

Abstract
Objective
There is a growing need to identify cerebrospinal fluid (CSF) markers that can detect Alzheimer’s disease (AD) pathology in cognitively normal individuals since it is in this population that disease-modifying therapies may have the greatest chance of success. While AD pathology is estimated to begin ~10–15 years prior to the onset of cognitive decline, substantial neuronal loss is present by the time the earliest signs of cognitive impairment appear. Visinin-like protein −1 (VILIP-1) has demonstrated potential utility as a marker of neuronal injury. We here investigate CSF VILIP-1 and VILIP-1/amyloid-β42 (Aβ42) ratio as diagnostic and prognostic markers in early AD.
Methods
We assessed CSF levels of VILIP-1, tau, phosphorylated-tau181 (p-tau181), and Aβ42 in cognitively normal controls [CNC] (n=211), individuals with early symptomatic AD (n=98), and individuals with other dementias (n=19). Structural magnetic resonance imaging (n=192) and amyloid imaging with Pittsburgh Compound-B (n=156) were obtained in subsets of this cohort. Among the CNC cohort, 164 individuals had follow-up annual cognitive assessments for 2–3 years.
Results
CSF VILIP-1 levels differentiated individuals with AD from CNC and individuals with other dementias. CSF VILIP-1 levels correlated with CSF tau, p-tau181, and brain volumes in AD. VILIP-1 and VILIP-1/Aβ42 predicted future cognitive impairment in CNC over the follow-up period. Importantly, CSF VILIP-1/Aβ42 predicted future cognitive impairment at least as well as tau/Aβ42 and p-tau181/Aβ42.
Interpretation
These findings suggest that CSF VILIP-1 and VILIP-1/Aβ42 offer diagnostic utility for early AD, and can predict future cognitive impairment in cognitively normal individuals similarly to tau and tau/Aβ42, respectively.
Keywords: Visinin-like protein-1, Alzheimer’s disease, biomarkers, cerebrospinal fluid, neuronal injury
INTRODUCTION
The identification of amyloid-β (Aβ) and tau as two key proteins involved in Alzheimer’s disease (AD) pathogenesis have driven research efforts in search for suitable biomarkers for AD over the last decade 1. Cerebrospinal fluid (CSF) measures of Aβ42, tau, and phosphorylated-tau 181 (which correlate with the presence of amyloid plaques and neurofibrillary tangles [NFT], respectively), have demonstrated diagnostic and prognostic utility in the earliest symptomatic and preclinical stages of disease 2–10. However, as other aspects of the disease are being unraveled, CSF markers that reflect other disease mechanisms (e.g. neuronal and synaptic injury 11, oxidative stress 12, and inflammation 13) are gaining interest as potential diagnostic and prognostic markers. Such markers may provide insight into the different mechanisms implicated in AD pathogenesis and assist in identifying novel targets for therapies in the future.
Clinicopathological studies support the notion of a long “preclinical” stage of the disease, with amyloid and tau pathologies estimated to begin ~10–15 years prior to the onset of cognitive impairment 5, 14, 15. It is in these initial stages that disease-modifying therapies may have the greatest chance of preserving normal brain function. Therefore, CSF biomarkers that can detect preclinical AD and predict future cognitive impairment will be useful in the design of clinical trials, selection of research populations, and assessment of disease outcomes and response to therapy. Since the earliest clinical signs associated with AD pathology appear only after a threshold of neuronal damage has been reached in vulnerable brain regions16, CSF biomarkers that reflect neuronal injury may provide useful biomarker surrogates for cognitive decline and progression to symptomatic dementia.
Visinin-like protein-1 (VILIP-1) is a neuronal calcium-sensor protein17 which has been identified as a marker of neuronal injury in large-scale gene-array analyses and in brain injury models 18. In a large cohort of well characterized individuals with AD, we find that CSF VILIP-1 offers diagnostic and prognostic utility for early stage AD. Furthermore, in the cohort studied herein, the VILIP-1/Aβ42 ratio offers predictive value for the conversion from normal cognition to cognitive impairment over a 2–3 year follow-up period which is at least comparable to that of the tau/Aβ42 and p-tau181/Aβ42 ratios, the current “gold-standard” CSF biomarkers in AD.
MATERIALS AND METHODS
PARTICIPANTS
Participants (n=309) were community-dwelling volunteers (37 to 91 years of age) enrolled in longitudinal studies of healthy aging and dementia through the Washington University Alzheimer’s Disease Research Center (WU-ADRC). Participants were in good general health with no other medical illness that could contribute importantly to dementia and no contraindication to lumbar puncture (LP) or magnetic resonance imaging (MRI). Apolipoprotein E (APOE) genotypes were obtained as described 19.
The Clinical Dementia Rating (CDR) was used to denote the presence or absence of dementia, and, when present, its severity 20, 21. A CDR designation of 0 indicating no dementia characterizes individuals who are cognitively normal controls (CNC) (n=211). At the WU-ADRC, a CDR 0.5 designation denotes very mild dementia, whereas a CDR 1 and CDR 2 denote mild and moderate dementia, respectively. Cognitive assessments were performed annually and included assignment of CDR, CDR-sum of boxes (CDR-SB) 22, Mini-Mental State Exam (MMSE)23, and a 1.5-hour psychometric test battery 21. CDR scores and clinical diagnoses were based on the cognitive assessment closest to the time of the LP (median interval, 3.4 months). Of the 309 participants enrolled in WU-ADRC and included in this study, 224 participants (CNC n=164, and AD n=60) had more than one annual cognitive assessment.
All clinical diagnoses were made in accordance with standard criteria 24, 25. Individuals with CDR 0.5 or greater at baseline (n=98) included in this study were all given a clinical diagnosis of AD. We have previously demonstrated that our CDR 0.5 cohort includes many individuals who meet criteria for mild cognitive impairment (MCI), as well as those who are insufficiently impaired to meet MCI criteria and might be designated as “pre-MCI” 21.
Research participants (n=19) clinically diagnosed with frontotemporal lobar degeneration (FTLD) (n=11), progressive supranuclear palsy (PSP) (n=7), or Lewy body dementia (LBD) (n=1) at the University of California San Francisco (UCSF) Memory and Aging Center were also included in this study. Diagnoses were made according to published criteria 26, 27.
Studies were approved by the Human Studies Committee at Washington University (n=309), and the UCSF Committee on Human Research (n=19). Informed consent was obtained from all participants.
CSF AND PLASMA COLLECTION, PROCESSING AND ASSESSMENT
CSF samples (20–30 ml) were collected from all participants and analyzed for total tau, p-tau181, and Aβ42 by enzyme-linked immunosorbent assays (Innotest, Innogenetics, Ghent, Belgium) as described 28. Blood samples (10–15 ml) were obtained from a subset of participants (CNC n=149 and AD n=64) and processed to obtain plasma as described 29.
CSF and plasma samples were analyzed for VILIP-1 by a microparticle-based immunoassay (Erenna, Singulex, USA).
IN VIVO AMYLOID IMAGING
A subset (n=156) of the CNC and AD cohorts underwent amyloid imaging via positron emission tomography (PET) utilizing Pittsburgh Compound-B (PET-PIB) within 1.1 years of their LP (median interval, 2.7 months) as described30. (See Supplementary Methods).
REGIONAL AND WHOLE BRAIN VOLUMETRY
A subset (n=192) of the CNC and AD cohorts underwent MRI within 1.1 years of their LP (median interval, 1.7 months). (See Supplementary Methods).
VILIP-1 IMMUNOREACTIVITY IN NORMAL AND AD BRAIN
(see Supplementary Methods).
STATISTICAL ANALYSES
Student’s t-tests, analysis of variance (ANOVA) or chi-square (χ2) analyses were used to determine whether demographic, clinical, MRI, or CSF/plasma biomarker variables differed between the clinical groups. Bonferroni’s correction was performed for all multiple comparisons. Receiver operating curve (ROC) analyses assessed rates of agreement between CSF biomarkers and clinical diagnoses or PIB-positivity (SPSS v.15). In these analyses, the proposed cut-off value for each biomarker or ratio represents the value which provided the maximum rate of agreement with the clinical diagnoses.
Cox proportional hazard models tested the effect of demographic variables (age, gender, education, and APOE ε4 genotype) and CSF biomarker measures, individually or in combination (using Principal Components Analyses [PCA]), on the conversion rate from CDR 0 to CDR 0.5 or greater (SAS Inc, Cary, NC). CSF biomarker measures were analyzed as continuous and categorical variables. For illustrative purposes, Kaplan-Meier estimates of conversion rates as a function of CSF biomarker measures (dichotomized at the 85th percentile value) were performed. Survival analyses were conducted using baseline CDR scores at the clinical assessment prior to the time of the LP (median interval, 3.5 months). The bootstrap method was used to further examine the utility of CSF biomarker measures as predictors of future cognitive impairment (statistical software R). (See Supplementary Methods). Statistical significance was defined as p<0.05 for all analyses. Confidence intervals (CI) reported herein represent 95% CI.
RESULTS
PARTICIPANTS
The demographics and clinical summary of the study participants are presented in (Table 1). The CNC and AD cohorts differed significantly in age, mean educational level, percentage of individuals with the APOE ε4 genotype, percentage of individuals with amyloid binding on PET-PIB, mean CDR-SB, and mean MMSE. CSF VILIP-1 levels correlated with age in CNC (r=0.16, p=0.02), but not in AD (r= 0.10, p=0.30).
TABLE 1.
Demographic, Clinical, Genotype, and CSF Biomarker Characteristics of Study Participants.
| Characteristics | CNC (n=211) | AD (n=98) | p value |
|---|---|---|---|
| Age at LP, mean (SD), y | 72.1 (7.1) | 74.9 (8.1) | 0.0026* |
| APOE genotype, n ε4+/ε4− (% ε4+)€ | 62/149 (29%) | 58/40 (59%) | <0.0001*§ |
| Education, mean (SD), y | 15.6 (2.9) | 14.3 (3.1) | 0.0005* |
| Gender n F/M (%F)¥ | 129/82 (61%) | 54/44 (55%) | 0.3150§ |
| CDR-SB (range 0–18), mean (SD) | 0.03 (0.12) | 3.4 (2.4) | <0.0001* |
| MMSE (range 0–30), mean (SD) | 28.9 (1.3) | 25.3 (3.8) | <0.0001* |
| PIB+/PIB− n (%PIB+)¶ | 36/95 (27%) | 18/7 (72%) | <0.0001*§ |
| CSF VILIP-1 (pg/ml), mean (SD) | 396 (149) | 520 (171) | <0.0001* |
| CSF tau (pg/ml), mean (SD)† | 295 (159) | 602 (282) | <0.0001* |
| CSF p-tau181 (pg/ml), mean (SD)† | 54 (23) | 90 (42) | <0.0001* |
| CSF Aβ42 (pg/ml), mean (SD)†† | 615 (246) | 390 (175) | <0.0001* |
| CSF tau/Aβ42, mean (SD) †† | 0.60 (0.55) | 1.85 (1.27) | <0.0001* |
| CSF p-tau181/Aβ42, mean (SD) †† | 0.11 (0.08) | 0.27 (0.18) | <0.0001* |
| CSF VILIP-1/Aβ42, mean (SD) †† | 0.74 (0.46) | 1.55 (0.72) | <0.0001* |
| Plasma VILIP-1 (pg/ml), mean (SD)γ | 90 (26) | 102 (34) | 0.0038* |
Abbreviations: CNC, cognitively normal controls; AD, Alzheimer’s disease; LP, lumbar puncture; CDR, Clinical Dementia Rating; APOE, Apolipoprotein E; CDR-SB, Clinical Dementia Rating – sum of boxes; MMSE, Mini Mental State Examination; SD, standard deviation; PIB, Pittsburgh Compound B; CSF, cerebrospinal fluid; Aβ42, amyloid-β peptide 1–42; p-tau181, tau phosphorylated at threonine 181; VILIP-1, visinin-like protein-1.
p <0.05
APOE ε4+ genotype was defined by the presence of at least one APOE ε4 allele.
Mean CSF levels of VILIP-1 were 430 and 439 pg/ml for males and females, respectively.
Chi-square (χ2) tests were used for group comparisons. All other group comparisons were performed using Student’s t-tests.
Individuals who underwent PET-PIB (n=156) included CNC (n=131) and AD (n=25).
n=296,
n=295
Plasma samples were obtained from CNC (n=149) and AD (n=64).
DIAGNOSTIC PERFORMANCE OF CSF VILIP-1 AND VILIP-1/Aβ42 IN AD
Summary statistics of the CSF biomarker measures in CNC and AD are presented in (Table 1). Participants with very mild (CDR 0.5, n=72), mild (CDR 1, n=23), or moderate (CDR 2, n=3) AD exhibited the typical CSF biomarker phenotype for AD characterized by significantly lower mean levels of CSF Aβ42, higher mean levels of CSF tau, p-tau181, tau/Aβ42, and p-tau181/Aβ42.
Differences between CNC (n=211) and AD (n=98) were significant for all CSF biomarker measures (p<0.0001). Mean CSF VILIP-1 and VILIP-1/Aβ42 levels were significantly higher in AD compared to CNC and scaled appropriately with the CDR categories (Figure 1A and F). Consistent with previous reports from similar aged populations 30, our CNC cohort includes individuals (n=36) with evidence of preclinical AD as determined by amyloid binding on PET-PIB. When only CNC participants with negative PIB-status (n=95) were included in the analyses, mean CSF VILIP-1 levels were significantly higher in AD compared to PIB-negative CNC (Figure 1B).
FIGURE 1. Cerebrospinal Fluid (CSF) VILIP-1, tau, Aβ42, tau/Aβ42, and VILIP-1/Aβ42 levels by CDR Category in CNC, AD, and non-AD dementias.

(A) Mean (± SE) CSF VILIP-1 levels were significantly higher in CDR 0.5 (506 ± 20 pg/ml, n=72) and CDR ≥1 (558 ± 34 pg/ml, n=26) compared to CDR 0 (396 ± 10 pg/ml, n=211) and non-AD dementias (323 ± 40 pg/ml, n=19) (p<0.0001). (B) Mean (± SE) CSF VILIP-1 levels were significantly higher in CDR 0.5 and CDR ≥1 compared to PIB-negative CDR 0 (383 ± 14 pg/ml, n=95) and non-AD dementias (p<0.0001). (C–E) Mean CSF tau and tau/Aβ42 levels were significantly higher while mean CSF Aβ42 levels were significantly lower in CDR 0.5 and CDR ≥1 compared to CDR 0 (p<0.0001). (F) Mean (± SE) CSF VILIP-1/Aβ42 levels were significantly higher in CDR 0.5 (1.46 ± 0.08, n=69) and CDR ≥1 (1.79 ± 0.14, n=26) compared to CDR 0 (0.74 ± 0.03, n=200) and non-AD dementias (0.44 ± 0.03, n=19) (p<0.0001). One-Way ANOVA with Welch’s correction for unequal variances, Tukey post-hoc test was used for all group comparisons. (Similar results were obtained when Bonferroni’s correction was used for all group comparisons). Abbreviations: CNC, cognitively normal controls; AD, Alzheimer’s disease; SE, standard error.
To examine the specificity of VILIP-1 as a diagnostic marker for AD, we included a small cohort of individuals with non-AD dementias (n=19). Mean CSF VILIP-1 levels in the non-AD cohort were significantly lower than those in AD (p<0.0001) (Figure 1A and B). No significant differences in mean CSF VILIP-1 levels between PIB-negative CNC and non-AD dementias were observed (p=0.089) (Figure 1B). Scatter-plots of CSF VILIP-1, tau, Aβ42, tau/Aβ42 and VILIP-1/Aβ42 levels in CNC, AD, and non-AD dementias are illustrated in (Figure 1A–F).
ROC analyses for the CSF biomarkers and ratios in relation to clinical diagnoses (Figure 2A) and in relation to PIB-status (Figure 2B) were performed. Interestingly, VILIP-1 and VILIP-1/Aβ42 accurately predicted the presence or absence of PIB-positivity, regardless of clinical diagnoses, with comparable utility to that of the other CSF biomarkers or ratios. Rates of agreement between CSF biomarkers and clinical diagnoses, and between CSF biomarkers and PIB-status are summarized (Supplementary Table 1A and B). The VILIP-1/Aβ42, tau/Aβ42, and p-tau181/Aβ42 ratios provided higher diagnostic accuracy (Area Under the Curve [AUC]) and higher specificity in relation to PIB-status than in relation to clinical diagnoses.
FIGURE 2.
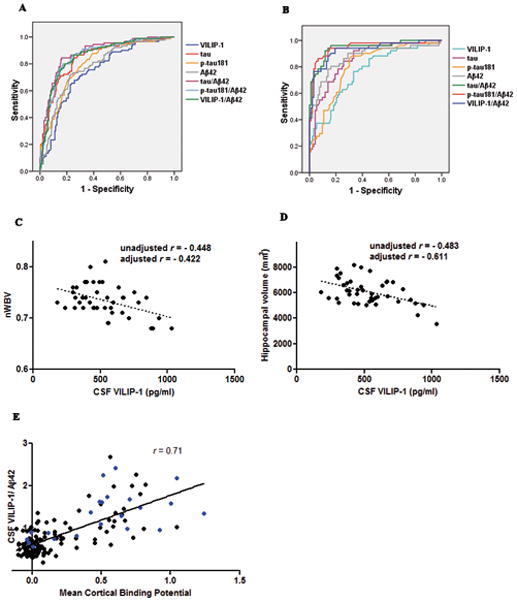
(A) Receiver Operator Curves (ROC) for the Diagnostic Utility of CSF Biomarkers and Ratios in Differentiating AD from CNC by Clinical Diagnosis. The area under the curve (AUC) ± standard error (SE) was 0.85 ± 0.02 for tau, 0.79 ± 0.03 for p-tau181, 0.79 ± 0.03 for Aβ42, 0.75 ± 0.03 for VILIP-1, 0.87 ± 0.02 for VILIP-1/Aβ42, 0.87 ± 0.02 for p-tau181/Aβ42, and 0.90 ± 0.02 for tau/Aβ42 (AD n=98, CNC n=211).
(B) Receiver Operator Curves (ROC) for the Diagnostic Utility of CSF Biomarkers and Ratios in Differentiating PIB-positive from PIB-negative Individuals. Study participants who underwent PET-PIB (n=156) were categorized by PIB status as PIB-positive (MCBP>0.18) (n=54) or PIB-negative (n=102) irrespective of clinical diagnoses. The area under the curve (AUC) ± standard error (SE) was 0.86 ± 0.03 for tau, 0.81 ± 0.04 for p-tau181, 0.87 ± 0.03 for Aβ42, 0.77 ± 0.04 for VILIP-1, 0.93 ± 0.02 for VILIP-1/Aβ42, 0.95 ± 0.02 for p-tau181/Aβ42, and 0.95 ± 0.02 for tau/Aβ42.
(C–D) Correlations of CSF VILIP-1 with nWBV and Hippocampal Volumes in AD. CSF VILIP-1 negatively correlated with (C) nWBV (unadjusted r= −0.448, p=0.003; adjusted r= −0.422, p=0.010), and (D) hippocampal volumes (unadjusted r= −0.483, p=0.001; adjusted r= −0.611, p=0.0001) in AD (n=43). Unadjusted linear regression lines are shown. Adjusted correlations included age, gender, and scanner type as co-variates. (E) Correlations Between CSF VILIP-1/Aβ42 and Amyloid Load by PET-PIB. CSF VILIP-1/Aβ42 (r=0.71, p<0.0001) correlated with PET-PIB mean cortical binding potential (MCBP) in the combined cohort (n=148). CSF VILIP-1/Aβ42 correlated with MCBP in AD (r=0.57, p=0.005) and CNC (r=0.67, p<0.0001) when examined separately. Individuals with AD (n=22) are represented by blue dots.
There were no significant differences in mean CSF VILIP-1 levels between individuals with AD who were on cholinesterase-inhibitors (521 pg/ml, n=42), NMDA-antagonists (e.g. Memantine) (465 pg/ml, n=3), both cholinesterase-inhibitors and NMDA-antagonists (536 pg/ml, n=20), or neither medication (513 pg/ml, n=33) at the time of their LP (p=0.91). In particular, no significant differences in mean CSF VILIP-1 levels were observed between individuals with AD who were on an NMDA-antagonist at the time of their LP (526 pg/ml, n=23) versus those who were not (517 pg/ml, n=75) (p=0.82).
Interestingly, mean plasma VILIP-1 levels were significantly elevated in AD compared to CNC by 13.3% as compared to the 31% increase seen in CSF in AD versus CNC (Table 1), and scaled appropriately with the CDR categories (Supplementary Figure 1).
CSF VILIP-1 CORRELATES WITH CSF TAU AND BRAIN VOLUMES IN AD
CSF VILIP-1 levels correlated with CSF tau and p-tau181, but not Aβ42 levels (Supplementary Figure 2A–C) in AD and CNC (when combined or examined separately). CSF VILIP-1 (Supplementary Figure 2D) and VILIP-1/Aβ42 (Figure 2E) showed significant correlations with PET-PIB mean cortical binding potential [MCBP] (reflective of amyloid load) in the combined (AD and CNC) cohort. Interestingly, CSF VILIP-1 correlated with MCBP in CNC but not in AD, while CSF VILIP-1/Aβ42 correlated with MCBP in both AD and CNC. Similarly to tau and p-tau18131, CSF VILIP-1 correlated with MCBP in CNC who had evidence of preclinical AD (i.e. MCBP ≥ 0.18) (Supplementary Figure 2E).
Summary statistics of the subset of participants who underwent MRI are shown (Supplementary Table 2A and B). CSF VILIP-1 negatively correlated with nWBV, hippocampal, entorhinal, and parahippocampal volumes in AD (n=43) (Figure 2C–D and Supplementary Table 3). When the AD cohort was sub-classified by CDR category, CSF VILIP-1 remained negatively correlated with nWBV, hippocampal and entorhinal volumes in the CDR 0.5 and CDR ≥ 1 cohorts (Supplementary Table 4). (See Supplementary Results).
CSF VILIP-1 AND VILIP-1/Aβ42 PREDICT FUTURE COGNITIVE IMPAIRMENT IN COGNITIVELY NORMAL INDIVIDUALS
We investigated whether CSF VILIP-1, tau, p-tau181, Aβ42, tau/Aβ42, p-tau181/Aβ42, and VILIP-1/Aβ42 predicted conversion from normal cognition (CDR 0) to cognitive impairment (CDR 0.5 or greater). Data from CNC(≥ 55 years of age) who have had one or more follow-up annual cognitive assessments (n=164) were included in these analyses. For individuals who converted from CDR 0 to CDR 0.5 or greater during follow-up (i.e. “converters”), follow-up time was calculated as the interval between the baseline clinical assessment (CDR 0) and the time of the first clinical assessment with a CDR 0.5 or greater. For individuals who did not convert on follow-up (i.e. “non-converters”), follow-up time was calculated as the interval from their baseline assessment (CDR 0) to their last annual assessment. Of the 164 participants meeting these criteria, 26 (16%) had 1 or more CDR ratings of 0.5 or greater at follow-up, which averaged 2 –3 years. This rate of conversion to cognitive impairment in cognitively normal individuals is consistent with previous population-based reports 32. Baseline demographic and clinical variables did not differ between the two groups, except converters were older than non-converters and included a higher percentage of individuals with amyloid binding on PET-PIB (Table 2).
TABLE 2.
Baseline Demographic, Clinical, Genotype, and CSF Biomarker Characteristics of Non-converters and Converters.
| Characteristics | Non-converters (CDR 0 at follow-up) (n=138) | Converters (CDR 0.5 or greater at follow-up) (n=26) | p value |
|---|---|---|---|
| Age at LP, mean (SD), y | 71.7 (6.7) | 77.4 (6.4) | <0.0001* |
| APOE genotype, ε4+/ε4− (%ε4+)€ | 45/93 (33%) | 6/20 (23%) | 0.34§ |
| Education, mean (SD), y | 15.45 (2.7) | 15.46 (4.3) | 0.98 |
| Gender n F/M (%F) | 88/50 (64%) | 16/10 (62%) | 0.83§ |
| Baseline MMSE (range 0–30), mean (SD) | 29.1 (1.1) | 29.0 (1.2) | 0.71 |
| MMSE at follow-up (range 0–30), mean (SD) | 29.0 (1.2) | 27.3 (2.2) | <0.0001* |
| Baseline CDR-SB, mean (SD, range) | 0.05 (0.2, 0–2) | 0.12 (0.3, 0–1.5) | 0.17 |
| CDR-SB at follow-up, mean (SD, range) | 0.02 (0.1, 0–1) | 1.27 (1.0, 0.5–4) | <0.0001* |
| Follow-up time, mean (SD), y | 2.8 (1.7) | 2.9 (1.4) | 0.91 |
| PIB+/PIB− (%PIB+)γ | 17/69 (20%) | 6/5 (55%) | 0.0107*§ |
| CSF VILIP-1 (pg/ml), mean (SD) | 376 (132) | 503 (177) | <0.0001* |
| CSF tau (pg/ml), mean (SD)† | 280 (144) | 412 (202) | <0.0001* |
| CSF p-tau181 (pg/ml), mean (SD) † | 52 (22) | 66 (31) | 0.0044* |
| CSF Aβ42 (pg/ml), mean (SD) † | 640 (241) | 527 (240) | 0.0331* |
| CSF tau/Aβ42, mean (SD) † | 0.52 (0.42) | 0.99 (0.77) | <0.0001* |
| CSF p-tau181/Aβ42, mean (SD) † | 0.10 (0.07) | 0.16 (0.12) | 0.0002* |
| CSF VILIP-1/Aβ42, mean (SD) † | 0.67 (0.40) | 1.12 (0.59) | <0.0001* |
Abbreviations: APOE, Apolipoprotein E; CDR, Clinical Dementia Rating; CDR-SB, Clinical Dementia Rating-sum of boxes; MMSE, Mini-Mental State Examination, PIB, Pittsburgh Compound-B; LP, lumbar puncture.
p <0.05
APOE ε4+ genotype was defined by the presence of at least one APOE ε4 allele.
Chi-square (χ2) tests were used for group comparisons.
These values represent the percentage of (PIB+) individuals among all individuals who were evaluated by PET-PIB in each of the “non-converters” (n=86) and “converters” (n=11) cohorts.
n=136 and n=25 for non-converters and converters, respectively.
Cox proportional hazard models were performed for each of the CSF markers or ratios as a continuous variable after adjusting for age, gender, education, and APOE ε4 genotype (Table 3). CSFVILIP-1 and VILIP-1/Aβ42 significantly predicted conversion from CDR 0 to CDR 0.5 or greater. Consistent with previous reports3, 9, CSF tau, p-tau181, tau/Aβ42, and p-tau181/Aβ42, but not Aβ42 alone, also predicted conversion over this follow-up period.
TABLE 3.
Baseline Demographic and CSF Biomarker Variables as Predictors of Time to Conversion from CDR 0 to CDR 0.5 or Greater.
| Characteristic or biomarker | Hazard ratio (95% CI) | Wald χ21 | p value |
|---|---|---|---|
| Age€ | 1.08 (1.030–1.132) | 10.57 | 0.001* |
| Education | 0.992 (0.877–1.122) | 0.016 | 0.899 |
| Female gender | 0.688 (0.313–1.512) | 0.87 | 0.349 |
| APOE ε4+ genotype | 0.789 (0.317–1.963) | 0.26 | 0.610 |
| tau | 1.003 (1.001–1.005) | 9.96 | 0.0016* |
| p-tau181 | 1.013 (1.000–1.026) | 4.11 | 0.0426* |
| Aβ42 | 0.998 (0.996–1.000) | 2.53 | 0.1117 |
| VILIP-1 | 1.004 (1.002–1.006) | 11.55 | 0.0007* |
| tau/Aβ42 | 2.613 (1.598–4.271) | 14.67 | 0.0002* |
| p-tau181/Aβ42† | 1.685 (1.216–2.335) | 9.83 | 0.0017* |
| VILIP-1/Aβ42 | 4.665 (2.453–8.871) | 22.05 | <0.0001* |
Cox proportional hazard models were used to assess the ability of demographic variables (age, gender, education, and APOE ε4+ genotype) and baseline levels of CSF biomarkers (as continuous variables) to predict conversion from CDR 0 to CDR 0.5 or greater over the 2–3 year follow-up period. Analyses for all CSF biomarker measures were adjusted for age, gender, education, and APOE ε4+ genotype. Abbreviations: CI, confidence intervals.
Hazard ratio for age after adjusting for the CSF biomarker measures tau, p-tau181, Aβ42, and VILIP-1, and demographic variables (education, gender, and APOE ε4+ genotype): 1.07 (CI: 1.01–1.145, p= 0.048).
Statistically significant (p value <0.05).
Because of the small values of the p-tau181/Aβ42 ratios, these values were transformed by multiplying each value by a constant of 10 prior to analysis.
Hazard ratios [HR] were then calculated for each of these markers or ratios as a dichotomous variable using the 85th percentile value as a cutoff (adjusting for age, gender, education, and APOE ε4 genotype). Individuals with high VILIP-1 (adjusted HR: 3.74, p=0.0023), corresponding to individuals whose VILIP-1 values were ≥ 535 pg/ml, progressed much more rapidly to cognitive impairment than individuals with lower values (<535 pg/ml, corresponding to the lower 85% of VILIP-1values). Individuals with high VILIP-1/Aβ42 (adjusted HR:13.00, p<0.0001), corresponding to individuals whose VILIP-1/Aβ42 values were ≥ 1.13, progressed much more rapidly to cognitive impairment than individuals with lower values (<1.13, corresponding to the lower 85% of VILIP-1/Aβ42 values). The adjusted hazard ratios for the CSF biomarkers and ratios (dichotomized at the 85th percentile value) as predictors of future cognitive impairment are illustrated (Figure 3). CSF VILIP-1 and Aβ42 values in cognitively normal elderly (n=164) included in these analyses are illustrated (Supplementary Figure 3).
FIGURE 3. Baseline CSF Measures of VILIP-1 (A), VILIP-1/Aβ42 (B), tau (C), tau/Aβ42 (D), p-tau181 (E), and p-tau181/Aβ42 (F) as Predictors of Conversion from CDR 0 to CDR 0.5 or Greater.

Kaplan-Meier estimates of the rates of conversion from CDR 0 to CDR 0.5 or greater as a function of CSF biomarker measures (dichotomized at the 85th percentile value) are shown. Analyses were adjusted for age, gender, education, and APOE ε4+ genotype. Cutoff values were 535 pg/ml, 440 pg/ml, 78 pg/ml, 1.13, 0.94, and 0.15 for VILIP-1, tau, p-tau181, VILIP-1/Aβ42, tau/Aβ42, and p-tau181/Aβ42, respectively. Adjusted hazard ratios were 3.74 (95% CI: 1.98–9.57, p=0.0023) for VILIP-1, 2.57 (95% CI: 1.31–6.97, p=0.0306) for tau, 1.72 (95% CI: 0.97–5.38, p=0.06) for p-tau181, 13.00 (95% CI: 4.38–30.90, p<0.0001) for VILIP-1/Aβ42, 9.82 (95% CI: 3.11–21.28, p<0.0001) for tau/Aβ42, and 7.83 (95% CI: 2.65–16.34, p<0.0001) for p-tau181/Aβ42.
In agreement with these findings, results from the bootstrap analyses indicate that the predictive ability for future cognitive impairment was 0.892 (p=0.0007) for VILIP-1, 0.866 (p=0.0016) for tau, 0.452 (p=0.0426) for p-tau181, 0.328 (p=0.1117) for Aβ42, 0.998 (p<0.0001) for VILIP-1/Aβ42, 0.974 (p=0.0002) for tau/Aβ42, and 0.902 (p=0.0017) for p-tau181/Aβ42. The combinations of CSF VILIP-1 and tau (0.904, p=0.0007), and of CSF VILIP-1 and p-tau181 (0.804, p=0.0031) were stronger predictors of conversion than either tau (0.866, p=0.0016) or p-tau181 (0.452, p=0.0426) alone, respectively. When VILIP-1 was added to the combination of tau and p-tau181, the three markers together were stronger predictors of conversion (0.844, p=0.0021) than the combination of tau and p-tau181 (0.714, p=0.0084). Importantly, the combination of VILIP-1, tau, p-tau181, and Aβ42 (0.826, p=0.0024) was a stronger predictor of conversion than the combination of tau, p-tau181, and Aβ42 (0.730, p=0.0078).
VILIP-1 IMMUNOREACTIVITY IN NORMAL AND AD BRAIN
[See Supplementary Results and (Supplementary Figure 4)].
DISCUSSION
We here confirm CSF VILIP-1 as a diagnostic marker for AD in a large cohort, and now show that CSF VILIP-1 and VILIP-1/Aβ42 are strong prognostic measures for early AD. To our knowledge, this is the first study to show that CSF VILIP-1 and VILIP-1/Aβ42 predict future cognitive impairment in cognitively normal individuals. Furthermore, we provide new data from a large cohort of well characterized individuals that CSF VILIP-1 negatively correlates with whole brain and regional brain volumes in AD, and positively correlates with amyloid load in preclinical AD.
We have previously demonstrated that CSF VILIP-1 is increased in a small cohort of individuals with AD33. We here validate these findings in a much larger cohort, including individuals in the earliest symptomatic stage of AD (CDR 0.5 or MCI). Our results suggest that CSF VILIP-1 and VILIP-1/Aβ42 offer diagnostic sensitivity for AD that is comparable to that of CSF tau, p-tau181, or Aβ42, and tau/Aβ42 or p-tau181/Aβ42, respectively. Notably, since our CDR 0.5 cohort includes individuals who may elsewhere be classified as MCI or pre-MCI, these findings reveal the promise of CSF VILIP-1 and VILIP-1/Aβ42 as diagnostic biomarkers for the earliest symptomatic stage of AD. Moreover, our study is the first to investigate the diagnostic utility of plasma VILIP-1 levels in AD. In agreement with the CSF findings, our plasma results suggest that significant, albeit smaller, differences in VILIP-1 levels between AD and non-demented controls can be detected peripherally.
Additionally, by including individuals with other neurodegenerative disorders (e.g. FTLD, PSP, and LBD), it appears that CSF VILIP-1 may confer some diagnostic specificity for AD. It is possible that elevated CSF VILIP-1 levels in the setting of chronic neurodegeneration may be influenced by relatively disease-specific mechanisms or alterations in signaling pathways 34–37. It will be of interest in the future to validate these findings in multiple larger cohorts, including more individuals with non-AD dementias, and to investigate the mechanisms by which VILIP-1 levels may be preferentially altered in AD.
While significant differences in mean CSF VILIP-1 levels exist between AD and CNC, some overlap between the two groups was observed. Overlap is likely due in part to the inclusion of cognitively normal individuals with preclinical AD as well as some individuals with AD who may have alternative diagnoses. Consistent with previous reports14, 30, our CNC cohort (60 years or older) includes individuals with amyloid binding on PET-PIB and some with elevated tau. Most of these individuals have CSF VILIP-1 levels that are comparable to those in AD (Figure 1A and B). Moreover, our AD cohort predominantly consists of individuals with very mild dementia (CDR 0.5), potentially decreasing the difference in CSF levels between the AD and CNC cohorts. Very similar overlap is observed with other CSF biomarkers such as tau, Aβ42, and tau/Aβ42 when used to compare AD and CNC based only on clinical diagnoses, not taking into account underlying AD pathology on PET-PIB (Figure 1). Importantly, our results suggest that VILIP-1/Aβ42, tau/Aβ42, and p-tau181/Aβ42 are better indicators of PIB-positivity than of clinical diagnoses (Figure 2A and B).
VILIP-1 is abundantly expressed in neurons, but not other cell types, in the brain (Supplementary Figure 4)35, and has demonstrated utility as a marker of neuronal injury in brain injury models 18. Our findings that CSF VILIP-1 negatively correlates with whole brain and regional brain volumes, at least as well as tau and p-tau181, in even the earliest symptomatic stages of AD, and with amyloid load in cognitively normal individuals, support the potential utility of CSF VILIP-1 as a biomarker surrogate for neurodegeneration in AD. The correlation between CSF VILIP-1 and CSF tau or p-tau181 levels likely reflects close associations of these proteins in AD (Supplementary Figure 4) 35, 38 and, perhaps, the ability of these markers to measure neurodegeneration since VILIP-1 is not a component of neurofibrillary tangles. In contrast, CSF Aβ42 levels appear to decrease years prior to the onset of cognitive impairment and remain relatively stable with further disease progression 28, 31, 39, 40. Similarly to tau and p-tau18131, the correlation between CSF VILIP-1 and amyloid load in preclinical AD suggests that increasing amyloid accumulation in these early stages 5, 6 is associated with greater neuronal injury. On the other hand, cortical amyloid deposition is likely close to reaching its maximal extent by the time individuals with AD become cognitively impaired 41, and does not appear to change considerably with further disease progression 42.
There is accumulating evidence that progressive neuronal and synaptic loss is the best surrogate for cognitive decline and disease progression in AD 41. Neuronal counts in the entorhinal and hippocampal regions are comparable in preclinical AD and “healthy aging”, but decrease substantially as the earliest signs of cognitive impairment appear16. We here demonstrate that CSF VILIP-1, alone or in combination with Aβ42 (VILIP-1/Aβ42), offers predictive value for future cognitive impairment in a large cohort of cognitively normal elderly. Together with CSF tau and Aβ42 levels, these data support the notion that increased tau levels (reflective of NFT, and perhaps neurodegeneration) and VILIP-1 levels (generally reflective of neuronal injury) occur following decreases in Aβ42 and increasing amyloid load over this short follow-up period, and herald the onset of symptomatic cognitive impairment. Furthermore, our findings suggest that CSF VILIP-1 complements the prognostic utility of CSF tau, p-tau181, and Aβ42 (collectively) in predicting future cognitive impairment over this follow-up period.
In the cohort studied herein, VILIP-1/Aβ42 appears to predict future cognitive impairment at least as well as tau/Aβ42 and p-tau181/Aβ42, the current “gold standard” prognostic biomarkers for AD (Table 3 and Figure 3). These results further suggest that while the initial deposition of amyloid plaques and neurofibrillary tangles probably begins a decade or longer prior to the onset of clinical symptoms 5, 14, 15, it is only after a threshold of neuronal injury has been reached in vulnerable brain regions that the clinical manifestations of cognitive impairment and later dementia appear 16. Therefore, it is likely that CSF markers of neuronal injury together with markers of amyloid may predict future cognitive impairment over short follow-up periods with comparable ability to that of CSF markers of tau together with amyloid.
Together, these findings highlight the potential use of CSF VILIP-1 and VILIP-1/Aβ42 in guiding trial design, treatment decisions, outcome assessments, and response to therapies in clinical trials of disease-modifying therapies. CSF VILIP-1 may have potential value as a secondary or tertiary outcome measure in such trials, and complement diagnostic and prognostic information provided by CSF tau, p-tau, and Aβ42. It will be important to validate these findings in larger cohorts of well characterized individuals with similar or longer durations of follow-up.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health [P50-AG05681, P01-AG03991, P01-26276, R01-NS6790, R01-NS67905, and P50-NS55977], Siemens Health Care Diagnostics [to JHL], and the Charles and Joanne Knight Alzheimer Research Initiative. FTLD/PSP/LBD CSF was generously provided by the University of California- San Francisco (UCSF) Memory and Aging Center (work supported by the National Institutes of Health/National Institute on Aging [R01 AG031278, K23-AG031861, P01 AG019724, P50 AG023501]; UCSF Alzheimer’s Disease Research Centers [P50 AG023501]; the CurePSP; and the Association for Frontotemporal Dementias).
We gratefully acknowledge the contributions of Dr. Dan Crimmins, Jay McQuillan, Floy Stewart, Jenny Gurney, Lisa Taylor-Reinwald, and the Clinical, Psychometrics, Biomarker, Imaging and Biostatistics Cores of the Charles F. and Joanne Knight Alzheimer’s Disease Research Center at Washington University.
References
- 1.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 2.Hansson O, Zetterberg H, Buchhave P, et al. Association between CSF biomarkers and incipient Alzheimer’s disease in patients with mild cognitive impairment: a follow-up study. Lancet Neurol. 2006;5:228–234. doi: 10.1016/S1474-4422(06)70355-6. [DOI] [PubMed] [Google Scholar]
- 3.Fagan AM, Roe CM, Xiong C, et al. Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol. 2007;64:343–349. doi: 10.1001/archneur.64.3.noc60123. [DOI] [PubMed] [Google Scholar]
- 4.Tarawneh R, Holtzman DM. Biomarkers in translational research of Alzheimer’s disease. Neuropharmacology. 2010;59:310–322. doi: 10.1016/j.neuropharm.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jack CR, Jr, Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9:119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perrin RJ, Fagan AM, Holtzman DM. Multimodal techniques for diagnosis and prognosis of Alzheimer’s disease. Nature. 2009;461:916–922. doi: 10.1038/nature08538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zetterberg H, Mattsson N, Shaw LM, Blennow K. Biochemical markers in Alzheimer’s disease clinical trials. Biomark Med. 2010;4:91–98. doi: 10.2217/bmm.09.80. [DOI] [PubMed] [Google Scholar]
- 8.De Meyer G, Shapiro F, Vanderstichele H, et al. Diagnosis-independent Alzheimer disease biomarker signature in cognitively normal elderly people. Arch Neurol. 2010;67:949–956. doi: 10.1001/archneurol.2010.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li G, Sokal I, Quinn JF, et al. CSF tau/Abeta42 ratio for increased risk of mild cognitive impairment: a follow-up study. Neurology. 2007;69:631–639. doi: 10.1212/01.wnl.0000267428.62582.aa. [DOI] [PubMed] [Google Scholar]
- 10.Jagust WJ, Landau SM, Shaw LM, et al. Relationships between biomarkers in aging and dementia. Neurology. 2009;73:1193–1199. doi: 10.1212/WNL.0b013e3181bc010c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chaves ML, Camozzato AL, Ferreira ED, et al. Serum levels of S100B and NSE proteins in Alzheimer’s disease patients. J Neuroinflammation. 2010;7:6. doi: 10.1186/1742-2094-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Montine TJ, Kaye JA, Montine KS, et al. Cerebrospinal fluid abeta42, tau, and f2-isoprostane concentrations in patients with Alzheimer disease, other dementias, and in age-matched controls. Arch Pathol Lab Med. 2001;125:510–512. doi: 10.5858/2001-125-0510-CFATAF. [DOI] [PubMed] [Google Scholar]
- 13.Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann Neurol. 1999;45:358–368. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 15.Tarawneh R, Holtzman DM. Critical issues for successful immunotherapy in Alzheimer’s disease: development of biomarkers and methods for early detection and intervention. CNS Neurol Disord Drug Targets. 2009;8:144–159. doi: 10.2174/187152709787847324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Price JL, Ko AI, Wade MJ, et al. Neuron number in the entorhinal cortex and CA1 in preclinical Alzheimer disease. Arch Neurol. 2001;58:1395–1402. doi: 10.1001/archneur.58.9.1395. [DOI] [PubMed] [Google Scholar]
- 17.Braunewell KH, Klein-Szanto AJ. Visinin-like proteins (VSNLs): interaction partners and emerging functions in signal transduction of a subfamily of neuronal Ca2+-sensor proteins. Cell Tissue Res. 2009;335:301–316. doi: 10.1007/s00441-008-0716-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laterza OF, Modur VR, Crimmins DL, et al. Identification of novel brain biomarkers. Clin Chem. 2006;52:1713–1721. doi: 10.1373/clinchem.2006.070912. [DOI] [PubMed] [Google Scholar]
- 19.Talbot C, Lendon C, Craddock N, et al. Protection against Alzheimer’s disease with apoE epsilon 2. Lancet. 1994;343:1432–1433. doi: 10.1016/s0140-6736(94)92557-7. [DOI] [PubMed] [Google Scholar]
- 20.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43:2412–2414. doi: 10.1212/wnl.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- 21.Storandt M, Grant EA, Miller JP, Morris JC. Longitudinal course and neuropathologic outcomes in original vs revised MCI and in pre-MCI. Neurology. 2006;67:467–473. doi: 10.1212/01.wnl.0000228231.26111.6e. [DOI] [PubMed] [Google Scholar]
- 22.Berg L, Miller JP, Baty J, et al. Mild senile dementia of the Alzheimer type. 4. Evaluation of intervention. Ann Neurol. 1992;31:242–249. doi: 10.1002/ana.410310303. [DOI] [PubMed] [Google Scholar]
- 23.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 24.Morris JC, Weintraub S, Chui HC, et al. The Uniform Data Set (UDS): clinical and cognitive variables and descriptive data from Alzheimer Disease Centers. Alzheimer Dis Assoc Disord. 2006;20:210–216. doi: 10.1097/01.wad.0000213865.09806.92. [DOI] [PubMed] [Google Scholar]
- 25.Berg L, McKeel DW, Jr, Miller JP, et al. Clinicopathologic studies in cognitively healthy aging and Alzheimer’s disease: relation of histologic markers to dementia severity, age, sex, and apolipoprotein E genotype. Arch Neurol. 1998;55:326–335. doi: 10.1001/archneur.55.3.326. [DOI] [PubMed] [Google Scholar]
- 26.Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–1554. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- 27.Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996;47:1–9. doi: 10.1212/wnl.47.1.1. [DOI] [PubMed] [Google Scholar]
- 28.Fagan AM, Mintun MA, Mach RH, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol. 2006;59:512–519. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- 29.Fagan AM, Head D, Shah AR, et al. Decreased cerebrospinal fluid Abeta(42) correlates with brain atrophy in cognitively normal elderly. Ann Neurol. 2009;65:176–183. doi: 10.1002/ana.21559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mintun MA, Larossa GN, Sheline YI, et al. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology. 2006;67:446–452. doi: 10.1212/01.wnl.0000228230.26044.a4. [DOI] [PubMed] [Google Scholar]
- 31.Fagan AM, Mintun MA, Shah AR, et al. Cerebrospinal fluid tau and ptau(181) increase with cortical amyloid deposition in cognitively normal individuals: implications for future clinical trials of Alzheimer’s disease. EMBO Mol Med. 2009;1:371–380. doi: 10.1002/emmm.200900048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Andersen K, Launer LJ, Dewey ME, et al. Gender differences in the incidence of AD and vascular dementia: The EURODEM Studies. EURODEM Incidence Research Group. Neurology. 1999;53:1992–1997. doi: 10.1212/wnl.53.9.1992. [DOI] [PubMed] [Google Scholar]
- 33.Lee JM, Blennow K, Andreasen N, et al. The brain injury biomarker VLP-1 is increased in the cerebrospinal fluid of Alzheimer disease patients. Clin Chem. 2008;54:1617–1623. doi: 10.1373/clinchem.2008.104497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Allen SJ, Wilcock GK, Dawbarn D. Profound and selective loss of catalytic TrkB immunoreactivity in Alzheimer’s disease. Biochem Biophys Res Commun. 1999;264:648–651. doi: 10.1006/bbrc.1999.1561. [DOI] [PubMed] [Google Scholar]
- 35.Braunewell K, Riederer P, Spilker C, et al. Abnormal localization of two neuronal calcium sensor proteins, visinin-like proteins (vilips)-1 and -3, in neocortical brain areas of Alzheimer disease patients. Dement Geriatr Cogn Disord. 2001;12:110–116. doi: 10.1159/000051244. [DOI] [PubMed] [Google Scholar]
- 36.Friedel RH, Schnurch H, Stubbusch J, Barde YA. Identification of genes differentially expressed by nerve growth factor- and neurotrophin-3-dependent sensory neurons. Proc Natl Acad Sci U S A. 1997;94:12670–12675. doi: 10.1073/pnas.94.23.12670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Braunewell KH. The darker side of Ca2+ signaling by neuronal Ca2+-sensor proteins: from Alzheimer’s disease to cancer. Trends Pharmacol Sci. 2005;26:345–351. doi: 10.1016/j.tips.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 38.Schnurra I, Bernstein HG, Riederer P, Braunewell KH. The neuronal calcium sensor protein VILIP-1 is associated with amyloid plaques and extracellular tangles in Alzheimer’s disease and promotes cell death and tau phosphorylation in vitro: a link between calcium sensors and Alzheimer’s disease? Neurobiol Dis. 2001;8:900–909. doi: 10.1006/nbdi.2001.0432. [DOI] [PubMed] [Google Scholar]
- 39.Blennow K, Zetterberg H, Minthon L, et al. Longitudinal stability of CSF biomarkers in Alzheimer’s disease. Neurosci Lett. 2007;419:18–22. doi: 10.1016/j.neulet.2007.03.064. [DOI] [PubMed] [Google Scholar]
- 40.Andreasen N, Hesse C, Davidsson P, et al. Cerebrospinal fluid beta-amyloid(1–42) in Alzheimer disease: differences between early- and late-onset Alzheimer disease and stability during the course of disease. Arch Neurol. 1999;56:673–680. doi: 10.1001/archneur.56.6.673. [DOI] [PubMed] [Google Scholar]
- 41.Ingelsson M, Fukumoto H, Newell KL, et al. Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology. 2004;62:925–931. doi: 10.1212/01.wnl.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- 42.Rowe CC, Ng S, Ackermann U, et al. Imaging beta-amyloid burden in aging and dementia. Neurology. 2007;68:1718–1725. doi: 10.1212/01.wnl.0000261919.22630.ea. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.