

Abstract
Genome-wide association (GWA) studies have identified numerous, replicable, genetic associations between common single nucleotide polymorphisms (SNPs) and risk of common autoimmune and inflammatory (immune-mediated) diseases, some of which are shared between two diseases. Along with epidemiological and clinical evidence, this suggests that some genetic risk factors may be shared across diseases—as is the case with alleles in the Major Histocompatibility Locus. In this work we evaluate the extent of this sharing for 107 immune disease-risk SNPs in seven diseases: celiac disease, Crohn's disease, multiple sclerosis, psoriasis, rheumatoid arthritis, systemic lupus erythematosus, and type 1 diabetes. We have developed a novel statistic for Cross Phenotype Meta-Analysis (CPMA) which detects association of a SNP to multiple, but not necessarily all, phenotypes. With it, we find evidence that 47/107 (44%) immune-mediated disease risk SNPs are associated to multiple—but not all—immune-mediated diseases (SNP-wise P CPMA<0.01). We also show that distinct groups of interacting proteins are encoded near SNPs which predispose to the same subsets of diseases; we propose these as the mechanistic basis of shared disease risk. We are thus able to leverage genetic data across diseases to construct biological hypotheses about the underlying mechanism of pathogenesis.
Author Summary
Over the last five years we have found over 100 genetic variants predisposing to common diseases affecting the immune system. In this study we analyze 107 such variants across seven diseases and find that almost half are shared across diseases. We also find that the patterns of sharing across diseases cluster these variants into groups; proteins encoded near variants in the same group tend to interact. This suggests that genetic variation may influence entire pathways to create risk to multiple diseases.
Introduction
The human immune-mediated diseases are the result of aberrant immune responses. These immune responses may lead to chronic inflammation and tissue destruction, often targeting a specific organ site. The outcome of this process is immune-mediated inflammatory and autoimmune disease, affecting approximately 5% of the population [1].
Extensive clinical and epidemiologic observations have shown that immune-mediated inflammatory and autoimmune diseases can occur either in the same individual or in closely related family members. This clustering of multiple diseases appears more frequently than expected if disease processes were independent. As each of the immune-mediated inflammatory and autoimmune diseases has strong genetic influences on disease risk [2]–[7], the observed clustering of multiple diseases could be due to an overlap in the causal genes and pathways [8], [9].
The patterns of clustering of diseases across the population are complex [10] – each disease has a prevalence between 0.01%–3%, so direct assessment of co-aggregation within individuals or families does not result in the very large samples required for genetic or epidemiological investigation. Thus it is unsurprising that to date, these observations have yet to be translated into determinants of the shared molecular etiologies of disease.
Recent GWA studies in immune-mediated and autoimmune diseases have identified 140 regions of the genome with statistically significant and robust evidence of presence of disease susceptibility loci. A subset of these loci have been shown to modulate risk of multiple diseases [3], [6], [11]–[14]. In addition, there is evidence that loci predisposing to one disease can have effects on risk of a second disease [15], although the risk allele for one disease may not be the same as for the second [16].
Together, these observations support the hypothesis of a common genetic basis of immune-mediated and autoimmune diseases [17]. There is now the ability to estimate both the number of loci contributing to risk of multiple diseases and the spectrum of diseases that each locus influences. In addition, grouping variants by the diseases they influence should provide insight into the specific biological processes underlying co-morbidity and disease risk.
In this report, we systematically investigate the genetic commonality in immune-mediated inflammatory and autoimmune diseases by examining the contributions of associated genomic risk regions in seven diseases: celiac disease (CeD), Crohn's disease (CD), multiple sclerosis (MS), psoriasis (Ps), rheumatoid arthritis (RA), systemic lupus erythematosus (SLE) and type 1 diabetes (T1D). We find that nearly half of loci identified in GWAS studies of an individual disease influence risk to at least two diseases, arguing for a genetic basis to co-morbidity. We also find several variants with opposing risk profiles in different diseases. Supporting the idea that common patterns of association implicate shared biological processes, we further demonstrate that loci clustered by the pattern of diseases they affect harbor genes encoding interacting proteins at a much higher rate than by chance. These results suggest that multi-phenotype mapping will identify the molecular mechanisms underlying co-morbid immune-mediated inflammatory and autoimmune diseases.
Results
We first test our hypothesis of common genetic determinants by examining evidence of association of genetic variants in known immune-mediated and autoimmune disease susceptibility loci to multiple disease phenotypes. We collated a list of 140 single nucleotide polymorphisms (SNPs) representing reported associations to at least one immune-mediated disease at genome-wide significance levels. Where data for the reported SNP itself were not available in our GWA studies (Table 1), we chose a proxy in high linkage disequilibrium to the reported marker (r2>0.9 in HapMap/CEU). We did not consider SNPs in the human Major Histocompatibility Complex (MHC) from this analysis, as its role in many of these diseases is well-established and the classically associated alleles in the HLA region are not well captured by SNPs [18]. We were able to acquire data for either the reported SNP or a good proxy in 107 of 140 cases, and assembled genotype test summaries for these from previously described GWA studies representing over 26,000 disease cases (Table 1).
Table 1. Participating studies.
| Disease | Cases | Controls | Reference |
| Celiac disease | 3796 | 8154 | 22 |
| Crohn's disease | 3230 | 4829 | 1 |
| Multiple sclerosis | 2624 | 7220 | 4 |
| Psoriasis | 1359 | 1400 | 5 |
| Rheumatoid arthritis | 5539 | 20169 | 6 |
| Systemic Lupus Erythematosus | 1963 | 4329 | 23 |
| Type 1 diabetes | 7514 | 9045 | 24 |
Data were collated for seven phenotypes from meta-analyses incorporating all known genome-wide association studies. SLE is the exception as no comprehensive meta-analysis has yet been published; data were instead obtained from a recent meta-analysis including some, but not all, known genome-wide association studies. Note that controls overlap in some cases due to the use of common shared sample genotypes.
We have developed a cross-phenotype meta-analysis (CPMA) statistic to assess association across multiple phenotypes. The CPMA statistic determines evidence for the hypothesis that each independent SNP has multiple phenotypic associations. Support for this hypothesis would be shown by deviations from expected uniformity of the distribution of association p-values, indicative of multiple associations. The likelihood of the observed rate of exponential decay of −log10(p) is calculated and compared to the null expectation (the decay rate should be unity) as a likelihood ratio test (see Materials and Methods for details). This CPMA statistic has one degree of freedom, as it measures a deviation in p-value behavior instead of testing all possible combinations of diseases for association to each SNP.
A total of 47 of the 107 SNPs tested have evidence of association to multiple diseases (SNP-wise PCPMA<0.01; expectation roughly 1 by chance; binomial probability of observing this result p = 3×10−64). This highly significant result confirms widespread sharing of genetic loci between immune-mediated inflammatory and autoimmune diseases. Further, these “multi-phenotype” SNPs include many loci not previously known to be shared across diseases, as well as new predictions of association for previously known shared loci (Table 2).
Table 2. SNPs associated with multiple phenotypes.
| SNP | RA | Psoriasis | MS | SLE | Crohn | Coeliac | T1D | CPMA | Reference | ||||||||||||
| Name | Chr | Position | Aj | Am | Genes | Z | p | Z | p | Z | p | Z | p | Z | p | Z | p | Z | p | p | |
| rs10889677 | 1 | 67437141 | C | A | IL23R | 0.0 | 9.8E-01 | 5.1 | 3.5E-07 | −2.6 | 4.9E-03 | −0.3 | 6.2E-01 | 10.3 | 9.0E-25 | 1.1 | 8.7E-01 | 0.1 | 5.3E-01 | 6.9E-25 | 37 |
| rs3087243 | 2 | 204564425 | G | A | CTLA4 | −5.7 | 1.2E-08 | 0.3 | 8.0E-01 | −0.7 | 2.5E-01 | −0.5 | 3.0E-01 | −1.2 | 2.3E-01 | 2.9 | 2.2E-03 | −8.5 | 1.1E-17 | 2.8E-21 | 36 |
| rs2542151 | 18 | 12769947 | T | G | PTPN2 | 4.2 | 3.0E-05 | 2.0 | 4.1E-02 | −0.8 | 2.2E-01 | 1.1 | 1.5E-01 | 6.8 | 1.2E-11 | 4.8 | 6.9E-07 | 7.1 | 5.9E-13 | 5.3E-19 | 1 |
| rs2201841 | 1 | 67406223 | A | G | IL23R | 0.0 | 9.9E-01 | 5.2 | 2.7E-07 | −2.2 | 1.3E-02 | −0.3 | 6.3E-01 | 9.9 | 3.5E-23 | 1.0 | 8.4E-01 | 0.2 | 5.7E-01 | 3.7E-18 | 5 |
| rs11209032 | 1 | 67452113 | G | A | IL23R | −0.6 | 5.2E-01 | 5.3 | 1.3E-07 | −1.2 | 1.2E-01 | −0.7 | 2.3E-01 | 8.4 | 3.1E-17 | 1.2 | 8.8E-01 | 0.2 | 4.4E-01 | 6.4E-18 | 33 |
| rs1893217 | 18 | 12799340 | A | G | PTPN2 | 4.2 | 2.4E-05 | 2.1 | 3.9E-02 | −0.6 | 2.8E-01 | 1.0 | 1.5E-01 | 6.5 | 6.5E-11 | 4.9 | 6.1E-07 | 7.4 | 8.2E-14 | 3.4E-17 | 24 |
| rs917997 | 2 | 102529086 | C | T | IL18RAP | 0.3 | 7.8E-01 | −0.2 | 8.8E-01 | 0.3 | 3.7E-01 | 1.1 | 1.3E-01 | 4.2 | 2.2E-05 | 7.6 | 1.1E-14 | −1.5 | 9.4E-01 | 4.9E-13 | 3 |
| rs12708716 | 16 | 11087374 | A | G | CLEC16A | 0.2 | 8.3E-01 | −0.3 | 8.1E-01 | 3.7 | 1.1E-04 | −1.0 | 1.5E-01 | −0.6 | 5.5E-01 | 1.6 | 5.3E-02 | −8.2 | 1.2E-16 | 2.0E-12 | 32 |
| rs2872507 | 17 | 35294289 | G | A | ORMDL3 | 4.1 | 4.7E-05 | 0.3 | 8.0E-01 | −3.3 | 5.5E-04 | −0.3 | 6.3E-01 | 4.7 | 2.1E-06 | 0.7 | 7.6E-01 | 5.0 | 2.5E-07 | 4.1E-09 | 1 |
| rs3821236 | 2 | 191728264 | G | A | STAT4 | 4.7 | 2.5E-06 | −1.7 | 9.5E-02 | 0.1 | 4.6E-01 | 6.2 | 2.1E-10 | −1.7 | 9.6E-02 | 1.9 | 3.1E-02 | 3.4 | 3.5E-04 | 9.8E-08 | 2 |
| rs6441961 | 3 | 46327388 | C | T | CCR1 | 1.1 | 2.7E-01 | −0.3 | 7.5E-01 | 0.1 | 4.7E-01 | 1.3 | 9.1E-01 | −0.5 | 6.1E-01 | 5.7 | 6.9E-09 | 3.7 | 1.0E-04 | 1.9E-07 | 3 |
| rs2290400 | 17 | 35319766 | C | T | ORMDL3 | −3.9 | 1.1E-04 | 0.4 | 7.2E-01 | 3.3 | 5.5E-04 | 0.2 | 5.7E-01 | 3.7 | 2.4E-04 | 0.2 | 4.1E-01 | −5.1 | 1.4E-07 | 1.2E-06 | 24 |
| rs7197475 | 16 | 30550368 | C | T | 16p11.2 | 2.9 | 3.8E-03 | 2.9 | 4.3E-03 | -0.9 | 1.7E-01 | 0.7 | 2.4E-01 | −4.2 | 2.5E-05 | 0.4 | 3.4E-01 | −0.9 | 1.9E-01 | 2.0E-06 | 35 |
| rs4917014 | 7 | 50083124 | T | G | IKZF1 | −2.5 | 1.3E-02 | 0.9 | 3.8E-01 | 3.5 | 2.6E-04 | −2.9 | 1.9E-03 | 1.6 | 1.1E-01 | 0.9 | 1.8E-01 | −3.3 | 5.2E-04 | 4.9E-06 | 35 |
| rs6822844 | 4 | 123867026 | G | T | IL2-IL21 | −3.4 | 6.5E-04 | −1.7 | 8.7E-02 | 1.2 | 1.1E-01 | -0.2 | 4.3E-01 | −2.4 | 1.5E-02 | 3.3 | 4.9E-04 | −1.1 | 1.5E-01 | 6.2E-06 | 38 |
| rs10517086 | 4 | 25761780 | G | A | 4p15.2 | 5.1 | 2.8E-07 | 0.6 | 5.7E-01 | 0.4 | 3.6E-01 | 0.1 | 5.5E-01 | 2.8 | 5.5E-03 | 0.2 | 5.6E-01 | 4.9 | 5.7E-07 | 2.3E-05 | 24 |
| rs11203203 | 21 | 42709255 | G | A | UBASH3A | 4.2 | 2.5E-05 | 1.3 | 1.9E-01 | 0.2 | 4.4E-01 | 1.6 | 5.3E-02 | 0.0 | 9.8E-01 | 3.1 | 1.0E-03 | 6.6 | 1.8E-11 | 2.5E-05 | 24 |
| rs4728142 | 7 | 128167918 | G | A | IRF5 | 4.5 | 7.1E-06 | 1.4 | 1.8E-01 | −1.5 | 6.4E-02 | 6.2 | 2.9E-10 | 0.8 | 4.4E-01 | 2.1 | 1.6E-02 | −0.2 | 4.1E-01 | 4.4E-05 | 35 |
| rs11755527 | 6 | 91014952 | C | G | BACH2 | −1.4 | 1.6E-01 | −0.7 | 5.1E-01 | −2.8 | 2.7E-03 | 1.0 | 8.5E-01 | −2.6 | 1.0E-02 | 3.5 | 2.8E-04 | 5.6 | 1.0E-08 | 8.0E-05 | 24 |
| rs7709212 | 5 | 158696755 | T | C | IL12B | −0.6 | 5.7E-01 | −6.3 | 3.8E-10 | 3.6 | 1.9E-04 | −2.3 | 1.1E-02 | 3.3 | 9.8E-04 | 0.1 | 5.5E-01 | 1.2 | 8.9E-01 | 8.8E-05 | 39 |
| rs947474 | 10 | 6430456 | A | G | PRKCQ | −4.4 | 8.7E-06 | −1.0 | 3.4E-01 | −0.3 | 6.3E-01 | −1.0 | 8.5E-01 | −2.4 | 1.9E-02 | 2.0 | 2.1E-02 | −3.7 | 1.1E-04 | 9.1E-05 | 34 |
| rs2188962 | 5 | 131798704 | C | T | 5q31 | −0.3 | 7.7E-01 | 3.0 | 3.2E-03 | 1.3 | 9.6E-02 | 1.5 | 7.0E-02 | 5.9 | 4.6E-09 | 1.8 | 3.7E-02 | 2.9 | 2.0E-03 | 1.6E-04 | 1 |
| rs744166 | 17 | 37767727 | A | G | STAT3 | −0.7 | 4.7E-01 | 1.3 | 1.9E-01 | -4.4 | 6.4E-06 | 0.7 | 7.6E-01 | −4.5 | 5.9E-06 | 2.2 | 9.8E-01 | −2.6 | 4.6E-03 | 2.2E-04 | 1 |
| rs4788084 | 16 | 28447349 | C | T | IL27 | 2.6 | 1.0E-02 | 1.3 | 2.1E-01 | 0.3 | 6.1E-01 | 1.3 | 9.1E-02 | 2.9 | 3.5E-03 | 2.3 | 1.1E-02 | −6.5 | 5.1E-11 | 4.1E-04 | 24 |
| rs2082412 | 5 | 158650367 | G | A | IL12B | −1.2 | 2.3E-01 | −6.2 | 8.8E-10 | NA | NA | −3.8 | 7.1E-05 | 2.6 | 9.6E-03 | 0.4 | 3.6E-01 | 0.0 | 5.0E-01 | 4.6E-04 | 5 |
| rs11465804 | 1 | 67414547 | T | G | IL23R | −0.4 | 6.8E-01 | −4.9 | 1.3E-06 | −1.7 | 9.5E-01 | −0.4 | 3.6E-01 | −12.5 | 1.0E-35 | 1.2 | 1.2E-01 | −1.0 | 1.6E-01 | 5.2E-04 | 1 |
| rs463426 | 22 | 20133739 | C | T | HIC2-UBE2L3 | 2.2 | 2.9E-02 | 1.4 | 1.6E-01 | 1.0 | 1.7E-01 | 0.3 | 3.7E-01 | 1.8 | 6.7E-02 | 3.3 | 4.1E-04 | 0.5 | 2.9E-01 | 5.2E-04 | 35 |
| rs763361 | 18 | 65682622 | C | T | CD226 | 2.1 | 3.3E-02 | 1.9 | 6.5E-02 | −1.7 | 4.5E-02 | 0.0 | 4.9E-01 | 2.0 | 4.1E-02 | 2.5 | 6.7E-03 | 5.1 | 1.6E-07 | 9.0E-04 | 24 |
| rs11584383 | 1 | 197667523 | T | C | KIF21B | 2.3 | 2.3E-02 | 0.0 | 9.7E-01 | 3.3 | 4.6E-04 | −0.3 | 6.2E-01 | −5.0 | 6.8E-07 | 0.8 | 2.0E-01 | −2.3 | 1.0E-02 | 9.0E-04 | 1 |
| rs6590330 | 11 | 127816269 | G | A | ETS1 | 3.2 | 1.5E-03 | −0.5 | 6.0E-01 | −2.7 | 3.7E-03 | 2.1 | 1.9E-02 | 0.6 | 5.2E-01 | 1.4 | 9.2E-01 | 1.8 | 3.9E-02 | 2.6E-03 | 35 |
| rs4900384 | 14 | 97568704 | A | G | 14q32.2 | 1.3 | 2.0E-01 | 0.8 | 4.4E-01 | 1.8 | 3.9E-02 | −1.9 | 3.0E-02 | 0.2 | 8.1E-01 | 3.2 | 7.6E-04 | 5.2 | 8.9E-08 | 2.8E-03 | 24 |
| rs10758669 | 9 | 4971602 | A | C | JAK2 | 1.0 | 3.3E-01 | 1.2 | 2.4E-01 | −3.3 | 4.2E-04 | 0.3 | 4.0E-01 | 5.0 | 6.8E-07 | 1.8 | 3.7E-02 | 1.0 | 1.7E-01 | 3.4E-03 | 1 |
| rs1913517 | 10 | 49789060 | A | G | LRRC18-WDFY4 | −4.4 | 1.1E-05 | 0.8 | 4.4E-01 | 0.4 | 3.5E-01 | −2.4 | 8.7E-03 | −0.4 | 6.9E-01 | 0.1 | 4.4E-01 | -0.8 | 2.2E-01 | 4.1E-03 | 35 |
| rs4505848 | 4 | 123490097 | A | G | IL2 | −0.5 | 6.0E-01 | 0.1 | 9.0E-01 | 0.5 | 7.0E-01 | 1.1 | 1.3E-01 | 3.0 | 2.6E-03 | 3.1 | 9.6E-04 | 6.6 | 2.3E-11 | 4.4E-03 | 24 |
| rs7804356 | 7 | 26664905 | T | C | 7p15.2 | −1.3 | 1.9E-01 | 1.2 | 2.2E-01 | 1.0 | 1.5E-01 | −0.1 | 4.8E-01 | 3.4 | 5.9E-04 | 1.3 | 9.9E-02 | −5.6 | 9.5E-09 | 5.6E-03 | 24 |
| rs11258747 | 10 | 6512897 | G | T | PRKCQ | 3.0 | 2.4E-03 | 1.9 | 6.0E-02 | -2.5 | 6.7E-03 | 0.6 | 7.3E-01 | 0.1 | 9.2E-01 | 0.2 | 4.2E-01 | 4.8 | 7.7E-07 | 7.5E-03 | 24 |
| rs703842 | 12 | 56449006 | A | G | CYP27B1 | −2.7 | 6.1E-03 | −0.1 | 8.9E-01 | 4.1 | 1.7E-05 | −0.4 | 6.7E-01 | −1.1 | 2.5E-01 | 2.0 | 2.3E-02 | −2.2 | 1.5E-02 | 8.3E-03 | 31 |
| rs1990760 | 2 | 162949558 | T | C | IFIH1 | −1.0 | 3.0E-01 | −2.4 | 1.7E-02 | 0.1 | 5.4E-01 | −3.4 | 3.2E-04 | 0.6 | 5.6E-01 | 0.4 | 6.5E-01 | −6.2 | 2.5E-10 | 8.6E-03 | 24 |
| rs2476601 | 1 | 114089610 | G | A | PTPN22 | 18.2 | 9.1E-74 | 0.0 | 1.0E+00 | 0.4 | 3.5E-01 | 4.0 | 3.3E-05 | −4.3 | 1.8E-05 | 1.7 | 4.2E-02 | 20.4 | 1.5E-92 | 6.3E-160 | 1 |
| rs3184504 | 12 | 110347328 | T | C | SH2B3 | −2.9 | 3.6E-03 | −2.0 | 4.1E-02 | 3.4 | 3.3E-04 | −2.7 | 3.6E-03 | −3.4 | 6.2E-04 | 7.3 | 1.2E-13 | −11.9 | 7.7E-33 | 4.3E-19 | 24 |
| rs11865121 | 16 | 11074189 | C | A | CLEC16A | 0.3 | 7.7E-01 | 0.6 | 5.2E-01 | 4.3 | 8.7E-06 | −0.7 | 2.3E-01 | 0.9 | 3.8E-01 | 1.1 | 1.4E-01 | −8.9 | 2.1E-19 | 1.1E-14 | 4 |
| rs2816316 | 1 | 189268470 | A | C | RGS1 | −1.0 | 3.3E-01 | −0.4 | 6.7E-01 | 3.1 | 9.0E-04 | −0.1 | 5.3E-01 | −0.5 | 6.4E-01 | 6.9 | 2.7E-12 | −3.9 | 4.2E-05 | 5.2E-13 | 3 |
| rs2104286 | 10 | 6139051 | T | C | IL2RA | −3.1 | 1.8E-03 | −0.1 | 9.5E-01 | 6.2 | 3.5E-10 | 0.4 | 6.5E-01 | −0.8 | 4.4E-01 | 0.5 | 3.1E-01 | −6.4 | 5.9E-11 | 1.2E-08 | 32 |
| rs3024505 | 1 | 203328299 | G | A | IL10 | −1.0 | 3.3E-01 | 0.9 | 3.8E-01 | 1.5 | 7.3E-02 | 4.2 | 1.3E-05 | 2.4 | 1.6E-02 | 1.6 | 5.4E-02 | −4.8 | 6.2E-07 | 2.2E-06 | 23 |
| rs10045431 | 5 | 158747111 | C | A | IL12B | 0.4 | 6.5E-01 | 4.5 | 6.6E-06 | 0.3 | 6.3E-01 | 2.4 | 8.8E-03 | −5.8 | 8.8E-09 | 0.5 | 6.8E-01 | 0.1 | 4.6E-01 | 6.0E-04 | 1 |
| rs610604 | 6 | 138241110 | T | G | TNFAIP3 | −4.2 | 3.3E-05 | 4.5 | 8.0E-06 | 0.3 | 3.8E-01 | −1.3 | 9.9E-02 | 1.4 | 1.8E-01 | 0.4 | 6.5E-01 | −0.2 | 4.3E-01 | 2.7E-03 | 5 |
| rs4613763 | 5 | 40428485 | T | C | PTGER4 | 0.9 | 3.9E-01 | 0.7 | 4.7E-01 | −4.2 | 1.1E-05 | 0.4 | 3.4E-01 | 9.6 | 5.0E-22 | 0.1 | 5.2E-01 | −0.5 | 3.1E-01 | 4.0E-03 | 1 |
47/107 SNPs tested showed significant evidence of association to multiple diseases (Pcpma<0.01), where only one is expected by chance. These SNPs are therefore candidate drivers for the shared genetic architecture between diseases. The SNPs shown in the lower panel also have strong evidence of association in opposite directions across phenotypes and may be crucial decision points in pathogenesis. Aj = major allele; Am = minor allele. Z scores are reported from published GWA studies and arbitrarily signed relative to the direction of effect in celiac disease. Note that no MS data were available for rs2082412 as it had not been imputed accurately in the participating MS study. Data for all SNPs is presented in Dataset S1.
Although our CPMA statistic is agnostic to effect direction in each disease, a subset of the 47 multi-phenotype (CPMA positive) SNPs appeared to have strong allelic effects in opposite directions in different diseases [16]. A total of 9 SNPs had strong evidence of such directional association (an association p<1×10−4 with at least one protective and one risk effect; lower panel in Table 2). This suggests that shared associations have complex effects on disease outcomes and may be of particular importance in pathogenic processes.
We next examined the patterns of association across 47 multi-phenotype SNPs to determine evidence of either a global autoimmune process or biological pathways influencing sets of diseases. On visual inspection of these data we found a striking patterning of associations across diseases: only one SNP (rs3184504, in an exon of SH2B3) exhibited evidence of association to all seven diseases; the others appeared to associate only to subsets of diseases (Table 2).
To formalize the analysis of association patterns across diseases, we determined specific patterns of associations across SNPs by computing SNP-SNP distances based on the level of association to each disease followed by hierarchical clustering to group them (Figure 1A; see Materials and Methods section for clustering details). SNPs in loci encoding proteins known to interact clustered together: for example, the independent effects at IL12B and IL23R, which encode subunits of a ligand-receptor pair are in the same region of the dendrogram. We next partitioned the dendrogram “tree” into four clusters and summarized the cumulative association of each cluster to each disease by combining our underlying dataset of association p-values per cluster, per disease using Fisher's omnibus test (Figure 1B; see Materials and Methods for details). Each cluster had a different pattern of associations across diseases; these patterns suggest that the clusters represent distinct co-morbid mechanisms.
Figure 1. Patterns of association across diseases correlate with protein-protein interactions.

A: 47 SNPs with evidence of association to multiple diseases (Pcpma<0.01) fall into groups clustered by the pattern of association across diseases. Clusters are numbered arbitrarily. B: Clusters show different patterns of association across diseases. We summarize the differential disease effects of each cluster with a cumulative association statistic (Fisher's method for combining p values). These patterns are different for each cluster, suggesting each represents a different co-morbid mechanism. Note that these figures are based on the same underlying association statistics the clustering in the first panel is derived from. C: proteins encoded within the linkage disequilibrium scope around SNPs in the same cluster interact either directly or via common intermediates. Three of our four clusters have significant protein inter-connectivity (permuted P<0.05; see Materials and Methods and [19] for details).
Our underlying hypothesis has been that phenotype-driven clusters represent distinct molecular mechanisms. This leads to the prediction that components of these clusters/pathways are encoded in associated loci; in other words, proteins encoded around SNPs in the same cluster should interact. We test this prediction by looking for connectivity between proteins encoded around SNPs within each cluster as described elsewhere [19]. Briefly, we define a genomic region around each SNP in terms of linkage disequilibrium and consider any protein overlapping that region. We then ask if proteins encoded around SNPs in the same cluster interact using protein-protein interaction maps, excluding interactions between proteins in the same region (see Materials and Methods and [19]). We find that three of the four clusters we define by patterns of association have significant connectivity (Figure 1C; permuted P<0.05) by this method, suggesting that these represent distinct molecular mechanisms affected by genetic risk variants. Two of these groups of interacting proteins are also preferentially expressed [19] in immune cell subtypes compared to other tissue types (Figure S1), supporting our hypothesis that these represent true pathways underlying pathogenesis.
Discussion
Immune-mediated inflammatory and autoimmune diseases have been known to cluster in families, suggesting a strong genetic component to risk. The genes in the human MHC (HLA complex) have been associated with disease risk, suggesting a common immune pathway. Less clear is whether other genetic variants associated with individual diseases also form common pathways/mechanisms for autoimmunity. Recent results from GWA studies suggest that common genetic mechanisms may underlie the observed clustering of multiple autoimmune diseases within a person or family. In this work we have tested the hypothesis that immunologically relevant genetic variation will either (1) underlie risk to all immune-mediated diseases, implicating a global immunological process; (2) influence risk to a discrete subset of diseases, implicating molecular entities underlying that co-morbidity; or (3) modulate risk for only one disorder thereby implying a disease-specific process.
A central goal of complex disease genetics is to uncover the pathways perturbed in disease and shed light on the underlying cellular processes. Despite a wealth of molecular insight into immune function few key pathways underlying genetic susceptibility to immune-mediated diseases have been elucidated. To identify these processes in immune-mediated inflammatory and autoimmune disease, we tested genetic variation contributing to seven diseases. We observed an overwhelming abundance of commonality across these phenotypes, assorting into cohesive phenotype-genotype groups that appear to underlie co-morbidities. By analyzing loci known to associate to at least one disease, we are able to identify groups of diseases that should be considered as a unified phenotype and analyzed together. We further demonstrate that this approach generates novel biological insights into pathogenesis, often difficult to obtain from genomic studies of single traits [20].
We have described a novel statistic, CPMA, which assesses evidence for multiple associations to a marker. Rather than perform a meta-analysis, which would only detect association to all phenotypes (or suffer from heterogeneity) or test all combinations of phenotypes which would increase the multiple testing burden, we look for deviation in the distribution of association p values. Our statistic thus detects markers associated to at least some, but not necessarily all, phenotypes; we note that this is a single degree of freedom test, providing high power to reject the null hypothesis. This power comes at the price of not knowing to which phenotypes the marker is associate; we overcome this with our clustering analysis, which resolves groups of markers associating to the same diseases. Thus our analytic strategy is able to both detect shared associations and identify the relevant phenotypes.
Our approach appears capable of distinguishing distinct genetic effects in the same locus in addition to validated shared associations. For example, it is now clear that the two signals in the IL2/IL21 locus on chromosome 4q27 are distinct, with T1D mapping to IL2 and other diseases to IL21 [21]. Our analysis detects this difference, clustering the two SNPs representing these associations separately (Figure 1, labeled “IL2” and “IL2/IL21”, respectively). Conversely, previous reports of an overlap in association between T1D and celiac disease [15] were in regions encoding genes highly expressed in T lymphocytes (RGS1, PTPN2 and CTLA4 in celiac; PTPN2 and CTLA4 in T1D). Our analysis identifies all these regions as CPMA-positive and highlights the second associations in T1D and celiac shown by Smyth et al. [15], indicating that our approach could be used to prioritize marginal associations for replication. We also observe other potential associations. For example, rs2816316 on near RGS1 exhibits evidence of association to MS; rs2542151 and rs1893217 on near PTPN2 has modest association to psoriasis. These last observations, whilst suggestive, require further investigation given the known effects of these regions on other diseases.
In summary, our multi-disease approach is applicable beyond the immune-mediated inflammatory and autoimmune diseases, to current studies of related traits in pharmacology, metabolic and psychiatric disease and in genetic studies of cellular phenotypes such as gene expression. For most studies of the genetic basis of complex human phenotypes, the pathogenic processes are still far from understood and biological pathways may be identified using these methods. Ultimately, these results will contribute to an improved molecular nosology of mechanistic definitions and, ultimately, towards improving clinical care and human health.
Materials and Methods
Ethics statement
All data were drawn from previously published genome-wide association studies from consortia with appropriate ethics oversight from their respective institutional review boards. As only summary data from a small number of markers across the genome were used here no further ethical issues arise.
Patient cohorts
Data were obtained from previously described case/control GWA studies of celiac disease [22], Crohn's disease [2], multiple sclerosis [5], psoriasis [6], rheumatoid arthritis [7], systemic lupus erythematosus [23] and type I diabetes [24] as shown in Table 1. We note that, with the exception of psoriasis, in these cohorts diagnosis of a second immune-mediated disease is a criterion for exclusion, thereby minimizing co-morbidity as a source of bias in our study.
Locus selection
For our analysis we selected 140 independent SNPs (r2<0.2) with reported associations to an immune-mediated disease in a genome-wide association scan and replicated in independent samples in that disease to combined genome-wide significance [25]. We then chose proxies for those SNPs present on the major versions of Affymetrix and Illumina genome-wide genotyping platforms [26]; 107 SNPs had sufficient data coverage to be included. Where possible we used the SNP originally reported; if data were unavailable for that marker, we chose a high LD proxy (HapMap/CEU r2>0.9) to represent the region.
Cross-phenotype meta-analysis
Our CPMA analysis relies on the expected distribution of p-values for each SNP across diseases. Under the null hypothesis of no additional associations beyond those already known, we expect association values to be uniformly distributed and hence -ln(p) to be exponentially decaying with a decay rate λ = 1. We calculate the likelihood of the observed and expected values of λ and express these as a likelihood ratio test:
This statistic therefore measures the likelihood of the null hypothesis given the data; we can reject the null hypothesis if sufficient evidence to the contrary is present. We note that, because we only estimate a single parameter, our test is asymptotically distributed as 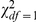 . This gives us more statistical power than relying on strategies combining association statistics, which would consume multiple degrees of freedom.
. This gives us more statistical power than relying on strategies combining association statistics, which would consume multiple degrees of freedom.
SNP–SNP distance calculation and clustering
To compare the patterns of association for multi-phenotype SNPs we first calculate SNP-SNP distances and then use hierarchical clustering on that distance matrix to assess relative relationships between SNP association patterns.
Calculating distances based directly on p values or the underlying association statistics is problematic, as each contributing study has slightly different sample sizes and therefore different statistical power to detect associations. Thus, distance functions based on numeric data – which incorporate magnitude differences between observations – would be biased if studies have systematically different data. Normalization procedures can account for such systematic differences but may fail to remove all bias. To reduce the impact such systematic irregularities might have on our comparison, we bin associations into informal “levels of evidence” categories. We define four classes (1<p<0.05, 0.05<p<0.001, 0.001<p<1×10-6, 1×10−6<p) and thus reduce our data to a SNP x disease matrix where entries are categorical variables describing these classes. We then calculate the Euclidean distances between pairs of SNPs using Gower's method for categorical data [27], which accounts for the discrete nature of the data.
To compare the distance relationships between SNPs we use hierarchical agglomerative clustering. This process joins single entities (in this case, SNPs) or groups of entities together if certain criteria are met. Successive rounds of clustering are preformed in an iterative way until all groups are joined, resulting in a tree of relationships where similar entities cluster on the same branches. In this analysis we cluster SNPs based on the Gower distance matrix using Ward's method for joining entities [28]. In contrast to linkage clustering methods, Ward's method seeks to minimize the information lost during the clustering process, calculated as the error sum of squares (ESS). The higher the ESS the more information is being lost due to inaccuracy of grouping entities together. This method thus seeks compact, spherical clusters of data which are maximally similar.
All distance and clustering analysis was done using the StatMatch and stats packages in the R programming language [29].
Cumulative association statistics
We compute per-cluster, per-disease cumulative association statistics by combining p values using Fisher's omnibus test, where the cumulative statistic Scum on N p-values is defined as:
and Scum follows the 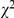 distribution with 2N degrees of freedom.
distribution with 2N degrees of freedom.
Protein–protein interaction analysis
We use previously described methodology [19] to assess whether proteins encoded around SNPs in each cluster interact. Briefly, we first compile lists of all proteins that an association may affect by defining locus boundaries around each SNP in terms of linkage disequilibrium and including all proteins overlapping this region. We then use a high-confidence protein-protein interaction map ([30] as modified in [19]) to ask whether proteins encoded around SNPs in each cluster interact either directly or via a common intermediary and assess the significance of such observations relative to the local structure of the protein-protein network as described elsewhere [19], using 4000 permutations. These data and methodology are publicly available for download and via a webserver (http://www.broadinstitute.org/mpg/dapple).
Supporting Information
Complete SNP-wise association data. Here we present the complete dataset on which we base our analysis. All data have been previously published as detailed in the main manuscript and in the key below, and are based on chi-square (1 df) or Z association statistics. Where not provided, we computed Z scores as the square root of the cognate chi-squared statistic. Sign was assigned with reference to the minor allele declared in the psoriasis GWAS (chosen arbitrarily). SNP - marker name. CHR – chromosome. POS - physical position (hg18). major_al - major SNP allele. minor_al - minor SNP allele. RA.Z - association Z score for rheumatoid arthritis (Stahl et al.Nat Genet 2010) [7]. RA.P - association p value for rheumatoid arthritis (Stahl et al.Nat Genet 2010) [7]. PS.Z - ditto for psoriasis (Nair et al. Nat Genet 2009) [6]. PS.P - ditto for psoriasis (Nair et al. Nat Genet 2009) [6]. MS.Z - ditto for multiple sclerosis (De Jager et al. Nat Genet 2009) [5]. MS.P - ditto for multiple sclerosis (De Jager et al. Nat Genet 2009) [5]. SLE.Z - ditto for systemic lupus erythematosus (Gateva et al. Nat Genet 2009) [23]. SLE.P - ditto for systemic lupus erythematosus (Gateva et al. Nat Genet 2009) [23]. CD.Z - ditto for Crohn's disease (Barrett et al. Nat Genet 2008) [2]. CD.P - ditto for Crohn's disease (Barrett et al. Nat Genet 2008) [2]. CeD.Z - ditto for celiac disease (Hunt et al. Nat Genet 2008) [4]. CeD.P - ditto for celiac disease (Hunt et al. Nat Genet 2008) [4]. T1D.Z - ditto for type I diabetes (Barrett et al. Nat Genet 2009) [24]. T1D.P - ditto for type I diabetes (Barrett et al. Nat Genet 2009) [24]. Disease - disease in which the SNP was originally reported: AITD autoimmune thyroid disease; AS ankylosing spondylitis; BD CD Crohn's disease; MS multiple sclerosis; PS psoriasis; SLE systemic lupus erythematosus; T1D type 1 diabetes; UC ulcerative colitis. cpma.p - p value for CPMA statistic (chi-squared, 1 df). Genes - nearby notable genes.
(TAB)
Enrichment in immune tissue expression for interacting genes encoded close to SNPs in (A) cluster 1 and (B) cluster 4. Following Rossin et al.[19] we looked for preferential expression of significant network genes in tissue subsets. Of the genes encoded around SNPs in clusters 1 and 4 (as defined in Figure 1), we found that those participating in significant networks are enriched in expression (purple circles) in immune tissues (red bars). Other genes encoded around those SNPs are not enriched in the same tissues (black circles). Thus interacting genes encoded around SNPs associated to the same immune diseases are preferentially expressed in immune tissues. Interacting genes for the remaining significant group, cluster 2, were not enriched.
(PDF)
Footnotes
The authors have declared that no competing interests exist.
The authors received no specific funding for this article.
References
- 1.Vyse T, Todd J. Genetic analysis of autoimmune disease. Cell. 1996;85:311–318. doi: 10.1016/s0092-8674(00)81110-1. [DOI] [PubMed] [Google Scholar]
- 2.Barrett JC, Hansoul S, Nicolae D, Cho J, Duerr RH, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Graham R, Cotsapas C, Davies L, Hackett R. Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nat Genet. 2008;40:1059–1061. doi: 10.1038/ng.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hunt KA, Zhernakova A, Turner G, Heap GAR, Franke L, et al. Newly identified genetic risk variants for celiac disease related to the immune response. Nat Genet. 2008;40:395–402. doi: 10.1038/ng.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Jager PL, Jia X, Wang J, De Bakker PIWd, Ottoboni L, et al. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat Genet. 2009;41:776. doi: 10.1038/ng.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nair RP, Duffin KC, Helms C, Ding J, Stuart PE, et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-κB pathways. Nat Genet. 2009;41:199–204. doi: 10.1038/ng.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stahl EA, Raychaudhuri S, Remmers EF, Xie G, Eyre S, et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet. 2010;42:508–514. doi: 10.1038/ng.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Criswell L, Pfeiffer K, Lum R, Gonzales B, Novitzke J, et al. Analysis of families in the multiple autoimmune disease genetics consortium (MADGC) collection: the PTPN22 620W allele associates with multiple autoimmune phenotypes. The American Journal of Human Genetics. 2005;76:561–571. doi: 10.1086/429096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wandstrat A, Wakeland E. The genetics of complex autoimmune diseases: non-MHC susceptibility genes. Nature immunology. 2001;2:802–809. doi: 10.1038/ni0901-802. [DOI] [PubMed] [Google Scholar]
- 10.Eaton W, Rose N, Kalaydjian A, Pedersen M, Mortensen P. Epidemiology of autoimmune diseases in Denmark. Journal of Autoimmunity. 2007;29:1–9. doi: 10.1016/j.jaut.2007.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maier L, Lowe C, Cooper J, Downes K, Anderson D, et al. IL2RA genetic heterogeneity in multiple sclerosis and type 1 diabetes susceptibility and soluble interleukin-2 receptor production. PLoS Genet. 2009;5:e1000322. doi: 10.1371/journal.pgen.1000322. doi: 10.1371/journal.pgen.1000322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Plenge RM, Cotsapas C, Davies L, Price AL, De Bakker PIWd, et al. Two independent alleles at 6q23 associated with risk of rheumatoid arthritis. Nat Genet. 2007;39:1477–1482. doi: 10.1038/ng.2007.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Remmers EF, Plenge RM, Lee AT, Graham R, Hom G, et al. STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. The New England Journal of Medicine. 2007;357:977–986. doi: 10.1056/NEJMoa073003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fung EY, Smyth DJ, Howson JM, Cooper JD, Walker NM, et al. Analysis of 17 autoimmune disease-associated variants in type 1 diabetes identifies 6q23/TNFAIP3 as a susceptibility locus. Genes Immun. 2009;10:188–191. doi: 10.1038/gene.2008.99. [DOI] [PubMed] [Google Scholar]
- 15.Smyth DJ, Plagnol V, Walker NM, Cooper JD, Downes K, et al. Shared and distinct genetic variants in type 1 diabetes and celiac disease. The New England Journal of Medicine. 2008;359:2767–2777. doi: 10.1056/NEJMoa0807917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sirota M, Schaub MA, Batzoglou S, Robinson WH, Butte AJ. Autoimmune disease classification by inverse association with SNP alleles. PLoS Genet. 2009;5:e1000792. doi: 10.1371/journal.pgen.1000792. doi: 10.1371/journal.pgen.1000792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhernakova A, van Diemen C, Wijmenga C. Detecting shared pathogenesis from the shared genetics of immune-related diseases. Nat Rev Genet. 2009;10:43–55. doi: 10.1038/nrg2489. [DOI] [PubMed] [Google Scholar]
- 18.de Bakker PIWd, Mcvean G, Sabeti PC, Miretti MM, Green T, et al. A high-resolution HLA and SNP haplotype map for disease association studies in the extended human MHC. Nat Genet. 2006;38:1166–1172. doi: 10.1038/ng1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rossin E, Lage K, Raychaudhuri S, Xavier R, Tatar D, et al. Proteins Encoded in Genomic Regions Associated with Immune-Mediated Disease Physically Interact and Suggest Underlying Biology. PLoS Genet. 2011;7:e1001273. doi: 10.1371/journal.pgen.1001273. doi: 10.1371/journal.pgen.1001273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khanna H, Davis EE, Murga-Zamalloa CA, Estrada-Cuzcano A, Lopez I, et al. A common allele in RPGRIP1L is a modifier of retinal degeneration in ciliopathies. Nat Genet. 2009;41:739–745. doi: 10.1038/ng.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Todd JA. Etiology of type 1 diabetes. Immunity. 2010;32:457–467. doi: 10.1016/j.immuni.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 22.Dubois PCA, Trynka G, Franke L, Hunt KA, Romanos J, et al. Multiple common variants for celiac disease influencing immune gene expression. Nature Publishing Group. 2010;42:295–302. doi: 10.1038/ng.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gateva V, Sandling JK, Hom G, Taylor KE, Chung SA, et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systematic lupus erythematosus. Nat Genet. 2009;41:1228–1233. doi: 10.1038/ng.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. 2009;41:703. doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hindorff L, Sethupathy P, Junkins H, Ramos E, Mehta J, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proceedings of the National Academy of Sciences. 2009;106:9362. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson AD, Handsaker R, Pulit S, Nizzari M, O'Donnell CJ, et al. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 2008;24:2938–2939. doi: 10.1093/bioinformatics/btn564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gower J. A general coefficient of similarity and some of its properties. Biometrics. 1971;27:857–874. [Google Scholar]
- 28.Ward J., Jr . Journal of the American Statistical Association; 1963. Hierarchical grouping to optimize an objective function. pp. 236–244. [Google Scholar]
- 29.Team RDC. Vienna, Austria: 2010. R: A Language and Environment for Statistical Computing. [Google Scholar]
- 30.Lage K, Karlberg EO, Størling ZM, Ólason PÍ, Pedersen AG, et al. A human phenome-interactome network of protein complexes implicated in genetic disorder. Nat Biotechnol. 2007;25:309–316. doi: 10.1038/nbt1295. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Complete SNP-wise association data. Here we present the complete dataset on which we base our analysis. All data have been previously published as detailed in the main manuscript and in the key below, and are based on chi-square (1 df) or Z association statistics. Where not provided, we computed Z scores as the square root of the cognate chi-squared statistic. Sign was assigned with reference to the minor allele declared in the psoriasis GWAS (chosen arbitrarily). SNP - marker name. CHR – chromosome. POS - physical position (hg18). major_al - major SNP allele. minor_al - minor SNP allele. RA.Z - association Z score for rheumatoid arthritis (Stahl et al.Nat Genet 2010) [7]. RA.P - association p value for rheumatoid arthritis (Stahl et al.Nat Genet 2010) [7]. PS.Z - ditto for psoriasis (Nair et al. Nat Genet 2009) [6]. PS.P - ditto for psoriasis (Nair et al. Nat Genet 2009) [6]. MS.Z - ditto for multiple sclerosis (De Jager et al. Nat Genet 2009) [5]. MS.P - ditto for multiple sclerosis (De Jager et al. Nat Genet 2009) [5]. SLE.Z - ditto for systemic lupus erythematosus (Gateva et al. Nat Genet 2009) [23]. SLE.P - ditto for systemic lupus erythematosus (Gateva et al. Nat Genet 2009) [23]. CD.Z - ditto for Crohn's disease (Barrett et al. Nat Genet 2008) [2]. CD.P - ditto for Crohn's disease (Barrett et al. Nat Genet 2008) [2]. CeD.Z - ditto for celiac disease (Hunt et al. Nat Genet 2008) [4]. CeD.P - ditto for celiac disease (Hunt et al. Nat Genet 2008) [4]. T1D.Z - ditto for type I diabetes (Barrett et al. Nat Genet 2009) [24]. T1D.P - ditto for type I diabetes (Barrett et al. Nat Genet 2009) [24]. Disease - disease in which the SNP was originally reported: AITD autoimmune thyroid disease; AS ankylosing spondylitis; BD CD Crohn's disease; MS multiple sclerosis; PS psoriasis; SLE systemic lupus erythematosus; T1D type 1 diabetes; UC ulcerative colitis. cpma.p - p value for CPMA statistic (chi-squared, 1 df). Genes - nearby notable genes.
(TAB)
Enrichment in immune tissue expression for interacting genes encoded close to SNPs in (A) cluster 1 and (B) cluster 4. Following Rossin et al.[19] we looked for preferential expression of significant network genes in tissue subsets. Of the genes encoded around SNPs in clusters 1 and 4 (as defined in Figure 1), we found that those participating in significant networks are enriched in expression (purple circles) in immune tissues (red bars). Other genes encoded around those SNPs are not enriched in the same tissues (black circles). Thus interacting genes encoded around SNPs associated to the same immune diseases are preferentially expressed in immune tissues. Interacting genes for the remaining significant group, cluster 2, were not enriched.
(PDF)