl, mα6::Cre;TrkBfl/fl and control mice (Fig. 4A–D,G–J). However, the mean number of inhibitory synapses per glomerulus and per Golgi terminal was reduced by 45% and 43%, respectively, in P42 Wnt1::Cre;TrkBfl/fl mice, largely consistent with previous findings (Fig. 4E,F) (Rico et al., 2002). Interestingly, the mean number of inhibitory synapses per glomerulus and per Golgi terminal was reduced by 43% and 49%, respectively, in P42 mα6::Cre;TrkBfl/fl mice compared with control (Fig. 4K,L). We did not observe noticeable differences in the organization of glomeruli or the number of glomeruli in the IGL in mice lacking TrkB in the cerebellum or specifically in granule cells compared with control (Fig. 4; data not shown). Since mice lacking TrkB in Golgi cells (mGluR2::Cre;TrkBfl/fl) die shortly after P21 due to seizures (data not shown), we were unable to perform ultrastructural analysis on these mice at comparable stages. These results indicate that TrkB, in both Golgi and granule cells, is important for formation of inhibitory synaptic junctions and that loss of postsynaptic TrkB (in granule cells) is sufficient to perturb the formation of inhibitory junctions. However, our data do not rule out the possibility that TrkB promotes stabilization, not the initial formation of these synapses.
TrkB is required for the assembly and maintenance of GABAergic synaptic proteins
To examine whether TrkB is required to maintain GABAergic synapses, we used a TrkB mouse-line that permits the use of a highly specific kinase inhibitor. In mice carrying a TrkB allele that contains a mutation within the kinase domain (TrkBF616A), the kinase activity of TrkB is inhibited in the presence of 1NMPP1, a membrane-permeable kinase inhibitor (Chen et al., 2005).
We first examined the effect of 1NMPP1 on the assembly of GABAergic synapses in homozygous TrkBF616A mice during the period of the initial formation of GABAergic synapses by treating neonatal TrkBF616A mice (P0) and their mother with 1NMPP1 for 4 weeks. Quantification of the mean area ratio of GAD65:vGluT1 and GAD67:vGluT1 revealed a 75% and 61% reduction, respectively, in P28 TrkBF616A mice treated with 1NMPP1 compared with control (Fig. 5A–F; supplemental Table S2B, available at www.jneurosci.org as supplemental material). We also observed a 69% reduction of gephyrin localization in P28 TrkBF616A mice treated with 1NMPP1 (Fig. 5G–I; supplemental Table S2B, available at www.jneurosci.org as supplemental material). Treatment of wild-type mice with 1NMPP1 from P0-P30 did not influence the localization of GABAergic synaptic proteins (supplemental Fig. S5A–I, available at www.jneurosci.org as supplemental material). These results indicate that the inactivation of TrkB kinase activity during P0 to P28, the period of initial formation of GABAergic synapses (Altman and Bayer, 1997), results in a reduction in the localization of GABAergic synaptic proteins consistent with the reduction in mice lacking TrkB in all cerebellar cells (Fig. 2).
Figure 5.

Inactivation of TrkB kinase activity disrupts the localization of GABAergic synaptic proteins. A–I, Homozygous mice carrying TrkBF616A allele were treated with water or 1NMPP1 from P0 to P28 and analyzed at P28. The localization of GAD65 (green; B), GAD67 (green; E) and gephyrin (red; H) in the IGL is reduced in TrkBF616A mice treated with 1NMPP1 compared with TrkBF616A mice treated with water (A, D, G). C, F, I, Quantification of the area ratio of GAD65:vGluT1 (C), GAD67:vGluT1 (F) and gephyrin:vGluT1 expression (I) in control and 1NMPP1-treated mice. J–R, Homozygous mice carrying TrkBF616A allele were treated with water or 1NMPP1 from P30 to P50 and analyzed at P50. The localization of GAD65 (green; K), GAD67 (green; N) and gephyrin (red; Q) is reduced in TrkBF616A mice treated with 1NMPP1 compared with control (J, M, P). L, O, R, Quantification of the area ratio of GAD65:vGluT1 (L), GAD67:vGluT1 (O) and gephyrin:vGluT1 expression (R) in control and 1NMPP1-treated mice. Scale bar, 10 μm.
Next, we examined the requirement of TrkB in the maintenance of GABAergic synapses in homozygous TrkBF616A mice, after completion of the establishment of GABAergic synapses, by treating mature P30 TrkBF616A mice for 20 d with 1NMPP1. Quantification of the mean area ratio of GAD65:vGluT1 and GAD67:vGluT1 revealed 39% and 54% reduction, respectively, in P50 TrkBF616A mice treated with 1NMPP1 compared with control (Fig. 5J–O; supplemental Table S2B, available at www.jneurosci.org as supplemental material). We also observed a 44% reduction of gephyrin localization in P50 1NMPP1-treated mice (Fig. 5P–R; supplemental Table S2B, available at www.jneurosci.org as supplemental material). Treatment of wild-type mice with 1NMPP1 from P30-P52 did not influence the localization of GABAergic synaptic proteins (supplemental Fig. S5J–R, available at www.jneurosci.org as supplemental material). These findings revealed that the inactivation of TrkB kinase activity after GABAergic synapses have formed still results in a reduction in the localization of GABAergic synaptic proteins, albeit to a lesser extent, providing strong evidence that TrkB kinase activity continues to be important for the maintenance of GABAergic synapses in adult mice.
GABAergic synaptic differentiation depends on the PLC-γ1-binding site in TrkB
The activities of TrkB are mediated, in part, by the recruitment of the adaptor protein Shc to phosphotyrosine-515 which initiates signaling cascades such as the Ras/MAPK and phosphatidylinositide 3-kinase pathways (Reichardt, 2006). Additionally, the recruitment of phospholipase C-γ1 (PLC-γ1), through phosphotyrosine-816, results in generation of diacylglycerol and IP3, release of Ca2+ stores, and activation of calcium-regulated protein kinases and phosphatases (Reichardt, 2006). To define the signaling pathway(s) downstream of TrkB responsible for regulation of GABAergic synapses, we examined the cerebella of mice with mutations in the Shc or PLC-γ1 docking sites of TrkB (Minichiello et al., 1998, 2002).
In mice carrying a point mutation in the Shc docking site of TrkB (TrkBSHC/SHC), we found that the mean area ratio of GAD67:vGluT1 and gephyrin:vGluT1 and the mean intensity of their expression was comparable to control (Fig. 6A–H; supplemental Table S2C, available at www.jneurosci.org as supplemental material; data not shown). These results show that TrkB-mediated recruitment of Shc at phosphotyrosine-515 is not necessary for the development and maintenance of GABAergic synapses. In contrast, when we examined mice homozygous for the PLC-γ1 docking site of TrkB (TrkBPLC/PLC), we found that the mean area ratio of GAD67:vGluT1 and the mean intensity of GAD67 expression were reduced by 25% and 5%, respectively, in P21 TrkBPLC/PLC mice (Fig. 6I–L; supplemental Table S2C, available at www.jneurosci.org as supplemental material; data not shown). Additionally, the mean area ratio of gephyrin:vGluT1 and the mean intensity of gephyrin expression were reduced by 80% and 12%, respectively, in TrkBPLC/PLC mice (Fig. 6M–P; supplemental Table S2C, available at www.jneurosci.org as supplemental material). Together, these results suggest that TrkB promotes the development and maintenance of GABAergic synapses via recruitment of PLC-γ1 and activation of Ca2+-regulated pathways.
Figure 6.

Disruption of the PLC-γ1-binding site, but not Shc, perturbs the localization of GABAergic synaptic proteins. A–H, The localization of GAD67 (green; B) and gephyrin (red; F) in the IGL of P21 TrkBSHC/SHC mice was comparable to control mice (A, E). C, D, G, H, Quantification of the area ratio of GAD67:vGluT1 (C), gephyrin:vGluT1 expression (G), and the intensity of GAD67 (D) and gephyrin expression (H) in control and TrkBSHC/SHC mice. I–P, The localization of GAD67 (green; J) and gephyrin (red; N) in the IGL of P21 TrkBPLC/PLC mice is reduced compared with control (I, M). Wild-type TrkB knock-in mice were used as control (TrkBWT/WT) (Minichiello et al., 2002). K, L, O, P, Quantification of the area ratio of GAD67:vGluT1 (K), gephyrin:vGluT1 expression (O) and the intensity of GAD67 (L) and gephyrin expression (P) in control and TrkBPLC/PLC mice. Scale bar, 10 μm.
TrkB regulates the localization of Contactin-1 on Golgi and granule cells
To explore potential effector proteins that could mediate the activities of TrkB in Golgi and/or granule cells, we examined the expression pattern of different families of adhesion molecules that have been implicated in neural connectivity and synaptic differentiation. In particular, we found that Contactin-1 (CNTN), a member of the Contactin/F3 family of proteins, is highly expressed in the IGL at P14, P21 and P90 (Fig. 7A; supplemental Fig. S6, available at www.jneurosci.org as supplemental material). Previous work has shown that this protein is important for the differentiation of cerebellar neurons and motor function (Berglund et al., 1999). Additionally, BDNF has been shown to influence the translocation of Contactin-1 to cell surface (Maruyama et al., 2007). Contactin-1 is expressed on Neurogranin+ Golgi cell axons (Fig. 7E) and on the dendrites of YFP+ granule cells, using a Thy1::YFP mouse line (H) that labels a subset of granule cells (Feng et al., 2000) (Fig. 7I). In addition we detected Contactin-1 on vGluT+ MF endings and GFAP+ astroglia fibers (Fig. 7M,Q).
Figure 7.
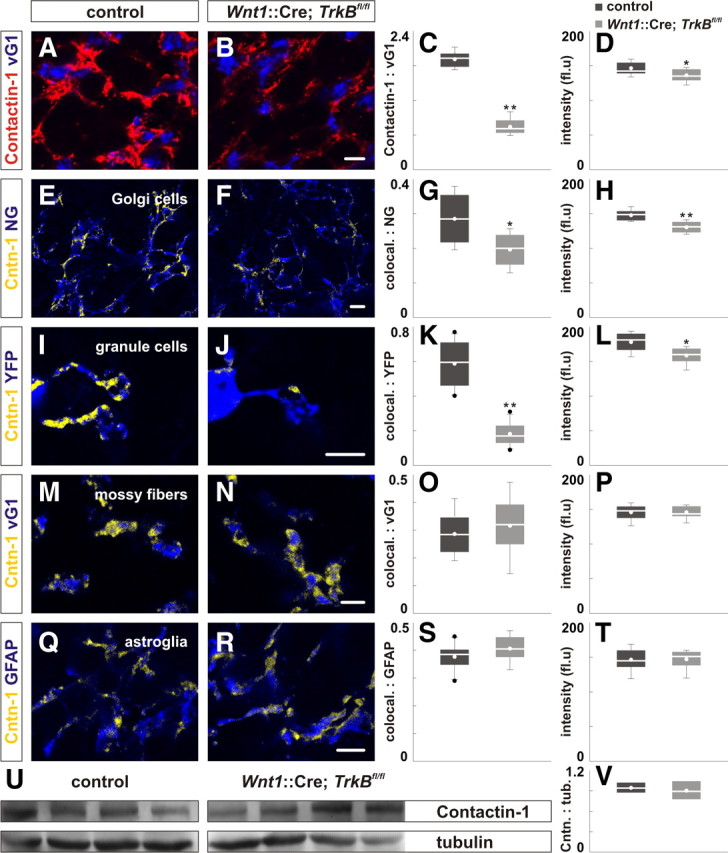
Loss of TrkB in the cerebellum results in reduction of Contactin-1 localization on Golgi and granule cells. A, B, Loss of TrkB results in reduced localization of Contactin-1 in the IGL of P21 Wnt1::Cre;TrkBfl/fl mice (B) compared with control (A). C, D, Quantification of the area ratio of Contactin-1:vGluT1 (C) and the intensity of Contactin-1 expression (D) expression in control and Wnt1::Cre;TrkBfl/fl mice. For E–T, yellow represents only colocalized Contactin-1 on individual markers in blue (see Materials and Methods). E–L, Loss of TrkB results in reduced Contactin-1 localization (yellow) on Neurogranin+ Golgi cell axons (blue) and YFP+ granule cell dendrites (blue) in Wnt1::Cre;TrkBfl/fl; Thy1::YFP mice (F, J) compared with control (E, I). G, H, K, L, Quantification of the area ratio of colocalized Contactin-1:Neurogranin (colocal.:NG) (G), Contactin-1:YFP (colocal.:YFP) (K), and the intensity of colocalized Contactin-1 expression (K, L). M–T, The colocalization of Contactin-1 (yellow) on vGluT+ mossy fibers (blue) and on GFAP+ astroglia is not perturbed in P21 Wnt1::Cre;TrkBfl/fl mice (N, R) compared with control (M, Q). O, P, S, T, Quantification of the area ratio of colocalized Contactin-1:vGluT1 (colocal.:vGluT1) (O), Contactin-1:GFAP (colocal.:GFAP) (S), and the intensity of colocalized Contactin-1 expression (P, T). Scale bar, 10 μm. U, Immunoblot analysis shows that the expression of Contactin-1 was not perturbed in lysates from four P21 Wnt1::Cre;TrkBfl/fl mice compared with control. V, Quantification of the intensity of the band for Contactin-1 (135 kDa) normalized to the intensity of tubulin in control (0.94 ± 0.04) and Wnt1::Cre;TrkBfl/fl lysates (0.90 ± 0.07; p = 0.885).
The presence of Contactin-1 on Golgi cell axons and granule cell dendrites suggested that Contactin-1 might be involved in the development of inhibitory synapses. To explore this possibility, we first examined the consequences of cerebellar TrkB deletion on the localization of Contactin-1. We found that the mean area ratio of Contactin-1:vGluT1 and the mean intensity of Contactin-1 expression were reduced by 61% and 8%, respectively, in Wnt1::Cre;TrkBfl/fl mice (Fig. 7A–D; supplemental Table S3A, available at www.jneurosci.org as supplemental material). We next analyzed the mean area and intensity of Contactin-1 expression colocalized on individual cell-types (yellow; Fig. 7; supplemental Fig. S6, available at www.jneurosci.org as supplemental material) (see Materials and Methods). We found that the mean area ratio of Contactin-1:NG and mean intensity of Contactin-1 expression on Golgi cell axons were reduced by 32% and 12%, respectively, in Wnt1::Cre;TrkBfl/fl mice (Fig. 7E–H; supplemental Table S3A, available at www.jneurosci.org as supplemental material). Additionally, the mean area ratio of Contactin-1:YFP and mean intensity of Contactin-1 expression on granule cell dendrites were reduced by 69% and 11% in Wnt1::Cre;TrkBfl/fl mice (Fig. 7I–L; supplemental Table S3A, available at www.jneurosci.org as supplemental material). However, the mean area ratio of Contactin-1:vGluT1 and Contactin-1:GFAP and the mean intensity of Contactin-1 expression on vGluT1+ MFs and GFAP+ astroglial processes were not affected in Wnt1::Cre;TrkBfl/fl mice (Fig. 7M–T; supplemental Table S3A, available at www.jneurosci.org as supplemental material). Immunoblot analysis revealed that the expression of Contactin-1 is not reduced in Wnt1::Cre;TrkBfl/fl mice (Fig. 7U,V). Together, we provide evidence that TrkB regulates the Contactin-1 localization on both Golgi cell axons and granule cell dendrites, consistent with the possibility that Contactin-1 mediates the activities of TrkB in the assembly and maintenance of GABAergic synapses.
Contactin-1 is required for presynaptic and postsynaptic differentiation at GABAergic synapses
Since the localization of Contactin-1 on Golgi cell axons and granule cell dendrites is reduced in mice lacking TrkB, we set out to assess the consequences of the loss of Contactin-1 on localization of GABAergic synaptic proteins. To achieve this, we analyzed P18 mice that lack Contactin-1 (Berglund et al., 1999). We observed a 55% and 73% reduction, respectively, of the mean area ratio of GAD65:vGluT1 and GAD67:vGluT1 in Contactin-1−/− mice compared with control (Fig. 8A–F; supplemental Table S3B, available at www.jneurosci.org as supplemental material). We also observed an 80% reduction of the mean area ratio of gephyrin:vGluT1 in Contactin-1−/− mice (Fig. 8G–I; supplemental Table S3B, available at www.jneurosci.org as supplemental material). Contactin-1−/− mice die around P18, so we could not analyze the consequences of Contactin-1 deletion on synaptic protein localization at P21 and beyond (Berglund et al., 1999). Together, these findings show that the loss of Contactin-1 results in similar perturbations in the localization of GABAergic synaptic proteins observed in mice lacking TrkB and raises the possibility that Contactin-1 may be an important effector that mediates TrkB-dependent organization of GABAergic synapses.
Discussion
In this paper, we present findings demonstrating that TrkB controls important features of differentiation within both the presynaptic and postsynaptic compartments of cerebellar GABAergic synapses. Importantly, through specific inhibition of the kinase activity of TrkB, we show that the kinase activity of TrkB is required not simply to initiate GABAergic synapse formation, but also to maintain these synapses in adulthood. We also show that the localization of Contactin-1 at the synaptic contacts between Golgi and granule cells requires TrkB suggesting that TrkB promotes synapse formation and maintenance, in part, by controlling the localization of cell adhesion molecules.
Role of TrkB in the assembly and maintenance of cerebellar GABAergic synapses
Recent work has provided many examples of structural plasticity in the adult CNS and revealed that plasticity is regulated through activity and proteins that control the F-actin cytoskeleton, cell adhesion molecule, as well as effectors of receptor tyrosine kinases (Holtmaat and Svoboda, 2009; Matter et al., 2009; Wu et al., 2009; Yang et al., 2009). Studies have demonstrated that individual protein constituents at synapses are in dynamic equilibrium and hence subject to regulation through changes in gene expression, protein phosphorylation and proteolysis (Gray et al., 2006; Bingol et al., 2010). Our results using a highly specific inhibitor to block the kinase activity of TrkB demonstrate that TrkB kinase activity is required to maintain GABAergic synaptic differentiation in the adult cerebellum.
Consistent with our findings, several recent studies using the same genetic-pharmacological approach have shown that TrkB continues to be required for normal neuronal function in mature animals (Johnson et al., 2008; Kaneko et al., 2008; Wang et al., 2009). TrkB is important for motivational learning, such as conditioned reinforcement (Johnson et al., 2008), processing of thermal and mechanical pain (Wang et al., 2009), and remodeling of visual cortical function in response to visual stimuli (Kaneko et al., 2008). Our data raise the possibility that TrkB is required to maintain the inhibitory synaptic connectivity necessary for coordinating the activities of neural circuits underlying these processes and suggests that BDNF-TrkB signaling is an important molecular regulator of structural plasticity in adult animals.
Regulation of synaptic proteins that control GABAergic and glycinergic synaptic function by TrkB
We previously reported that in mice lacking TrkB in the cerebellum there is a dramatic reduction in the number of synaptic specializations at Golgi-granule cell contacts, as assessed by electron microscopy, with a similar reduction in the localization of presynaptic GABAergic molecular markers throughout the cerebellum (Rico et al., 2002). The results in this paper document that loss of TrkB in the cerebellum leads to a reduction of presynaptic GABAergic proteins, including a dual GABA/glycine transporter (vGAT), but not a plasmalemmal glycine transporter (GlyT2). In addition, the loss of TrkB in the cerebellum leads to a reduction in the localization of gephyrin, a postsynaptic protein required for normal GABA and glycine receptor clustering (Moss and Smart, 2001). Together, our data suggest that both GABAergic and glycinergic function at Golgi-granule cell synapses are dependent on TrkB. Prior work has shown that there are several classes of Golgi-granule cell synapses with some purely GABAergic or glycinergic, and a majority with dual GABAergic-glycinergic properties (Simat et al., 2007). Our data has not determined whether these different classes of synapses are equally dependent on TrkB.
Role of PLC-γ1- and Ca2+-regulated pathways in controlling GABAergic synapses
To begin to define the signaling pathway(s) downstream of TrkB responsible for regulation of GABAergic synaptic development, we examined the synapses between Golgi and granule cells in mice carrying disrupted docking sites for specific effectors of activated TrkB. No deficit in the formation of GABAergic synapses was observed in mice expressing TrkB mutated in the Shc-binding site (Y515) even though binding of Shc has been shown to promote activation of both Ras and PI-3 kinase (Fig. 6) (Reichardt, 2006). Other mechanisms have been discovered, however, for activation of Ras-Erk and PI-3 kinase by Trk receptors. For instance, Grb2, an adaptor bound to a Ras exchange factor, can interact with other tyrosine residues in the kinase activation loop of Trk receptors (Qian et al., 1998; MacDonald et al., 2000). In addition, c-Abl, an activator of Ras and MAPK pathways, can interact with Trk receptors at different sites that are not phosphotyrosines (Yano et al., 2000; Robinson et al., 2005). Thus our results show that the Shc-binding site (Y515) is not necessary, but do not rule out the possibility that PI-3 Kinase and MAPK pathways are important for the development of Golgi-granule cell synapses.
Studies using mice carrying a mutation in the PLC-γ1-binding site (Y816) have documented striking deficits in BDNF-dependent CaMKinase IV and CREB activation (Minichiello et al., 2002). Our observation that these mice exhibit deficits in localization of both presynaptic and postsynaptic proteins at Golgi-granule cell synapses suggests that Ca2+-regulated pathways control GABAergic synapse formation. Phosphorylation of PLC-γ1 by activated TrkB generates diacylglycerol and inositol-tris-phosphate (Reichardt, 2006). Inositol-tris-phosphate activates IP3 receptors and increases cytoplasmic Ca2+ levels (Reichardt, 2006). Therefore, it seems plausible that TrkB-mediated phosphorylation of PLC-γ1 results in activation of a variety of Ca2+-regulated kinases and phosphatases, one or more of which could regulate the localization of GAD67, gephyrin and other synaptic proteins. In addition, activation of Ca2+-calmodulin-kinases results in induction of CREB and other transcription factors (Lonze and Ginty, 2002), which provides a plausible pathway through which TrkB promotes the expression of GAD65. In turn, CREB has important functions in controlling many adaptive neuronal responses (Lonze and Ginty, 2002). The potential involvement of Ca2+ suggests that the formation and maintenance of cerebellar GABAergic synapses are controlled by activity-dependent processes. Interestingly, the expression of BDNF is Ca2+ and CREB-dependent (West et al., 2001; Lonze and Ginty, 2002) and mutation of the major CREB-regulated promoter of BDNF results in reduced inhibitory neuron and synapse formation in the cortex (Hong et al., 2008; Sakata et al., 2009).
Contactin/F3 family of IgSF cell adhesion molecules and the assembly of GABAergic synapses
The neuronal cell adhesion molecules of the Ig superfamily (IgSF) play important roles in the assembly of neural circuits (Ango et al., 2004; Hattori et al., 2007; Yamagata and Sanes, 2008). Our identification of Contactin-1, and potentially other IgSF cell adhesion molecules, as possible effectors of TrkB in the cerebellum suggests IgSF cell adhesion molecules have important functions in the development of cerebellar circuits. Previous work has provided evidence that members of the Contactin family as well as other IgSF members control formation of specific synapses (Huang et al., 2007; Sakurai et al., 2009). Some of these proteins mediate trans-interactions with IgSF family members (Kuhn et al., 1991; Laursen et al., 2009), protein tyrosine phosphatases (Peles et al., 1995; Bouyain and Watkins, 2009) and tenascin (Michele and Faissner, 2009). Others mediate cis-interactions with IgSF members and gangliosides (Kasahara et al., 2000). Contactins also have been shown to associate with the cytoskeleton and to activate Src-family and Ca2+-dependent signaling pathways (Laursen et al., 2009; Michele and Faissner, 2009).
In addition to promoting formation of synapses between Golgi and granule cells, Contactin-1 has been shown to regulate arborization of presynaptic and postsynaptic processes (Berglund et al., 1999) and could have additional roles in target recognition or stabilization and maintenance of synaptic contacts. Because Contactin-1 can exist anchored in the cell membrane through a glycosylphosphatidyl inositol moiety (Berglund and Ranscht, 1994) and can occur in a soluble form (Durbec et al., 1992), Contactin-1 can potentially act both as a neuronal receptor and as a substrate for neurite growth. A better understanding of how F3/Contactin family of molecules mediate the development of synapses may reveal an IgSF molecular code for synaptic specificity in the cerebellum similar to other regions of the CNS (Hattori et al., 2007; Huang et al., 2007; Yamagata and Sanes, 2008).
Footnotes
A.I.C. was supported by a Sandler Postdoctoral Research Fellowship. D.R.C. was supported by grants from the National Eye Institute (EY01869), the March of Dimes, and Research to Prevent Blindness. B.R. was supported by a grant from the National Institutes of Health (NS038297). L.F.R. was supported by a grant from the National Institute of Neurological Disorders and Stroke (NS16033) and was a Howard Hughes Medical Institute investigator. We thank K. Zang for technical assistance. We are grateful to C. Chun for samples from 1NMPP1-treated TrkBF616A mice, A. Brzozowska-Prechtl for samples from Contactinl−/− mice, T. Jessell for antibodies, and S. Nakanishi for the mGluR2 promoter. We are grateful to J. LaVail and P. Ohara for advice on EM analysis. We thank J. N. Betley, J. Kaltschmidt, and T. Jessell for discussion and comments on the manuscript.
References
- Altman J, Bayer SA. Development of the cerebellar system: in relation to its evolution, structure, and functions. Boca Raton, FL: CRC; 1997. [Google Scholar]
- Ango F, di Cristo G, Higashiyama H, Bennett V, Wu P, Huang ZJ. Ankyrin-based subcellular gradient of neurofascin, an immunoglobulin family protein, directs GABAergic innervation at Purkinje axon initial segment. Cell. 2004;119:257–272. doi: 10.1016/j.cell.2004.10.004. [DOI] [PubMed] [Google Scholar]
- Bao S, Chen L, Qiao X, Thompson RF. Transgenic brain-derived neurotrophic factor modulates a developing cerebellar inhibitory synapse. Learn Mem. 1999;6:276–283. [PMC free article] [PubMed] [Google Scholar]
- Berglund EO, Ranscht B. Molecular cloning and in situ localization of the human contactin gene (CNTN1) on chromosome 12q11–q12. Genomics. 1994;21:571–582. doi: 10.1006/geno.1994.1316. [DOI] [PubMed] [Google Scholar]
- Berglund EO, Murai KK, Fredette B, Sekerková G, Marturano B, Weber L, Mugnaini E, Ranscht B. Ataxia and abnormal cerebellar microorganization in mice with ablated contactin gene expression. Neuron. 1999;24:739–750. doi: 10.1016/s0896-6273(00)81126-5. [DOI] [PubMed] [Google Scholar]
- Betley JN, Wright CV, Kawaguchi Y, Erdélyi F, Szabó G, Jessell TM, Kaltschmidt JA. Stringent specificity in the construction of a GABAergic presynaptic inhibitory circuit. Cell. 2009;139:161–174. doi: 10.1016/j.cell.2009.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingol B, Wang CF, Arnott D, Cheng D, Peng J, Sheng M. Autophosphorylated CaMKIIalpha acts as a scaffold to recruit proteasomes to dendritic spines. Cell. 2010;140:567–578. doi: 10.1016/j.cell.2010.01.024. [DOI] [PubMed] [Google Scholar]
- Bouyain S, Watkins DJ. The protein tyrosine phosphatases PTPRZ and PTPRG bind to distinct members of the contactin family of neural recognition molecules. Proc Natl Acad Sci U S A. 2009;107:2443–2448. doi: 10.1073/pnas.0911235107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Ye H, Kuruvilla R, Ramanan N, Scangos KW, Zhang C, Johnson NM, England PM, Shokat KM, Ginty DD. A chemical-genetic approach to studying neurotrophin signaling. Neuron. 2005;46:13–21. doi: 10.1016/j.neuron.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Danielian PS, Muccino D, Rowitch DH, Michael SK, McMahon AP. Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr Biol. 1998;8:1323–1326. doi: 10.1016/s0960-9822(07)00562-3. [DOI] [PubMed] [Google Scholar]
- Durbec P, Gennarini G, Goridis C, Rougon G. A soluble form of the F3 neuronal cell adhesion molecule promotes neurite outgrowth. J Cell Biol. 1992;117:877–887. doi: 10.1083/jcb.117.4.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles JC, Ito M, Szentágothai J. The cerebellum as a neuronal machine. Berlin: Springer; 1967. [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Fritschy JM, Harvey RJ, Schwarz G. Gephyrin: where do we stand, where do we go? Trends Neurosci. 2008;31:257–264. doi: 10.1016/j.tins.2008.02.006. [DOI] [PubMed] [Google Scholar]
- Fünfschilling U, Reichardt LF. Cre-mediated recombination in rhombic lip derivatives. Genesis. 2002;33:160–169. doi: 10.1002/gene.10104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray NW, Weimer RM, Bureau I, Svoboda K. Rapid redistribution of synaptic PSD-95 in the neocortex in vivo. PLoS Biol. 2006;4:e370. doi: 10.1371/journal.pbio.0040370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishanin RN, Yang H, Liu X, Donohue-Rolfe K, Nune GC, Zang K, Xu B, Duncan JL, Lavail MM, Copenhagen DR, Reichardt LF. Retinal TrkB receptors regulate neural development in the inner, but not outer, retina. Mol Cell Neurosci. 2008;38:431–443. doi: 10.1016/j.mcn.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori D, Demir E, Kim HW, Viragh E, Zipursky SL, Dickson BJ. Dscam diversity is essential for neuronal wiring and self-recognition. Nature. 2007;449:223–227. doi: 10.1038/nature06099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat A, Svoboda K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci. 2009;10:647–658. doi: 10.1038/nrn2699. [DOI] [PubMed] [Google Scholar]
- Hong EJ, McCord AE, Greenberg ME. A biological function for the neuronal activity-dependent component of Bdnf transcription in the development of cortical inhibition. Neuron. 2008;60:610–624. doi: 10.1016/j.neuron.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Wilkinson GA, Fariñas I, Backus C, Zang K, Wong SL, Reichardt LF. Expression of Trk receptors in the developing mouse trigeminal ganglion: in vivo evidence for NT-3 activation of TrkA and TrkB in addition to TrkC. Development. 1999;126:2191–2203. doi: 10.1242/dev.126.10.2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZJ, Scheiffele P. GABA and neuroligin signaling: linking synaptic activity and adhesion in inhibitory synapse development. Curr Opin Neurobiol. 2008;18:77–83. doi: 10.1016/j.conb.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZJ, Kirkwood A, Pizzorusso T, Porciatti V, Morales B, Bear MF, Maffei L, Tonegawa S. BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell. 1999;98:739–755. doi: 10.1016/s0092-8674(00)81509-3. [DOI] [PubMed] [Google Scholar]
- Huang ZJ, Di Cristo G, Ango F. Development of GABA innervation in the cerebral and cerebellar cortices. Nat Rev Neurosci. 2007;8:673–686. doi: 10.1038/nrn2188. [DOI] [PubMed] [Google Scholar]
- Ito M. The cerebellum and neural control. New York: Raven; 1984. [Google Scholar]
- Jakab RL, Hámori J. Quantitative morphology and synaptology of cerebellar glomeruli in the rat. Anat Embryol (Berl) 1988;179:81–88. doi: 10.1007/BF00305102. [DOI] [PubMed] [Google Scholar]
- Johnson AW, Chen X, Crombag HS, Zhang C, Smith DR, Shokat KM, Gallagher M, Holland PC, Ginty DD. The brain-derived neurotrophic factor receptor TrkB is critical for the acquisition but not expression of conditioned incentive value. Eur J Neurosci. 2008;28:997–1002. doi: 10.1111/j.1460-9568.2008.06383.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko M, Hanover JL, England PM, Stryker MP. TrkB kinase is required for recovery, but not loss, of cortical responses following monocular deprivation. Nat Neurosci. 2008;11:497–504. doi: 10.1038/nn2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara K, Watanabe K, Takeuchi K, Kaneko H, Oohira A, Yamamoto T, Sanai Y. Involvement of gangliosides in glycosylphosphatidylinositol-anchored neuronal cell adhesion molecule TAG-1 signaling in lipid rafts. J Biol Chem. 2000;275:34701–34709. doi: 10.1074/jbc.M003163200. [DOI] [PubMed] [Google Scholar]
- Klein R, Smeyne RJ, Wurst W, Long LK, Auerbach BA, Joyner AL, Barbacid M. Targeted disruption of the trkB neurotrophin receptor gene results in nervous system lesions and neonatal death. Cell. 1993;75:113–122. [PubMed] [Google Scholar]
- Kuhn TB, Stoeckli ET, Condrau MA, Rathjen FG, Sonderegger P. Neurite outgrowth on immobilized axonin-1 is mediated by a heterophilic interaction with L1(G4) J Cell Biol. 1991;115:1113–1126. doi: 10.1083/jcb.115.4.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landis DM, Weinstein LA, Halperin JJ. Development of synaptic junctions in cerebellar glomeruli. Brain Res. 1983;284:231–245. doi: 10.1016/0165-3806(83)90008-1. [DOI] [PubMed] [Google Scholar]
- Laursen LS, Chan CW, ffrench-Constant C. An integrin-contactin complex regulates CNS myelination by differential Fyn phosphorylation. J Neurosci. 2009;29:9174–9185. doi: 10.1523/JNEUROSCI.5942-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–623. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- Luikart BW, Parada LF. Receptor tyrosine kinase B-mediated excitatory synaptogenesis. Prog Brain Res. 2006;157:15–24. doi: 10.1016/s0079-6123(06)57002-5. [DOI] [PubMed] [Google Scholar]
- MacDonald JI, Gryz EA, Kubu CJ, Verdi JM, Meakin SO. Direct binding of the signaling adapter protein Grb2 to the activation loop tyrosines on the nerve growth factor receptor tyrosine kinase, TrkA. J Biol Chem. 2000;275:18225–18233. doi: 10.1074/jbc.M001862200. [DOI] [PubMed] [Google Scholar]
- Maruyama E, Ogawa K, Endo S, Tsujimoto M, Hashikawa T, Nabetani T, Tsugita A. Brain-derived neurotrophic factor induces cell surface expression of short-form tenascin R complex in hippocampal postsynapses. Int J Biochem Cell Biol. 2007;39:1930–1942. doi: 10.1016/j.biocel.2007.05.012. [DOI] [PubMed] [Google Scholar]
- Mason CA, Gregory E. Postnatal maturation of cerebellar mossy and climbing fibers: transient expression of dual features on single axons. J Neurosci. 1984;4:1715–1735. doi: 10.1523/JNEUROSCI.04-07-01715.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matter C, Pribadi M, Liu X, Trachtenberg JT. Delta-catenin is required for the maintenance of neural structure and function in mature cortex in vivo. Neuron. 2009;64:320–327. doi: 10.1016/j.neuron.2009.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC. Neurotrophins and synaptic plasticity. Annu Rev Neurosci. 1999;22:295–318. doi: 10.1146/annurev.neuro.22.1.295. [DOI] [PubMed] [Google Scholar]
- Michele M, Faissner A. Tenascin-C stimulates contactin-dependent neurite outgrowth via activation of phospholipase C. Mol Cell Neurosci. 2009;41:397–408. doi: 10.1016/j.mcn.2009.04.004. [DOI] [PubMed] [Google Scholar]
- Minichiello L, Casagranda F, Tatche RS, Stucky CL, Postigo A, Lewin GR, Davies AM, Klein R. Point mutation in trkB causes loss of NT4-dependent neurons without major effects on diverse BDNF responses. Neuron. 1998;21:335–345. doi: 10.1016/s0896-6273(00)80543-7. [DOI] [PubMed] [Google Scholar]
- Minichiello L, Calella AM, Medina DL, Bonhoeffer T, Klein R, Korte M. Mechanism of TrkB-mediated hippocampal long-term potentiation. Neuron. 2002;36:121–137. doi: 10.1016/s0896-6273(02)00942-x. [DOI] [PubMed] [Google Scholar]
- Moss SJ, Smart TG. Constructing inhibitory synapses. Nat Rev. 2001;2:240–250. doi: 10.1038/35067500. [DOI] [PubMed] [Google Scholar]
- Nagappan G, Lu B. Activity-dependent modulation of the BDNF receptor TrkB: mechanisms and implications. Trends Neurosci. 2005;28:464–471. doi: 10.1016/j.tins.2005.07.003. [DOI] [PubMed] [Google Scholar]
- O'Sullivan GA, Hofer W, Betz H. Inhibitory postsynaptic membrane specializations are formed in gephyrin-deficient mice. Neurosci Lett. 2009;458:106–110. doi: 10.1016/j.neulet.2009.04.036. [DOI] [PubMed] [Google Scholar]
- Palay SL, Chan-Palay V. Cerebellar cortex: cytology and organization. New York: Springer; 1974. [Google Scholar]
- Peles E, Nativ M, Campbell PL, Sakurai T, Martinez R, Lev S, Clary DO, Schilling J, Barnea G, Plowman GD, Grumet M, Schlessinger J. The carbonic anhydrase domain of receptor tyrosine phosphatase beta is a functional ligand for the axonal cell recognition molecule contactin. Cell. 1995;82:251–260. doi: 10.1016/0092-8674(95)90312-7. [DOI] [PubMed] [Google Scholar]
- Poo MM. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- Qian X, Riccio A, Zhang Y, Ginty DD. Identification and characterization of novel substrates of Trk receptors in developing neurons. Neuron. 1998;21:1017–1029. doi: 10.1016/s0896-6273(00)80620-0. [DOI] [PubMed] [Google Scholar]
- Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci. 2006;361:1545–1564. doi: 10.1098/rstb.2006.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rico B, Xu B, Reichardt LF. TrkB receptor signaling is required for establishment of GABAergic synapses in the cerebellum. Nat Neurosci. 2002;5:225–233. doi: 10.1038/nn808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson KN, Manto K, Buchsbaum RJ, MacDonald JI, Meakin SO. Neurotrophin-dependent tyrosine phosphorylation of Ras guanine-releasing factor 1 and associated neurite outgrowth is dependent on the HIKE domain of TrkA. J Biol Chem. 2005;280:225–235. doi: 10.1074/jbc.M410454200. [DOI] [PubMed] [Google Scholar]
- Sakata K, Woo NH, Martinowich K, Greene JS, Schloesser RJ, Shen L, Lu B. Critical role of promoter IV-driven BDNF transcription in GABAergic transmission and synaptic plasticity in the prefrontal cortex. Proc Natl Acad Sci U S A. 2009;106:5942–5947. doi: 10.1073/pnas.0811431106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai K, Toyoshima M, Ueda H, Matsubara K, Takeda Y, Karagogeos D, Shimoda Y, Watanabe K. Contribution of the neural cell recognition molecule NB-3 to synapse formation between parallel fibers and Purkinje cells in mouse. Dev Neurobiol. 2009;69:811–824. doi: 10.1002/dneu.20742. [DOI] [PubMed] [Google Scholar]
- Seil FJ, Drake-Baumann R. TrkB receptor ligands promote activity-dependent inhibitory synaptogenesis. J Neurosci. 2000;20:5367–5373. doi: 10.1523/JNEUROSCI.20-14-05367.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simat M, Parpan F, Fritschy JM. Heterogeneity of glycinergic and gabaergic interneurons in the granule cell layer of mouse cerebellum. J Comp Neurol. 2007;500:71–83. doi: 10.1002/cne.21142. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Wang X, Ratnam J, Zou B, England PM, Basbaum AI. TrkB signaling is required for both the induction and maintenance of tissue and nerve injury-induced persistent pain. J Neurosci. 2009;29:5508–5515. doi: 10.1523/JNEUROSCI.4288-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe D, Inokawa H, Hashimoto K, Suzuki N, Kano M, Shigemoto R, Hirano T, Toyama K, Kaneko S, Yokoi M, Moriyoshi K, Suzuki M, Kobayashi K, Nagatsu T, Kreitman RJ, Pastan I, Nakanishi S. Ablation of cerebellar Golgi cells disrupts synaptic integration involving GABA inhibition and NMDA receptor activation in motor coordination. Cell. 1998;95:17–27. doi: 10.1016/s0092-8674(00)81779-1. [DOI] [PubMed] [Google Scholar]
- West AE, Chen WG, Dalva MB, Dolmetsch RE, Kornhauser JM, Shaywitz AJ, Takasu MA, Tao X, Greenberg ME. Calcium regulation of neuronal gene expression. Proc Natl Acad Sci U S A. 2001;98:11024–11031. doi: 10.1073/pnas.191352298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik SM, Katsurabayashi S, Guillemin I, Friauf E, Rosenmund C, Brose N, Rhee JS. A shared vesicular carrier allows synaptic corelease of GABA and glycine. Neuron. 2006;50:575–587. doi: 10.1016/j.neuron.2006.04.016. [DOI] [PubMed] [Google Scholar]
- Wu SH, Arévalo JC, Sarti F, Tessarollo L, Gan WB, Chao MV. Ankyrin Repeat-rich Membrane Spanning/Kidins220 protein regulates dendritic branching and spine stability in vivo. Dev Neurobiol. 2009;69:547–557. doi: 10.1002/dneu.20723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata M, Sanes JR. Dscam and Sidekick proteins direct lamina-specific synaptic connections in vertebrate retina. Nature. 2008;451:465–469. doi: 10.1038/nature06469. [DOI] [PubMed] [Google Scholar]
- Yan Q, Radeke MJ, Matheson CR, Talvenheimo J, Welcher AA, Feinstein SC. Immunocytochemical localization of TrkB in the central nervous system of the adult rat. J Comp Neurol. 1997;378:135–157. [PubMed] [Google Scholar]
- Yang G, Pan F, Gan WB. Stably maintained dendritic spines are associated with lifelong memories. Nature. 2009;462:920–924. doi: 10.1038/nature08577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano H, Cong F, Birge RB, Goff SP, Chao MV. Association of the Abl tyrosine kinase with the Trk nerve growth factor receptor. J Neurosci Res. 2000;59:356–364. doi: 10.1002/(sici)1097-4547(20000201)59:3<356::aid-jnr9>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]