

Abstract
Purpose
To determine the cause of Leber congenital amaurosis (LCA) and developmental cataracts in a consanguineous Pakistani family.
Methods
The diagnosis was established in all affected individuals of a Pakistani LCA family by medical history, funduscopy, and standard ERG. We performed genome-wide linkage analysis for mapping the disease locus in this family.
Results
Congenitally severely reduced visual acuity and nystagmus were reported for all patients who, in the later phase of the disease, also developed cataracts. LCA in the family cosegregated with homozygosity for a single nucleotide polymorphism (SNP) haplotype on chromosome 6p14.1. The respective candidate region contained Leber congenital amaurosis 5 (LCA5), a gene previously reported to underlie LCA. We subsequently identified a novel truncating mutation in exon 4 of LCA5, c.642delC, in homozygous state in all affected persons of the family.
Conclusions
We report a novel LCA5 mutation causing LCA in a Pakistani family. Developmental cataracts were present in two of the four patients, raising the possibility that LCA5 mutations may predispose to this additional ocular pathology.
Introduction
Leber congenital amaurosis (LCA, OMIM 204000) accounts for at least 5% of all retinal dystrophies and approximately 20% of children attending schools for the blind. LCA is the most severe retinal dystrophy causing blindness or severe visual impairment before the age of 1 year. Inheritance is autosomal recessive in most cases. Clinically, LCA is characterized by the presence of four key features, namely severe and early visual loss (usually around the age of 6 weeks), sensory nystagmus, amaurotic pupils, and minimal or absent responses in the electroretinogram (ERG). LCA can be observed as part of syndromes such as Joubert syndrome. To date, 15 genes associated with LCA have been identified [1]. These genes are involved in various genetic pathways, including retina development (crumbs homolog-1 [CRB1] and cone-rod homeobox-containing gene [CRX]), phototransduction (guanylate cyclase 2D [GUCY2D] and aryl hydrocarbon receptor interacting protein-like 1 [AIPL1]), vitamin A metabolism (retinal pigment epithelium-specific protein 65 kDa [RPE65]; lecithin retinol acyltransferase [LRAT], and retinol dehydrogenase 12 [RDH12]), ciliary formation and function (tubby like protein 1, [TULP1]; retinitis pigmentosa GTPase regulator interacting protein 1, [RPGRIP1]; centrosomal protein 290 kDa [CEP290], and Leber congenital amaurosis 5 [LCA5]), and RPE phagocytosis (c-mer proto-oncogene tyrosine kinase [MERTK]). The function of RD3 remains to be elucidated. In rare cases, certain mutations in CRX and inosine 5′-monophosphate dehydrogenase 1 (IMPDH1), which is involved in guanine nucleotide synthesis, have been shown to cause dominant LCA. Recently, another gene, SPATA7 was identified as the LCA3 gene [2]. Mutations in the known LCA genes account for ~70% of non-syndromic LCA cases. LCA5 mutations probably account for less than 3% of all cases.
Although many persons with LCA may have normal or near normal fundus appearance as infants, pigmentary retinopathy as in retinitis pigmentosa may develop at later stages (RP). Consistent with this observation, four known LCA disease genes, including CRX, CRB1, RPE65, and TULP1, have also been linked to RP. Mutations in LCA disease genes may lead to diverse phenotypes, e.g., cone rod dystrophy (CRX, AIPL1, GUCY2D, RPE65, and RPGRIP1), retinal dystrophy (RDH12), and Bardet-Biedl, Joubert or Meckel syndrome (CEP290). Therefore, the study of LCA provides potential insight into other retinal dystrophies and genetic syndromes [1].
Here, we report a consanguineous family from Pakistan with four affected individuals diagnosed with autosomal recessive LCA (Figure 1A). Genomewide linkage analysis mapped the disease region to chromosome 6p14.1 which contained LCA5 (Figure 1B,C). Mutational analysis identified a homozygous novel frameshift mutation (c.642delC) in exon 4 of LCA5 in all patients.
Figure 1.

Genetic analysis of LCA family BUIT-LA01. A: Family pedigree. Circles, females; squares, males. Black symbols, affected. Double horizontal line indicates consanguinity. The sample of IV:5 was not subjected to genomewide SNP mapping because this sample was initially not available. B: Graphical view of LOD score calculations of genomewide SNP mapping in the BUIT-LA01 family. LOD scores calculated with ALLEGRO are given along the y-axis relative to genomic position in cM (centi Morgan) on the x-axis. Note the highest peak in the region on chromosome 6 (LOD=2.66). C: Corresponding disease gene locus on chromosome 6q14.1 (4.5 Mb region, only 11 annotated genes; UCSC genome browser).
Methods
Family enrollment and clinical evaluation
A consanguineous family, BUIT-LA01, with four individuals affected by LCA was enrolled from a remote area in Pakistan (Figure 1A). The diagnosis was established in all patients (IV:2, IV:3, IV:4, and IV:5) by medical history, decreased visual acuity since birth, nystagmus, funduscopy and by standard ERG. Clinical history was obtained from participating members to rule out obvious environmental causes of vision impairment. There was no history of deafness, mental retardation, or any other associated disease related to the patients or other family members (IV:1, IV:6). The study was approved by the institutional review board (IRB) at the Department of Biotechnology and Informatics, BUITEMS, Quetta, Pakistan, and the institutional review board of the Ethics Committee of the University Hospital of Cologne, Germany. It was performed in adherence to the tenets of the declaration of Helsinki. After detailed explanation to the family members about the background of the study, written consent was obtained from all participants. Venous blood samples (5 ml) were obtained for DNA extraction and genomic DNA was isolated following standard protocols [3]. Standard electroretinograms (LKC Technologies) were measured under scotopic and photopic conditions in the elder affected patient (IV:3) according to the standard of the International Society for Clinical Electrophysiology of Vision beginning after 20 min of dark adaptation [4]. Pupils were fully dilated using phenylephrine HCL (10%) and tropicamide (1%). To study the retinal features, funduscopy was performed on two of the elder affected individuals (IV:3 and IV:5) of the family.
Linkage analysis
Samples from the parents and all children except IV:5 (this sample was collected at a later point of time) were subjected to a whole genome scan, using an Affymetrix GeneChip Human Mapping 10K Array, version 2.0 (Affymetrix, Santa Clara, CA). GRR and PedCheck were used to verify relationships and to identify Mendelian errors [5,6]. Non-parametric linkage analysis was performed with MERLIN [7]. Parametric linkage and haplotype analysis were performed using ALLEGRO [8], assuming autosomal recessive inheritance, full penetrance and a disease gene frequency of 0.0001. All data handling was performed using the graphical user interface ALOHOMORA [9]. Graphic output of haplotypes was generated with HaploPainter [10].
Mutational analysis
The seven coding exons and adjacent intronic sequences of the LCA5 gene (NM_181714) were amplified and sequenced (Table 1). PCR products were amplified using 100 ng of genomic DNA in a 25 µl reaction mixture containing 10 pmol of forward and reverse primers, 0.2 mM dNTP, 10 mM Tris-HCl, 50 mM KCl, 1.5 mM MgCl2, and 0.5 units of Taq polymerase (Invitrogen Corp., Carlsbad, CA). After initial denaturation at 95 °C for 4 min, 30 cycles were performed, which consisted of 95 °C for 1 min, 55–62 °C (depending on the fragment) for 1 min, and 72 °C for 1 min, with a final extension step of 72 °C for 10 min for all exons. PCR products were digested with exonuclease I and shrimp alkaline phosphatase (Fermentas Life Sciences, Glen Burnie, MD) and sequenced bi-directionally using BigDye Terminator v.3.1 kit (Applied Biosystems, Darmstadt, Germany). DNA mutation nomenclature of identified mutation was based on cDNA sequence of the longest isoform of LCA5 with +1 corresponding to the A of the ATG translation initiation codon (codon 1) in the respective reference sequence (GenBank NM_181714). For the description of sequence variation, we followed the recommendations of the Human Genome Variation Society (HGVS). Samples from 50 ethnically matched healthy controls from Pakistan were investigated by direct sequencing for the presence of the mutation.
Table 1. Primers used for PCR amplification and direct sequencing of LCA5 coding exons and adjacent intronic regions.
| Exon | Primer sequence | tm (°C) | cds (bp) | Product size (bp) |
|---|---|---|---|---|
| 3F |
TGTGGAGAAAATAGATTGCACAG |
59.28 |
|
|
| 3R |
CCTATAAAACGTAAATCAGCCCAC |
60.14 |
190 |
465 |
| 4F |
AGAATAATTCCGTATAAACTATTGGG |
57.42 |
|
|
| 4R |
TTTTCCCAAAATGACTATGATCC |
59.22 |
530 |
996 |
| 5F |
TGTACATGAATACTATGCCCAGTC |
58.10 |
|
|
| 5R |
TTATACCAACAAACCTTTTCTAAGTG |
57.26 |
138 |
437 |
| 6F |
CAAAAGGAAGCTGAACCAGG |
59.85 |
|
|
| 6R |
CAGCGTCACTTCAGGGG |
58.83 |
97 |
420 |
| 7F |
CCAAGCTGAGCAAAACATGC |
61.88 |
|
|
| 7R |
TTAGGTATATCTCCTAAAAGCCAAAG |
56.64 |
143 |
538 |
| 8F |
TTCAAGGAGTGATAACTTGTGATTTAC |
59.12 |
|
|
| 8R |
TAAGCCATCCCCTACCACTG |
59.95 |
133 |
437 |
| 9F |
TCTTTTCTCACTTGATTTATAATACCC |
57.59 |
|
|
| 9R | TTGGCAAACTATCTATGTGGTG | 57.72 | 863 | 1167 |
Results
All affected individuals of BUIT-LA01 displayed severe visual impairment since birth, bilateral keratoconus and nystagmus (Figure 1A). Slit lamp examination showed radial spoke-shaped cataracts with positive oculodigital reflex in the two older patients who were 13 and 15 years old (Figure 1A; IV:3, IV:5; and Figure 2). Cataracts were (until now) not present in the two younger patients (IV:2, IV:4; Figure 1A) who are 7 years and 9 years old. The fundus features showed moderately attenuated vessels, waxy pale disc appearance, pigmentary changes especially at macular areas in both eyes, and some bony spicule-like pigments clumping in the peripheral region and posterior pole of the retina (Figure 3). ERGs showed normal implicit time with reduced rods and cones function as a whole and markedly reduced response from rods. Results of scotopic ERGs were markedly abnormal, with the ISCEV standard, flash conducted by −25 dB, and the rod b-wave amplitude was severely reduced with a normal implicit time. Under photopic conditions, the b-wave amplitude was moderately reduced while photopic 30-Hz flicker stimulus revealed decreased amplitude with normal implicit time in both eyes (Figure 4 and Table 2).
Figure 2.

Photograph of patient IV:3 showing bilateral cataracts (radial spoke-shaped) and keratoconus.
Figure 3.
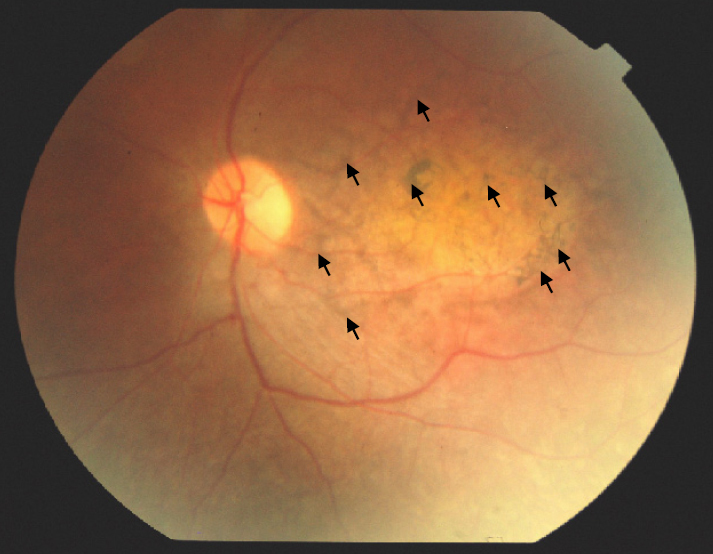
Fundus photograph of patient (IV:4) with waxy pale disc appearance and pigmentary changes (arrows) in the macular region and the posterior pole of the retina.
Figure 4.
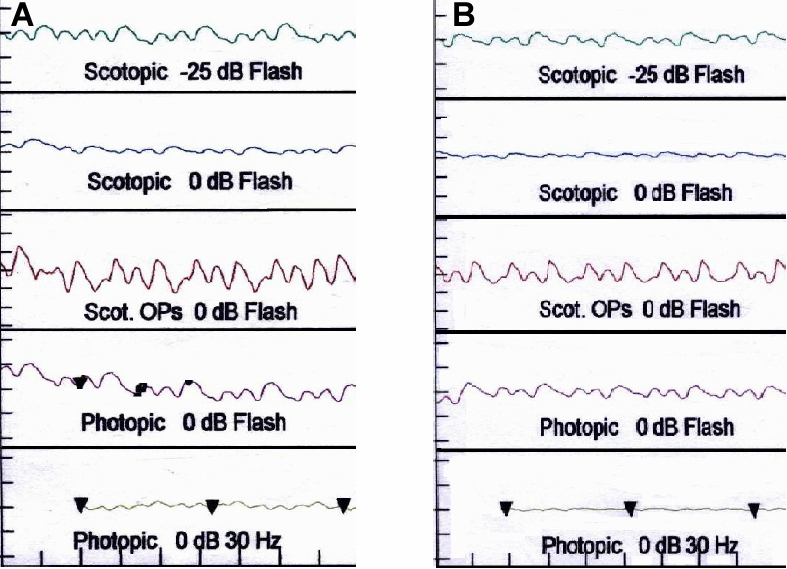
Electroretinogram (ERG) profile of IV:3. A: Right eye of IV:3. B: Left eye of IV:3.
Table 2. Standard flash ERG values in affected individual (IV:3) and normal values.
| Patient (IV:3) | Scotopic (−25 dB) | Scotopic (0 dB) | Photopic (0 dB) | Photopic/ flicker (30 Hz) |
|---|---|---|---|---|
| Right Eye |
46.5 |
68.7 |
64.6 |
3.6 |
| Left Eye |
126 |
89.1 |
76.1 |
4.8 |
| Normal control |
167 |
371 |
94 |
62 |
| Normal values | 185 | 419 | 102 | 70 |
Genomewide linkage analysis revealed a maximum parametric LOD (logarithm of the odds) score of 2.66 for a 4.5 Mb region on chromosome 6q14.1, flanked by single nucleotide polymorphisms (SNPs) rs2153886 and rs72968, that showed homozygosity by descent in all four affected siblings (Figure 1B). This critical interval contained 11 genes, including LCA5, the gene encoding lebercilin, a protein of the photoreceptor connecting cilium. Mutational analysis identified a novel frameshift mutation (c.642delC) in exon 4 of LCA5 in homozygous state in all patients from family BUIT-LA01. This variant should either lead to a subsequent truncation of the protein (225 instead of 697 residues in the wildtype), or an unstable mRNA molecule that is rapidly degraded. The mutation was absent in 50 unrelated control individuals from Pakistan.
Discussion
Mutations in the ciliary protein lebercilin, encoded by LCA5, have been shown to cause LCA. Recent data have shown that lebercilin functions in selective protein transport through the photoreceptor’s connecting cilium, suggesting defective intraflagellar transport as underlying disease mechanism [11]. To date, 10 mutations in the respective gene, LCA5, have been reported ([12–16], HGMD). Here, we report the identification of a novel mutation (c.642delC) in exon 4 of LCA5 in a consanguineous family from Pakistan. The affected individuals are between 7 and 15 years old and all presented with typical signs of LCA at birth. There were no signs for a syndromic entity at the age of examination (e.g., renal failure, neurologic symptoms, mental retardation, dysmorphic features). The phenotype of the family described herein is consistent with the phenotype described previously in LCA5 patients [13,15–17]. An additional phenotypic feature observed in our family were radial spoke-shaped developmental cataracts (illustrated in Figure 2 for individual IV:3) in the two older patients (13 years and 15 years old), while cataracts were not present in the two younger patients (IV:2, IV:4; Figure 1A) who were at 7 years and 9 years of age, respectively. Although LCA is a congenital abnormality, a previous report of a Pakistani LCA5 family suggested further progression of the phenotype, but not including cataracts [18]. The patients of that already-reported Pakistani LCA5 family had phenotypes that are different from that of the original LCA5 family of Swiss descent [17]. Visual acuity was already poor at birth (reduced to light perception in the second decade), and the disease progressed with pigmentary anomalies in the peripheral retina and increasing atrophy in the macular region [18]. The phenotype of the patients from three families from Maghreb resembled to that of the previously reported LCA5 families. There were no signs of cataracts, although the patient were between 17 and 40 years old [13]. In a recent study on 14 LCA families (3 families linked to LCA5) from Northern Pakistan, the prevalence of cataracts in LCA5 was estimated to be 8%. In that study, cataracts represent a late-onset feature of LCA5 and did not manifest before the second decade of life [15]. Our study suggests that cataracts may also be an early feature in LCA5-linked LCA. Alternatively, given the parental consanguinity in our LCA5 family, it cannot be excluded that the manifestation of cataracts in two patients reflects homozygosity for a mutation in an unlinked (cataract) gene. We have previously described such constellations with two overlapping recessive conditions, mimicking a single, syndromic disorder [19,20]. Furthermore, LCA-associated cataracts have been postulated to result from a combination of genetic and environmental factors [1].
In summary, we describe a Pakistani family with Leber congenital amaurosis linked to the LCA5 locus, and the identification of a novel frameshift mutation confirms LCA5 as contributor to congenital retinal degeneration. The finding of early-onset cataract in two patients from our family extends the LCA5 phenotype spectrum. Finally, our study confirms the potential of homozygosity mapping in recessive retinal degeneration families from inbred populations.
In the Table, tm indicates melting temperature; cds indicates coding sequence; bp indicates product size in basepairs.
Acknowledgments
We are grateful to the family members for their participation in this study. We also thank Dost Muhammad Baloch and Abdul Majeed Cheema for providing laboratory facilities at BUITEMS, Quetta. This study was supported by faculty funds at BUITEMS with the help of Ahmed Farooq Bazai, Vice Chancellor, BUITEMS, and by the Gertrud Kusen-Stiftung to H.J.B.
References
- 1.den Hollander AI, Roepman R, Koenekoop RK, Cremers FP. Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog Retin Eye Res. 2008;27:391–419. doi: 10.1016/j.preteyeres.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 2.Wang H, den Hollander AI, Moayedi Y, Abulimiti A, Li Y, Collin RW, Hoyng CB, Lopez I, Abboud EB, Al-Rajhi AA, Bray M, Lewis RA, Lupski JR, Mardon G, Koenekoop RK, Chen R. Mutations in SPATA7 cause Leber congenital amaurosis and juvenile retinitis pigmentosa. Am J Hum Genet. 2009;84:380–7. doi: 10.1016/j.ajhg.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grimberg J, Nawoschik S, Belluscio L, McKee R, Turck A, Eisenberg A. A simple and efficient non-organic procedure for the isolation of genomic DNA from blood. Nucleic Acids Res. 1989;17:8390. doi: 10.1093/nar/17.20.8390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marmor MF, Holder GE, Seeliger MW, Yamamoto S. Standard for clinical electroretinography (2004 update). Doc Ophthalmol. 2004;108:107–14. doi: 10.1023/b:doop.0000036793.44912.45. [DOI] [PubMed] [Google Scholar]
- 5.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. GRR: graphical representation of relationship errors. Bioinformatics. 2001;17:742–3. doi: 10.1093/bioinformatics/17.8.742. [DOI] [PubMed] [Google Scholar]
- 6.O'Connell JR, Weeks DE. PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet. 1998;63:259–66. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 8.Gudbjartsson DF, Jonasson K, Frigge ML, Kong A. Allegro, a new computer program for multipoint linkage analysis. Nat Genet. 2000;25:12–3. doi: 10.1038/75514. [DOI] [PubMed] [Google Scholar]
- 9.Rüschendorf F, Nurnberg P. ALOHOMORA: a tool for linkage analysis using 10K SNP array data. Bioinformatics. 2005;21:2123–5. doi: 10.1093/bioinformatics/bti264. [DOI] [PubMed] [Google Scholar]
- 10.Thiele H, Nurnberg P. HaploPainter: a tool for drawing pedigrees with complex haplotypes. Bioinformatics. 2005;21:1730–2. doi: 10.1093/bioinformatics/bth488. [DOI] [PubMed] [Google Scholar]
- 11.Boldt K, Mans DA, Won J, van Reeuwijk J, Vogt A, Kinkl N, Letteboer SJ, Hicks WL, Hurd RE, Naggert JK, Texier Y, den Hollander AI, Koenekoop RK, Bennett J, Cremers FP, Gloeckner CJ, Nishina PM, Roepman R, Ueffing M. Disruption of intraflagellar protein transport in photoreceptor cilia causes Leber congenital amaurosis in humans and mice. J Clin Invest. 2011;121:2169–80. doi: 10.1172/JCI45627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.den Hollander AI, Koenekoop RK, Mohamed MD, Arts HH, Boldt K, Towns KV, Sedmak T, Beer M, Nagel-Wolfrum K, McKibbin M, Dharmaraj S, Lopez I, Ivings L, Williams GA, Springell K, Woods CG, Jafri H, Rashid Y, Strom TM, van der Zwaag B, Gosens I, Kersten FF, van Wijk E, Veltman JA, Zonneveld MN, van Beersum SE, Maumenee IH, Wolfrum U, Cheetham ME, Ueffing M, Cremers FP, Inglehearn CF, Roepman R. Mutations in LCA5, encoding the ciliary protein lebercilin, cause Leber congenital amaurosis. Nat Genet. 2007;39:889–95. doi: 10.1038/ng2066. [DOI] [PubMed] [Google Scholar]
- 13.Gerber S, Hanein S, Perrault I, Delphin N, Aboussair N, Leowski C, Dufier JL, Roche O, Munnich A, Kaplan J, Rozet JM. Mutations in LCA5 are an uncommon cause of Leber congenital amaurosis (LCA) type II. Hum Mutat. 2007;28:1245. doi: 10.1002/humu.9513. [DOI] [PubMed] [Google Scholar]
- 14.Jacobson SG, Aleman TS, Cideciyan AV, Sumaroka A, Schwartz SB, Windsor EA, Swider M, Herrera W, Stone EM. Leber congenital amaurosis caused by Lebercilin (LCA5) mutation: retained photoreceptors adjacent to retinal disorganization. Mol Vis. 2009;15:1098–106. [PMC free article] [PubMed] [Google Scholar]
- 15.McKibbin M, Ali M, Mohamed MD, Booth AP, Bishop F, Pal B, Springell K, Raashid Y, Jafri H, Inglehearn CF. Genotype-phenotype correlation for leber congenital amaurosis in Northern Pakistan. Arch Ophthalmol. 2010;128:107–13. doi: 10.1001/archophthalmol.2010.309. [DOI] [PubMed] [Google Scholar]
- 16.Ramprasad VL, Soumittra N, Nancarrow D, Sen P, McKibbin M, Williams GA, Arokiasamy T, Lakshmipathy P, Inglehearn CF, Kumaramanickavel G. Identification of a novel splice-site mutation in the Lebercilin (LCA5) gene causing Leber congenital amaurosis. Mol Vis. 2008;14:481–6. [PMC free article] [PubMed] [Google Scholar]
- 17.Dharmaraj S, Li Y, Robitaille JM, Silva E, Zhu D, Mitchell TN, Maltby LP, Baffoe-Bonnie AB, Maumenee IH. A novel locus for Leber congenital amaurosis maps to chromosome 6q. Am J Hum Genet. 2000;66:319–26. doi: 10.1086/302719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mohamed MD, Topping NC, Jafri H, Raashed Y, McKibbin MA, Inglehearn CF. Progression of phenotype in Leber's congenital amaurosis with a mutation at the LCA5 locus. Br J Ophthalmol. 2003;87:473–5. doi: 10.1136/bjo.87.4.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ebermann I, Elsayed SM, Abdel-Ghaffar TY, Nurnberg G, Nurnberg P, Elsobky E, Bolz HJ. Double homozygosity for mutations of AGL and SCN9A mimicking neurohepatopathy syndrome. Neurology. 2008;70:2343–4. doi: 10.1212/01.wnl.0000314731.65875.5c. [DOI] [PubMed] [Google Scholar]
- 20.Ebermann I, Walger M, Scholl HP, Charbel Issa P, Luke C, Nurnberg G, Lang-Roth R, Becker C, Nurnberg P, Bolz HJ. Truncating mutation of the DFNB59 gene causes cochlear hearing impairment and central vestibular dysfunction. Hum Mutat. 2007;28:571–7. doi: 10.1002/humu.20478. [DOI] [PubMed] [Google Scholar]