

Abstract
Whole genome shotgun assembly is the process of taking many short sequenced segments (reads) and reconstructing the genome from which they originated. We demonstrate how the technique of bidirected network flow can be used to explicitly model the double-stranded nature of DNA for genome assembly. By combining an algorithm for the Chinese Postman Problem on bidirected graphs with the construction of a bidirected de Bruijn graph, we are able to find the shortest double-stranded DNA sequence that contains a given set of k-long DNA molecules. This is the first exact polynomial time algorithm for the assembly of a double-stranded genome. Furthermore, we propose a maximum likelihood framework for assembling the genome that is the most likely source of the reads, in lieu of the standard maximum parsimony approach (which finds the shortest genome subject to some constraints). In this setting, we give a bidirected network flow-based algorithm that, by taking advantage of high coverage, accurately estimates the copy counts of repeats in a genome. Our second algorithm combines these predicted copy counts with matepair data in order to assemble the reads into contigs. We run our algorithms on simulated read data from Escherichia coli and predict copy counts with extremely high accuracy, while assembling long contigs.
Key words: bidirected flow, bidirected graph, Chinese postman, de Bruijn graph, genome assembly, matepairs, sequence assembly
1. Introduction
Whole genome shotgun sequencing is the process by which a genome is broken into many small segments (reads) whose sequence is then determined. The problem of combining these reads to reconstruct the source genome is known as genome assembly and is one of the fundamental algorithmic problems within bioinformatics. Until recently, most sequencing has been done using Sanger-style technology, which generates long reads of length up to 700 nucleotides. However, the advent of Next Generation Sequencing (NGS) technologies has enabled sequencing with much greater coverage at only a fraction of the cost. The drawback, however, is that the read length is much shorter, sometimes as low as 25 nucleotides. The short read length posses significant challenges to the problem of de novo genome assembly—the determination of a completely unknown genome—which is the topic of this article.
Most assemblers can be grouped into two categories. Some, like the string graph approach of Myers, (2005), are based on an overlap graph—a graph where vertices correspond to reads and edges correspond to overlaps. Others, such as the EULER assembler (Pevzner et al., 2001), are based on de Bruijn graphs—graphs where the edges correspond to substrings of the reads, and the vertices correspond to overlaps between these strings. Both of the models share the key property that the edges of the graph “spell” some string, and by walking the edges of the graph, it is possible to recreate the genome. Because walks on graphs can be elegantly defined using the concept of balance around vertices (each vertex must be entered and left an equal number of times), the assembly algorithms based on these graphs use network flow as the underlying technique for finding the genome.
1.1. Bidirected flow
One difficulty that arises from using network flow on these graphs is that they only model the overlaps of strings. A read, on the other hand, is actually a DNA molecule—it consists of two strands that are reverse complements of each other (Fig. 1A). The start (called 5′) of one strand is complementing the end (called 3′) of the other. Whenever DNA is sequenced, the molecule is always read in the same direction, from 5′ to 3′, but it is impossible to know from which of the two strands the sequence is read.
FIG. 1.

(A) An example of double stranded DNA. The sequence read from this DNA can be either ATTGCC or GGCAAT. (B) Three possible types of overlaps between two reads: each read can be in either of two orientations, but two of the cases (both to the left and both to the right) are symmetric. (C) The three corresponding types of bidirected edges. The left node corresponds to the lower read. Note that the arrow points into a node if and only if the overlap covers the start (5′) of the read.
Kececioglu (1992) introduced an elegant method for dealing with double-strandedness by modeling overlaps between DNA molecules using a bidirected overlap graph. Each read is represented by a single node, and each overlap (edge) has an orientation at both endpoints. The three types of bidirected edges correspond to the three possible ways in which the overlap can occur (Fig. 1B,C). Just like a walk in the directed overlap graph spells a string composed of the substrings it traverses, so a walk in the bidirected overlap graph spells a double-stranded string (molecule) composed of the double-stranded reads it traverses.
However, the use of bidirected graphs in genome assembly has not been widespread. In this article, we “rediscover” the seminal work and algorithms for minimum cost flow in a bidirected graph by Edmonds (1967) and Gabow (1983), and show that it has wide applications in assembly. We give an exact polynomial time algorithm for finding the shortest double-stranded genome given its k-molecule spectrum (the set of all k-long DNA molecules that constitute the genome), a problem that has been solved for strings (Pevzner, 1989) but remained open for molecules. Furthermore, we use the bidirected overlap graph and bidirected flow as the underlying algorithms for assembling short read data. To the best of our knowledge, these are the first exact polynomial time assembly algorithms that explicitly model the double-stranded nature of DNA.
1.2. Maximum likelihood genome assembly
All of the previous work on genome assembly shares a major assumption: given a set of reads, the goal of the assembly algorithm is to minimize the length of the assembled genome. The problem of modeling genome assembly in this way is that most genomes have repeats—multiple identical, or nearly identical, stretches of DNA that are longer than the read length. These repeats will be under-represented in the assembly of the shortest genome, a problem known as over-collapsing the repeats. Most ways of addressing this problem are to enforce certain constraints on the genome, such as restricting the number of times a read is present or requiring some minimal overlap between two adjacent reads. Such methods help to alleviate some of the difficulty of assembling Sanger-style reads; however, the problem becomes exacerbated when the read length is drastically decreased and the number of repeats becomes huge.
We propose that the overall goal of an assembler should be not to minimize the length of the assembled genome, but to maximize the likelihood that it was the source of the set of reads. Because whole genome shotgun sequencing samples the genome at random locations, the k-mers that are present more often in the genome are more likely to be sampled. Sanger-style sequencing typically does not have sufficient coverage to take advantage of these frequencies. However, the high coverage of NGS makes it possible to not only determine whether a particular read is present in a genome, but also to statistically estimate its copy count (the number of times a read appears in the genome). We formulate the problem of genome assembly as maximizing the likelihood of the observed read frequencies, rather than minimizing the length of the genome. This problem can be formulated as a minimum cost bidirected flow (biflow) problem with convex costs, and we show that it can be effectively solved with a generic flow solver for the case of bacterial sized genomes, achieving copy counts that are accurate more than 99.99% of the time.
1.3. Extending contigs using matepairs
Network flow techniques alone are still insufficient to assemble a genome in the presence of long repeats. One of the key pieces that has allowed for whole genome shotgun assembly of mammalian genomes are matepairs—pairs of reads which come from opposite strands, at an approximately known distance in the source genome. Matepairs can be generated by taking a piece of DNA of a known size (called an insert) and generating reads from its two ends. Matepairs allow for the spanning of repeats, allowing the assembler to join together long genomic regions even in the presence of a repeat which is not spanned by any read. The typical approach for using matepairs in assembly is to build initial contigs (chains of edges in the overlap graph) and then attempt to join them using information from the matepairs.
In this article, we introduce a novel technique for taking advantage of matepair information, which is based on Dijkstra's shortest path algorithm. We avoid the costly search between mated reads in the graph and, instead, focus on searching locally for contigs whose proximity is supported by their matepairs. Because the paths we search for are bounded by a small length that is independent of the genome size (the maximum variation in the insert size), our algorithm scales extremely well for large genomes and high coverage. We use our method to improve the lengths of the contigs assembled by the bidirected flow algorithm above.
1.4. Organization
We will first present related work in Section 2 and give the necessary definitions in Section 3. In Section 4, we show how bidirected flow can be used to reconstruct a genome from k-molecules in the maximum parsimony setting. We then show how to efficiently solve minimum cost bidirected flow in Section 5, as well as introduce variants on the standard flow formulation. Next, we present the maximum likelihood approach by giving our algorithm for copy count prediction in Section 6. We show how to use these copy counts to build initial contigs and then extend them using matepair information in Section 7. Finally, we present the experimental results of our assembly algorithms on simulated data in Section 8.
2. Related Work
Most methods for genome assembly are based on either a de Bruijn graph or some variation of an overlap graph, like a string graph. The de Bruijn graph was first used in genome assembly by the EULER assembler (Pevzner et al., 2001). Pevzner et al. (2001) had the insight that, by dividing the reads into shorter k-mers, all of the instances of a repeat collapse into a single set of vertices. They then represent each read as a walk on the de Bruijn graph, and search for a superwalk that contains all the reads. This approach was later expanded to use A-Bruijn graphs (Pevzner et al., 2004), where the initial subdivision into k-mers is not necessary. The EULER assembler uses both the reads and their reverse-complements to construct the de Bruijn graph, but does not address the issue of choosing the correct orientation for the reads. The copy counts are determined based on the solution to a minimum cost circulation flow problem on the resulting de Bruijn Graph. The initial contigs based on these copy counts are then extended using additional matepair information (Pevzner and Tang, 2001). They search for all paths in the de Bruijn graph connecting the two reads of a matepair. If there exists only one with a length approximately equal to the length of the insert, it is replaced by a direct edge. This approach has the disadvantage that it requires an algorithm to find all paths of (approximately) a given length between two nodes, which is a difficult computational problem, and scales poorly with the size of the de Bruijn graph. In contrast, our algorithm does not search for all the paths between mated reads, but rather, we search only for the existence of short paths between some pairs of reads.
Myers introduced an alternative model of genome assembly based on a string graph (Myers, 2005). Instead of dividing the reads into k-mers, the algorithm starts by building an overlap graph. As in the Celera assembler, Myers determines whether a contig (represented by an edge) is present uniquely in the genome by modeling the reads on a contig as a Poisson arrival process and calculating the probability that the arrival rate for an edge is twice as high as for the genome as a whole. If this probability is low (p < 10−6), the edge (contig) is labeled unique, and the flow through this edge is constrained to be exactly one. Another kind of constraint is placed on every edge that has an interior vertex. Since it must be traversed at least once if the read corresponding to the interior vertex is to take part in the reconstruction of the genome, the flow is constrained to be at least one on this edge. Through this process, Myers (2005) classifies all edges as either unique, required, or optional, and the goal of the assembly is to find the shortest walk that respects all the edge constraints. Though Myers uses bidirected edges in his string graph, he does not address the problem of solving flow in a bidirected, rather than a directed graph.
Bidirected graphs were used for genome assembly in Myers (1995, 2005) and to model breakpoint graphs in Kececioglu and Sankoff (1995). Remarkably, however, they had already been studied within the context of graph theory in the 1960s, when Edmonds (1967) formulated the problem of bidirected flow showed it equivalent to perfect b-matchings. Edmonds' work was later extended by Gabow (1983), who gave the fastest to-date algorithm for bidirected flow on sparse graphs. Several textbooks have a thorough discussion of bidirected flow (Lawler, 2001; Schrijver, 2003).
Most of the methods for assembly were developed to work with Sanger-style data and do not give good results on NGS data. The rapid growth of NGS technologies has brought about a need for new methods that are specifically designed to work with short reads. SSAKE (Warren et al., 2007) is an assembler that uses a simple algorithm for building contigs by greedily extending existing overlaps. VCAKE (Jeck et al., 2007) extended SSAKE to work with error-prone, rather than perfect, data. Another approach based on elongating existing contigs is the SHARCGS assembler (Dohm et al., 2007). The Shorty assembler (Chen and Skiena, 2007) uses a de Bruijn graph approach in combination with matepairs to assemble a small bacteria—the 600-Kb Mycoplasma genitalium. Chaisson and Pevzner (2007) have adapted EULER to use reads generated by 454 sequencers, whose length is about 100 basepairs. Another promising tool is the Velvet assembler (Zerbino and Birney, 2008), which combines the traditional de Bruijn graph approach with a novel error-removal algorithm. ALLPATHS is a recent method that uses the idea of sequence graphs to assemble short reads but works for Sanger-style reads as well (Butler et al., 2008). EDENA is a method that is based on string graphs but implements additional heuristics (Hernandez et al., 2008).
3. Definitions
3.1. Strings, molecules, and de Bruijn graphs
Denote by 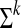 the set of all strings of length k over the alphabet Σ. The length of a string v is denoted by |v|. Denote by v[i, j] the substring of v beginning at the ith position and ending at the jth position, inclusive. A string of length k is called a k-mer. The k-spectrum of v is the set of all k-mers that are substrings of v. A string is called k-circular if its first k characters are the same as its last k characters.
the set of all strings of length k over the alphabet Σ. The length of a string v is denoted by |v|. Denote by v[i, j] the substring of v beginning at the ith position and ending at the jth position, inclusive. A string of length k is called a k-mer. The k-spectrum of v is the set of all k-mers that are substrings of v. A string is called k-circular if its first k characters are the same as its last k characters.
A DNA molecule is an unordered pair of strings (also called strands) that are reverse complements of each other. We say a molecule corresponds to each of its two constitutive strings, and vice-versa. A DNA molecule m is a submolecule of m′ if a corresponding string of m is a substring of a corresponding string of m′. A k-molecule is a DNA molecule whose corresponding strings have length k. The k-molecule-spectrum of a DNA molecule is the set of all k-molecules that are its submolecules. A DNA molecule is k-circular if the first k characters of its corresponding strings are the same as their last k characters, respectively.
Given a set of strings S, we define the de Bruijn graph Bk(S) as a directed graph, using a positive integer parameter k. The vertices of Bk(S) are  . We identify a vertex by the k-mer associated with it. The edges are
. We identify a vertex by the k-mer associated with it. The edges are 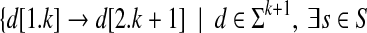 such that d is a substring of s}.
such that d is a substring of s}.
3.2. Bidirected graphs
Consider an undirected graph G with a set of vertices V and a set of edges E. In this article, we allow graphs to have multiple edges (in other words to be multigraphs), and the multiplicity of an edge e is the number of edges in G whose endpoints are the same as e's. If the endpoints are distinct, the edge is called a link; otherwise, it is a loop. Additionally, we assign two orientations to each of the edges, one with respect to each of its endpoints. There are two kinds of orientations—positive and negative—and thus we can say an edge is positive-incident or negative-incident to an endpoint. When taken together with the orientations of its edges, G is called a bidirected graph. If there is additionally a weight 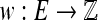 function on the edges, we say that G is weighted. The weight of a graph is the sum of the weights of its edges. A bidirected graph is connected if its underlying undirected graph is connected.
function on the edges, we say that G is weighted. The weight of a graph is the sum of the weights of its edges. A bidirected graph is connected if its underlying undirected graph is connected.
A bidirected graph G has an associated incidence matrix I G : V × E → {−2, −1, 0, 1, 2} (we omit G when it is obvious from the context). If an edge e is not incident to a vertex x, then I(x, e) = 0. For a link e and a vertex x, I(x, e) = +1 if e is positive-incident to x, and I(x, e) = −1 if e is negative-incident to x. For a loop e and a vertex x, I(x, e) has the value of +2 if e is twice positive-incident to x, −2 if e is twice negative-incident to x, and 0 otherwise.1 For an example of a bidirected graph and its incidence matrix, see Figure 2. The in-degree of a vertex x in graph G is defined as 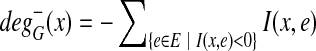 . Similarly, the out-degree is defined as
. Similarly, the out-degree is defined as 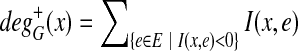 . Let
. Let 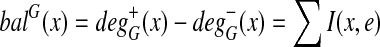 be the balance at each vertex. G is balanced if the balance of each vertex is 0.
be the balance at each vertex. G is balanced if the balance of each vertex is 0.
FIG. 2.

(A) This is an example of a bidirected graph. By convention, we draw edges that are positive(negative) incident to a vertex using an out(in) arrow at that vertex. The sequence W, A, X, B, Y, C, Y, B, X, D, Z is a walk, while W, A, X, D, Z is not. (B) The corresponding edge incidence matrix, with the zero entries ommited. (C) This is the associated monotonized directed graph.
A (x1, xk)-walk is a sequence 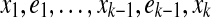 where ei is an edge incident to xi and xi+1, and for all 2 ≤ i ≤ k − 1, ei−1 and ei have opposite orientations at xi. The length of this walk is k−1, the number of edges it contains. Since the specification of vertices is redundant, we may omit them sometimes and specify a walk as just a sequence of edges. A walk is said to be a cycle or cyclical if its endpoints are the same and e1 and ek−1 have opposite orientations at x1. A bidirected graph is strongly connected if it is connected and for every edge there is a cycle containing it. Note that this is equivalent to saying that for every pair of vertices, there is a cycle containing them.
where ei is an edge incident to xi and xi+1, and for all 2 ≤ i ≤ k − 1, ei−1 and ei have opposite orientations at xi. The length of this walk is k−1, the number of edges it contains. Since the specification of vertices is redundant, we may omit them sometimes and specify a walk as just a sequence of edges. A walk is said to be a cycle or cyclical if its endpoints are the same and e1 and ek−1 have opposite orientations at x1. A bidirected graph is strongly connected if it is connected and for every edge there is a cycle containing it. Note that this is equivalent to saying that for every pair of vertices, there is a cycle containing them.
We can view a directed graph as a special kind of bidirected graph, where every edge is positive-incident to one of its endpoints and negative-incident to the other one. In this case, the definitions of walk and cycle reduce to their usual meanings in directed graphs. However, some properties of directed graphs do not translate to bidirected graphs. For example, it is possible for the shortest walk between two vertices to repeat a vertex in a bidirected graph. In Figure 2, observe that there does not exist a walk between W and Z, which does not repeat a vertex, something that is not possible in a directed graph.
3.3. Bidirected overlaps
Suppose that all k-molecules x have had one of their corresponding k-mers labeled as coming from the “positive” strand (p(x)), and the other as coming from the “negative” strand (n(x)). Let x and y be two k-molecules represented by vertices (they may be identical), and let e be a bidirected edge between x and y Then e is a bidirected overlap if one of the following holds
e is positive-incident to x and negative-incident to y and p(x) overlaps p(y).
e is positive-incident to x and positive-incident to y and p(x) overlaps n(y).
e is negative-incident to x and negative-incident to y and n(x) overlaps p(y).
e is negative-incident to x and positive-incident to y and n(x) overlaps n(y).
The length of this bidirected overlap is the length of the underlying string overlap. Note that this edge construction is identical to the one defined by Kececioglu (1992) for an overlap between two DNA molecules (Fig. 1).
3.4. CPP and flow
Let G be a weighted graph, either directed, undirected, or bidirected. A tour is a walk that traverses every edge of G at least once. A circuit is a tour that is cyclical. Given G, the Chinese Postman Problem (CPP) is to find a minimum weight circuit (called a Chinese postman circuit), or report that one doesn't exist. An Eulerian circuit of G is a circuit that contains every edge of the graph exactly once, and a graph which contains an Eulerian circuit is called Eulerian. The following is a generalization of a well-known fact for directed graphs whose proof is almost identical to the directed case and is therefore omitted.
Observation 1
A bidirected graph contains an Eulerian circuit if and only if it is connected and balanced.
Let G = (V, E) be a bidirected graph. Let 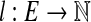 and
and 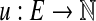 be lower and upper bounds associated with the edges, and
be lower and upper bounds associated with the edges, and 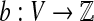 be a balance constraint associated with each vertex. The function
be a balance constraint associated with each vertex. The function 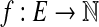 is called a flow if for every edge, l(e) ≤ f (e) ≤ u(e), and for every vertex v, the flow along the positive-incident edges minus the flow along the negative-incident edges is equal to b(v) (specifically,
is called a flow if for every edge, l(e) ≤ f (e) ≤ u(e), and for every vertex v, the flow along the positive-incident edges minus the flow along the negative-incident edges is equal to b(v) (specifically, 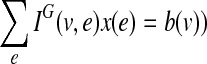 . Given a bidirected graph with l, u, b, and a cost
. Given a bidirected graph with l, u, b, and a cost 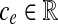 associated with each edge, the min-cost bidirected flow (biflow) problem is to find a flow that minimizes
associated with each edge, the min-cost bidirected flow (biflow) problem is to find a flow that minimizes 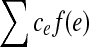 .
.
4. Reconstructing a Genome From k-Molecules
Consider the problem of assembling the shortest circular genome given its k-molecule spectrum. Formally stated, the problem is: given a set of k-molecules S, what is the shortest (k − 1)-circular DNA molecule whose k-molecule spectrum is S? Though the equivalent problem has been solved for strings (Pevzner, 1989), no exact polynomial time algorithm is known for this molecule version. In this section, we show that the problem reduces to solving the CPP on the associated bidirected de Bruijn graph, and present a polynomial time algorithm for solving both of these problems.
4.1. Bidirected de Bruijn graph
In an earlier work, Pevzner (1989) showed that if S is a set of k-mers then the (directed) Chinese postman circuit of the de Bruijn graph Bk−1(S) corresponds to the shortest (k − 1)-circular string with k-spectrum S. When working with DNA molecules, which are double-stranded, it is necessary to model k-molecules instead of k-mers in the de Bruijn graph. In a later work, Pevzner et al. (2001) attempt to do this by including both of the k-mers associated with every k-molecule in the de Bruijn graph. They then search for two “complementary” walks, each corresponding to one of the DNA strands (Fig. 3). Instead, we show how to construct a bidirected de Bruijn graph, where each k-molecule is represented only once.
FIG. 3.

Given the k-molecule-spectrum {ATT/AAT, TGC/GCA, GCC/GGC, CCA/TGG, CAA/TTG, AAC/GTT}, the approach of Pevzner et al. (2001) builds the graph on the left, and searches for two “complementary” walks. The bidirected de Bruijn graph is on the right. One walk that includes all of the edges spells ATTGCCAAC, and its reverse walk spells GTTGGCAAT.
Our input is a set of k-molecules S. We will arbitrarily label one of the k-mers corresponding to each k-molecule as coming from the “positive” strand and the other from the “negative” strand. Let the nodes of the bidirected de Bruijn graph be all (k − 1)-molecules that are submolecules of some molecule in S. For every k-molecule in the spectrum, let z be one of its corresponding k-mers. Let x and y be the (k − 1)-molecules corresponding to z[1.k − 1] and z[2.k], respectively. We make an edge between the vertices of x and y. This edge is positive-incident to x if z[1.k − 1] is the positive strand of x, and negative-incident otherwise. It is negative-incident to y if z[2.k] is the positive strand of y, and positive-incident otherwise. Note that the choice of z does not affect the orientation of the edge, and that the edge is a bidirected overlap of length k − 1. Intuitively, this graph is unweighted, but we can view all the edges as having weight one.
Now, consider a circular (w1,wm)-walk w in this bidirected de Bruijn graph G. We can imagine traversing w along the edges from w1 to wm, and spelling a string as we go along. We adopt the convention that if we enter a node on a positive-incident edge we read the negative k-mer, if on the negative incident we read the positive k-mer. At each consecutive vertex, the k-mer that is read must by definition overlap the last k − 1 characters of the previous k-mer read. In this way, we can spell a (k − 1)-circular string of k + m − 1 characters by traversing w and, at each vertex, adding a new character to our string. Moreover, w has a corresponding reverse walk, which is a walk with the same edges but in the reverse direction. It follows from the construction of G that the string spelled by w's reverse walk is the reverse complement of the string spelled by w, and thus every circular walk in G of length l corresponds to a (k − 1)-circular DNA molecule of length k + l. Observe that this method of traversing a walk in a bidirected graph to spell a molecule works for any graph whose edges are bidirected overlaps.
The k-molecule spectrum of the spelled DNA molecule is exactly the set of k-molecules corresponding to each edge of the walk. Therefore, every circuit of length l in G corresponds to a molecule of length k + l whose spectrum is S. Conversely, a (k − 1)-circular DNA molecule d that has spectrum S can be viewed as a sequence of d − k + 1 consecutive overlapping k-molecules. It is easy to see that this corresponds to a circuit of G that spells d and whose length is |d| − k. It follows that a Chinese postman circuit of G is the shortest (k − 1)-circular DNA molecule with S as its spectrum.
Theorem 2
Given a set of k-molecules S, we can find the shortest (k − 1)-circular DNA molecule whose k-molecule spectrum is S in time O(|S|2 log2(|S|)).
Proof
We will show in Theorem 6(next section) that we can solve CPP on a bidirected graph in time O(|E|2 log(|V|) log(|E|)). In the case of a bidirected de Bruijn graph, where |E| ≤ |S| and |V| ≤ 2|S|, this running time is actually O(|S|2 log2(|S|)). Since we can trivially construct and perform basic operations on the graph within this time, the time bound follows.
4.2. Chinese Postman Problem on bidirected graphs
Given a weighted bidirected graph G, recall that the CPP is to find a minimum weight circuit of G, or report that one does not exist. CPP is polynomially time solvable on both undirected and directed graphs (Edmonds and Johnson, 1973). For undirected graphs, CPP is reducible to minimum cost perfect matchings. For directed graphs, it is reducible to minimum cost network flow. It becomes NP-hard on mixed graphs, which are graphs with both directed and undirected edges (Garey and Johnson, 1979). We now present an efficient algorithm for solving CPP on bidirected graphs via a reduction to minimum cost bidirected flow.
To find a Chinese postman circuit, first consider the case G is Eulerian. Since an Eulerian circuit of G is has the smallest possible weight of any circuit, it is also a Chinese postman circuit. In the general case, however, when G is not Eulerian, our approach is to make the graph Eulerian by duplicating some of the edges, and then using a standard algorithm to find an Eulerian circuit. We shall prove that if we minimize the total weight of the duplicated edges, the Eulerian circuit we find in the modified graph will correspond to a Chinese postman circuit in the original graph.
Formally, we say a graph G′ is an extension of G if it can be obtained from G by duplicating some of its edges. The Eulerization Problem (EP) is to find a min-weight Eulerian extension of G, or report that one does not exist. The following theorem shows that CPP and EP are polynomially equivalent.
Theorem 3
Let G be a weighted bidirected graph. There exists a circuit of weight i if and only if there exists an Eulerian extension of weight i.
Proof
For the only if direction, let W be a circuit in G. Let 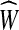 be the extension of G that is compromised from j copies of every edge e, where j is the number of times W contains e. Since W is an Eulerian circuit it follows that
be the extension of G that is compromised from j copies of every edge e, where j is the number of times W contains e. Since W is an Eulerian circuit it follows that 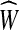 is an Eulerian graph. Finally, the weight of W and
is an Eulerian graph. Finally, the weight of W and 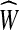 are the same by definition.
are the same by definition.
For the if direction, let G′ be an Eulerian extension of G. Let W′ be an Eulerian circuit in G′. Construct W from W′ by replacing every edge e′ ∉ G by an edge e ∈ G such that e′ is a duplicate of e (we refer to this as a relabeling of W′). W is thus a valid cycle in G which visits every edge at least once and whose weight is the same as that of W′ and of G′. ▪
We can now state the necessary and sufficient conditions for G to have a circuit:
Lemma 4
G has a circuit if and only if it is strongly connected.
Proof
We will prove that G has an Eulerian extension if and only if it is strongly connected. If G has an Eulerian extension, then it must be connected, and for every edge, there is a cycle containing it (namely, the one made by relabeling the Eulerian circuit of the extension).
Conversely, suppose that G is strongly connected. For every edge, we can duplicate all the other edges of a cycle that contains it. In this way, we get an extension of G whose edge set is a union of cycles, which means it is balanced. This proves the if part.
Now, we give an algorithm for the EP. First, we consider the case that G is not connected. Since any extension of G will also not be connected, our algorithm can safely report that there is no Eulerian extension of G, and hence no circuit. For the case that G is connected, we will formulate EP as a min-cost bidirected flow problem. First, we represent an extension G′ of G with a function 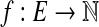 , where f (e) represents the number of additional copies of edge e in G′. Conversely, it is clear that any function
, where f (e) represents the number of additional copies of edge e in G′. Conversely, it is clear that any function  corresponds to an extension of G. Now, we would like to formulate EP in terms of f instead of in terms of an extension. The minimization criterion is the weight of the extension, which is
corresponds to an extension of G. Now, we would like to formulate EP in terms of f instead of in terms of an extension. The minimization criterion is the weight of the extension, which is 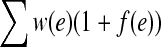 , where w is the weight function of G. The criterion that G′ is Eulerian is, by Observation 1, the criteria that it is connected and balanced. The connectivity criterion is redundant since G is connected and thus any extension of G must also be connected. The balance condition for each vertex x can be stated as:
, where w is the weight function of G. The criterion that G′ is Eulerian is, by Observation 1, the criteria that it is connected and balanced. The connectivity criterion is redundant since G is connected and thus any extension of G must also be connected. The balance condition for each vertex x can be stated as: 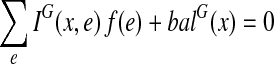 . That is, the balance of x in G′ is the balance of x in G plus the contribution of all the copied edges. We are now able to formulate EP as the following integer linear program:
. That is, the balance of x in G′ is the balance of x in G plus the contribution of all the copied edges. We are now able to formulate EP as the following integer linear program:
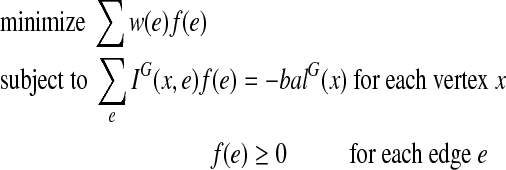 |
From the definition in Section 3, this is actually a minimum cost bidirected flow problem, which can be solved using the algorithm of Gabow (1983). Our final algorithm for CPP on bidirected graphs is given by Algorithm 1. For the running time, we need to bound the size of the solution:
Algorithm 1.
Chinese Postman Problem on bidirected graphs
| 1: ifG is not connected then return “no circuit exists” |
| 2: Use algorithm of (Gabow, 1983) to solve the corresponding minimum cost bidirected flow (see text). |
| 3: if there is no solution then return “no circuit exists” |
| 4: Let G′ be the graph G with f(e) copies of every edge e, in addition to e itself. |
| 5: Use a standard algorithm to find an Eulerian circuit W of G′. |
| 6: Relabel W according to Theorem 3. |
| 7: returnW |
Lemma 5
The minimum weight Eulerian extension of G has at most 2|E||V| edges.
Proof
Suppose G′ is a min-weight Eulerian extension of G. We can decompose the edge set of G′ into a set of minimal cycles. Each one must contain an edge that no other cycle contains, otherwise it can be removed from G′ to get a smaller weight Eulerian extension. Therefore, there are at most |E| cycles. By minimality, each cycle contains at most 2|V| edges, since otherwise it must be a union of two smaller cycles. The bound follows.
Theorem 6
The running time of Algorithm 1 is O(|E|2 log(|V|) log(|E|)).
Proof
Gabow's algorithm runs in time O(|E|2 log(|V|) log(max u(e)); recall that u is the flow upper bound function. Lemma 5 implies that f (e) ≤ 2|E||V| for every edge e, so that we can safely let u(e) = 2|E||V| in our flow formulation. The running time of Gabow's algorithm is thus O(|E|2 log(|V|) log(|E|)). Since all the other operations of Algorithm 1 can be trivially performed within this time, the time bound follows.
5. Minimum Cost Bidirected Flow
In this section, we give an algorithm for minimum cost bidirected flow and describe equivalent formulations of the biflow problem.
5.1. Efficient algorithm for min-cost biflow
Though Gabow's algorithm for min-cost biflow is polynomial, the algorithm is difficult to implement, and we were not able to find any existing efficient implementation. Here, we give a reduction that allows us to convert a bidirected flow problem into a directed flow problem, for which many efficient algorithms have been developed, for example, the network simplex algorithm (Ahuja et al., 1993). We are also able to take advantage of off-the-shelf packages for solving network flow within our implementation. Though this results in a 2-approximation algorithm in the worst case, it found the optimal solution on almost all our input instances.
One of the easiest ways that directed network flow for a graph G(V, E) can be solved is through a reduction to a linear program (LP). The reduction is based on building the |V| × |E| edge incidence matrix I|V||E| for the graph. Every column of I corresponds to e ∈ E, and every row corresponds to v ∈ V. The cell Im,n is 1 if the edge n is an in-edge of vertex m, it is −1 if it is an out-edge from m, and 0 if it is not incident on m. The edge incidence matrix of a graph can be viewed as the constraint matrix for an LP where the optimal LP solution corresponds to the minimum flow in the graph.
Incidence matrices based on directed graphs are Totally Unimodular (TU), leading to LPs that always have integral solutions. The incidence matrics of bidirected graphs, however, may have two ±1s or a ±2 in a column. The resulting matrices are known as binet matrices (Appa and Kotnyek, 2006) and have the property that the optimal solution of the LP is guaranteed to be half-integral (a multiple of 0.5).
Our algorithm is based on the result by Hochbaum (2004), who demonstrates a reduction from a binet matrix to a TU matrix by monotonization: doubling the number of columns and rows. Solving the LP defined by the new TU matrix is equivalent to solving it in the original binet matrix. However the new TU matrix corresponds to a directed graph, and one can find the min-cost flow in directed graphs using algorithms that are much faster than general LP solvers. We now formulate the monotonization procedure of Hochbaum in terms of the underlying bidirected graph.
For every vertex v of the original bidirected graph we introduce two vertices v1 and v2 in the new directed graph. For every in-edge of v we create two directed “twin” edges, one of which points into the v1 vertex and the other points out of the v2 vertex. For all out-edges of v, we again create twin edges, one of which points out of v1 and the other into v2. An example of the transformation is given in Figure 2C. We transfer all of the bounds and costs on the original edges to the respective twin edges, and after finding the min-cost flow in the directed graph we transfer the results to the original bidirected graph by adding the flows through the pairs of twin edges and dividing by two. Because the procedure above is equivalent to the monotonization procedure of Hochbaum, it has the same provable properties, for example, that the optimal result is half integral and that the monotonized flow is at worst a 2-approximation to the optimal integral flow.
5.2. Convex costs and vertex bounds
We will take advantage of two variations on the min-cost biflow problem, which we describe here. The first allows for having lower and upper bounds for the flow going through each vertex, as well as adding a cost function on the vertices as well as the edges. Such a problem can be reduced to the min-cost biflow problem as follows. Take every vertex v and split it into two vertices v+ and v−. Reconnect any edge that was pointing into v to be pointing into v−. Similarly, reconnect any edge that was pointing out of v to be pointing out of v+. As a final step, add an edge from v− to v+. Now, assign any lower/upper bounds, as well as any costs, associated with v to the edge from v− to v+. After repeating this procedure for every vertex, a flow on the transformed graph corresponds to a flow on the original graph, and vice-versa. This transformation is based on a similar transformation on directed graphs (Ahuja et al.,1993).
The second equivalent variation is called the convex min-cost biflow problem. Here, the cost ce associated with an edge e is no longer a real number but rather a convex function 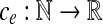 , and the goal is to minimize
, and the goal is to minimize 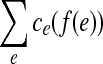 . Such a minimization function is called separable convex because it is a sum of convex functions on each of the variables, independently. In the directed case, this problem is polynomially equivalent to the linear min-cost flow problem, as each convex function can be modeled with piecewise-linear approximations. The same reduction holds in the bidirected case. For a thorough discussion of this reduction, we refer the reader to a text on network flow (Ahuja et al., 1993).
. Such a minimization function is called separable convex because it is a sum of convex functions on each of the variables, independently. In the directed case, this problem is polynomially equivalent to the linear min-cost flow problem, as each convex function can be modeled with piecewise-linear approximations. The same reduction holds in the bidirected case. For a thorough discussion of this reduction, we refer the reader to a text on network flow (Ahuja et al., 1993).
6. Predicting Copy-Counts Using Maximum Likelihood
In this section, we describe our maximum likelihood framework for genome assembly, and give an algorithm that, given a set of reads (DNA molecules), finds the genome that maximizes the global read-count likelihood.
6.1. Maximizing the global read-count likelihood
Let D be a circular genome of length N(D), and let di denote the number of times the k-molecule i appears in D. Probabilistically, the dataset of n reads corresponds to a set of outcomes from n independent trials. In each trial, a position is uniformly sampled from D and the outcome of the trial is the k-molecule beginning at that position. For a given i, the probability that the outcome of a single trial is i is simply 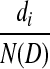 . Let the random variable Xi denote the number of trials whose outcome is i. There are 4k such variables, and when considered independently of each other, they each follow the binomial distribution. When taken together, their joint distribution is exactly the multinomial distribution, given by
. Let the random variable Xi denote the number of trials whose outcome is i. There are 4k such variables, and when considered independently of each other, they each follow the binomial distribution. When taken together, their joint distribution is exactly the multinomial distribution, given by
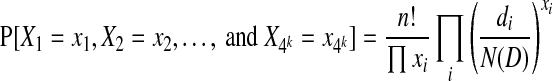 |
For the assembly problem, D is not known but the results of the n trials are known. Thus, we can consider the likelihood of the parameters of the distribution (di) given the outcome of the trials (xi), which we call the global read-count likelihood:
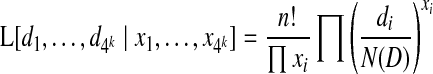 |
In our approach, we attempt to assemble the genome with the maximum global read-count likelihood. Equivalently, we minimize the negative log of this likelihood, − log L. We will eventually want to solve this using convex cost bidirected flow, so we need − log L to be a separable convex function in terms of the di's. That is, we need to find convex functions ci such that 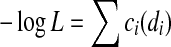 . Unfortunately, since the multinomial distribution has the constraint that
. Unfortunately, since the multinomial distribution has the constraint that 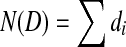 , this is not possible.
, this is not possible.
However, as the number of trials goes to infinity, the Xi random variables become independent. Because the number of trials (sampled k-molecules) is typically large, we can approximate the multinomial distribution as the product of the individual binomial distributions of each Xi. Since in the binomial approximation the length of the genome N(D) is a constant that is independent of each di, we can replace it by N, which is the length of the actual genome from which the reads were sampled. The approximate length of the actual genome can be ascertained through one of a number of biological experiments, or through an Expectation-Maximization type approach. For our experiments, we assume that the genome size is known.
The resulting approximation for L is thus
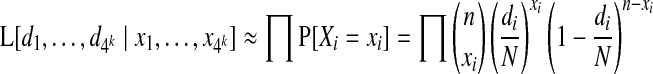 |
Now we can write 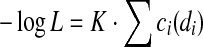 , where K is some positive constant independent of all di, and
, where K is some positive constant independent of all di, and
 |
6.2. Putting it all together
We are now ready to describe our algorithm for predicting copy counts. The first step is to build a bidirected overlap graph from the set of reads, which are DNA molecules. The vertices of this graph are the reads, and the edges are all possible bidirected overlaps of length at least omin, where omin is a parameter to our algorithm. We then perform transitive edge reduction, where we remove any overlap that is spelled by two shorter overlaps. This procedure is identical to the one described in Myers (2005), and we refer to the resulting graph as the transitively reduced bidirected overlap graph. While the set of possible DNA molecules spelled by the graph remains unchanged, the reduction drastically reduces the number of edges. Moreover, we have the following observation:
Observation 7
Let r be a read and W a walk in the transitively reduced bidirected overlap graph. The number of times W visits r is equal to the number of times r appears a submolecule of the molecule spelled by W.
In this graph, the original double-stranded genome corresponds to a circuit (assuming high enough coverage). We make a final change to the graph by adding a supersource and supersink to the graph. This is a standard modification that will allow us to convert a flow to a circulation problem (Ahuja et al., 1993). Next, we define a convex min-cost biflow problem on this graph, with bounds and costs on both the edges and the vertices. Each vertex has a lower bound of 1 since it represents a read that must be present in the genome at least once. All other lower bounds are 0 and all upper bounds are infinity. We add prohibitively large costs to the edges from/to the supersource/sink so that their usage is minimized. By Observation 7, the di's described above actually correspond to the value of the flow through vertex i, and we let ci be the convex cost functions for the vertices.
We finally solve the biflow problem by first applying the reductions of Section 5.2 and then using the efficient algorithm of Section 5.1. Since any flow can be decomposed into a collection of walks, our flow represents a (non-contiguous) assembly of the genome, and the flow going through each vertex represents the number of time the read is present in the assembly.
7. From Flow to Contigs and the Use of Matepairs
We now show how the results of the copy count prediction algorithm are used to build initial contigs, and give an algorithm that extends these contigs by resolving repeats using matepairs. At this point, we have found a flow on the overlap graph, as described above. In general, any flow can be decomposed into a collection of walks, which, in our case, correspond to the assembled contigs. Since there is an exponential number of decompositions possible, we use a heuristic to find one where the length of the walks (contigs) is large and the accuracy of the contigs is high.
7.1. Graph simplification
In many cases, it can be inferred that certain walks will appear as a subwalk in any decomposition. First, we remove all edges with flow zero from the overlap graph. Next, by applying the following the three rules to every vertex v, we can greatly simplify the overlap graph (Fig. 4):
FIG. 4.

(A–C) These demonstrate the three cases of graph simplification described in Section 7. Case A is a chain, case B a loop attached to a chain, and case C is a split vertex. A join vertex case is symmetrical and is not shown. The three simplifications are shown to the right. In all cases, the new graph can “spell” the exact same strings as the initial graph. (D) This is a conflict node. By iterative application of cases A, B, and C, we generate a graph where all remaining vertices are of type D.
Case A
There is exactly one edge going into v and exactly one edge going out of v. The flow on both edges is the same. We can merge the two edges and remove v from the graph.
Case B
There are exactly two edges going out of v and two edges going into v, and exactly one of the edges going out of v is also going into v (a loop). The flow on all three edges is the same. We can merge the three edges and remove v from the graph.
Case C
There is exactly one edge going into v and m > 1 edges going out of v (v is a split vertex), or there is exactly one edge going out of v and m > 1 edges going into v (v is a join vertex). The flow on the in(out) edge is equal to the sum of the flows on the out(in) edge. We can split the in(out) edge into m copies, merge each one with one of the out(in) edges, and remove v from the graph.
We call a vertex v removable if it falls into one of the above cases, and a conflict vertex otherwise. For every removable vertex in the graph, we perform one of the three operations above. It can be shown that after at most 2|V| operations, all the remaining vertices are conflict vertices. In practice, this process reduced the number of edges in the overlap graph by over 105 fold.
7.2. Conflict node resolution algorithm
Once the graph contains only conflict vertices, we attempt to resolve each in turn by finding pairs of edges that are incident on the vertex with opposite orientations and are supported by matepairs. For each vertex, we do a breadth first search in both the in and out directions, recording all of the reads that are within a specified distance threshold. We skip any read that was initially on an edge that had been split (during case C of the previous step), as it no longer has a unique position in the overlap graph. We now have two sets of vertices, L and R, corresponding to reads that were observed on the inside of a vertex and the outside of a vertex, respectively (Fig. 5). The high coverage provided for by NGS methods allows us to concentrate our analysis on reads only a short distance away from the conflict vertex. For each of the reads found, we locate their matepairs in the graph (treating the forward and reverse matepairs separately) and run an all-pairs bounded shortest path algorithm from all the mates of L to all the mates of R. Because the overlap graph is sparse, the most efficient algorithm for all-pairs shortest path is to run Dijkstra's algorithm from every vertex. Furthermore, we terminate Dijkstra's algorithm when all vertices within the bounding distance have been explored: if we expect that the true size of the insert will vary by at most E from the expected size, than the bounding distance is 2E.
FIG. 5.

Resolution of a conflict vertex: we take a incoming and an outgoing edge from the conflict vertex v, and take a pair of reads along them, such as A and B. If these edges belong together in our assembly, then A and B must be close to each other in the source genome. Consequently their mates A′ and B′ should also be close to each other (assuming A and B have the same orientation) in the source genome. We locate A′ and B′ in our graph, and look for the shortest path between them. If the distance is less than twice the error of the insert size, we consider it a evidence for joining the corresponding edges at v.
To resolve conflict vertices we implement a simple greedy matching algorithm. All of the edges incident on a particular vertex are separated into two classes depending on their direction at the node—in or out. For every pair of (in, out) edges, we compute the number of mates that are within the bounding distance from each other. If a significant fraction of one edge's matepairs are within this distance from the matepairs of another edge on the opposite side (a matching condition), the two edges are joined into a common edge. We handle any half-integral edges by allowing either of the edges to get matched to the integral edge incident to the conflict vertex. The process is repeated until no more pairs of edges that satisfy the matching condition are found at the current vertex.
After every conflict vertex has been considered, the graph simplification steps described in the previous section are run again, as new removable vertices may be created during the matching process. The matching procedure is then iterated for a set number of steps, or until convergence.
8. Results
We implemented a prototype assembly algorithm for short reads using the algorithms of Sections 6 and 7. We have experimented with both CPLEX and CS2 (Goldberg, 1997) for solving network flow, and found that while the running times are comparable, CS2 uses less memory; consequently we used it for all of the experiments below. To simplify the implementation, we model the convex cost function using a three-piece linear approximation (Ahuja et al., 1993). The overall running time of our algorithm is approximately 1 hour on a single machine.
8.1. Description of the dataset
To test our algorithms, we generated synthetic read data from the Escherichia coli genome, which has a total length of 4.6M basepairs. We uniformly sampled the genome to find the location of the first read of a matepair, and then sampled the second read at a distance within 10% of the expected insert size, also uniformly. The reads generated were always of length 25 and error-free (the importance of the assumptions of error-free reads and uniform coverage is elaborated upon in the Discussion, Section 9). The coverage rate used was varied from 50 to 100×, though we also tried one test with 200× coverage (a single run of the Solexa system can generate more than 200× coverage for E. coli). The minimum overlap length (omin) was varied from 17 to 21. The exact datasets used are summarized in Table 1.
Table 1.
Copy Count Results
| Dataset | Coverage | Insert size | omin | −2 | −1 | 0 | +1 | +2 | +3 |
|---|---|---|---|---|---|---|---|---|---|
| 50 × 3k | 50 | 3000 | 17 | 4 | 397 | 3937038 | 170 | 18 | 6 |
| 75 × 3k | 75 | 3000 | 19 | 0 | 9 | 4324061 | 28 | 3 | 0 |
| 75 × 6k | 75 | 6000 | 19 | 0 | 7 | 4324665 | 32 | 0 | 0 |
| 100 × 3k | 100 | 3000 | 21 | 0 | 2 | 4466328 | 6 | 0 | 0 |
| 100 × 6k | 100 | 6000 | 21 | 0 | 2 | 4466636 | 23 | 0 | 0 |
| 200 × 6k | 200 | 6000 | 19 | 0 | 0 | 4547426 | 4 | 0 | 0 |
The insert size was simulated with a uniform error of up to 10%. The right side shows the deviations of the predicted read copy counts from their true values. For instance, there were 397 reads whose true copy count was 1 less than the predicted copy count. While half-integral flows were observed with some parameter settings (too low coverage, too low omin), the flow was always integral over all runs with the parameters shown. The first four columns describe the datasets used in the evaluation of the assembly algorithm.
8.2. Copy count results
To evaluate the accuracy of our maximum likelihood flow solving algorithm, we compared the flow going through every vertex in the overlap graph to the number of times that the corresponding read appears in the original genome. The results are presented in Tables 1 and 2. For the vast majority of the reads, we correctly predicted their copy count in the genome, with the fraction of mis-estimated counts varying between 10−4 and 10−6, depending on the coverage. When our algorithm mispredicted the number of occurrences, the error was typically small compared to the true frequency of the read. We also note that the results show only slight improvement past 75 × coverage.
Table 2.
Copy Count Results on Repeats
| Dataset | −2 | −1 | 0 | +1 | +2 | +3 |
|---|---|---|---|---|---|---|
| 50 × 3k | 3 | 182 | 31371 | 170 | 18 | 6 |
| 75 × 3k | 0 | 5 | 31933 | 28 | 3 | 0 |
| 75 × 6k | 0 | 7 | 31927 | 32 | 0 | 0 |
| 100 × 3k | 0 | 1 | 31994 | 6 | 0 | 0 |
| 100 × 6k | 0 | 2 | 31974 | 23 | 0 | 0 |
| 200 × 6k | 0 | 0 | 31999 | 4 | 0 | 0 |
Similar to Table 1, except here we only show the statistics for reads whose true copy counts are greater than one (repeats).
Our algorithm relies on having an estimate on the length of the genome, and we tested to what extent the accuracy is affected when the length is mis-estimated. Running our algorithm using a length that was 10% shorter on the 75 × 3k dataset, we made 84 off-by-one errors (a 2.3-fold increase), and only one more off-by-two error (total of 4). For a length that was 10% longer, we made 351 off-by-one errors, a 9.5-fold increase, and 5 more off-by-two errors (total of 8). When the length was made 20% shorter, there were 537 off-by one errors (a 14.5-fold increase), 6 off-by-two, and 1 off-by-three errors. As expected, when the length estimate was made shorter, most of the errors were under-estimations of the actual counts, while the opposite occurred when the length was made longer. Despite these decreases in accuracy, the algorithm still performs better than on 50 × coverage with perfect length. Additionally, we believe that the length estimation can be iteratively corrected using an Expectation-Maximization type approach, similar to that of Myers (2005).
8.3. Overall assembly results
In order to estimate the quality of the assembly resulting from matepair information, we take every edge of the graph after the conflict node resolution and generate the sequence which it spells. As per convention, we compute the N50 and N90 scores as the length of the shortest contig such that 50% and 90% of the original genome is in longer contigs, and the number of such contigs. To check for the presence of errors in the assembly, each contig was aligned to the reference E. coli genome. The number of errors in a contig was computed as the number of local alignments that is required to completely tile it minus one. The results are summarized in Tables 3 and 4.
Table 3.
Assembly Results (N50)
| Data set | Length (kb) | Number of contigs | One error per | Total contigs |
|---|---|---|---|---|
| 50 × 3k | 23.3 | 55 | 385 kb | 916 |
| 75 × 3k | 23.9 | 51 | 257 kb | 832 |
| 75 × 6k | 23.5 | 53 | 330 kb | 910 |
| 100 × 3k | 24.8 | 51 | 385 kb | 814 |
| 100 × 6k | 23.5 | 53 | 331 kb | 899 |
| 200 × 6k | 23.5 | 53 | 463 kb | 896 |
Length is the N50 score, number is the number of contigs longer than the N50 length. The errors are computed as the total length of errors over the total size of all contigs. Evaluation of the assembly quality for the various datasets using N50.
Table 4.
Assembly Results (N90)
| Data set | Length (kb) | Number of contigs | One error per | Total contigs |
|---|---|---|---|---|
| 50 × 3k | 6.9 | 197 | 276 kb | 916 |
| 75 × 3k | 7.6 | 182 | 244 kb | 832 |
| 75 × 6k | 7.1 | 189 | 276 kb | 910 |
| 100 × 3k | 7.7 | 181 | 319 kb | 814 |
| 100 × 6k | 7.3 | 187 | 296 kb | 899 |
| 200 × 6k | 7.3 | 187 | 346 kb | 896 |
Evaluation of the assembly quality for the various datasets using N90.
Overall, the length of the contigs that contained 50% and 90% of the genome varied between 23–28k and 7–8k, respectively, while the error rate was about one error every 100–180k. These errors illustrate a weakness of a greedy matching algorithm, which may be mislead by two well-matched edges that contradict many other good matchings. While the contig sizes are short by the standard of whole genome assembly with Sanger reads, they compare favourably with the results that Chaisson et al. (2004) obtained on Neisseria meningitis (genome length 2.2Mb) with 70 nucleotide reads, albeit without matepairs: in their experiments, they required 344 contigs to achieve 95% coverage of the genome, while our algorithm required 206 contigs to cover 95% of E. coli, a genome which is twice as long as N. meningitis. These results demonstrate the power of matepair information in resolving the proper layout of the genome, even in the case of very short reads.
9. Discussion
In this article, we have demonstrated that the technique of bidirected flow is a powerful method with numerous applications to genome assembly. By unifying Pevzner's work on de Bruijn graphs, Kececioglu's and Myers' work on bidirected graphs in assembly, and Edmonds' and Gabow's work on bidirected flow, we are able to give an exact polynomial time assembly algorithm in the parsimony setting that explicitly deals with double-strandedness.
We have also introduced a maximum likelihood framework for sequence assembly, and shown that bidirected flow can be used to give a practical and efficient algorithm in this context. In our experiments, we make two major assumptions—that the reads are error-free and that the genome sequencing rate is uniform. Our algorithms, therefore, do not form a full assembly tool that handles real data, but if combined with error and sequencing bias correction components, can be used as ingredients for such a tool. We believe that the first of our assumptions is not fundamental, and a limited amount of error in the reads can be overcome using methods similar to the ones developed for the EULER assembler (Pevzner et al., 2001). Moreover, the high coverage rate should improve the correction accuracy of these methods. The second assumption, however, is more essential to the accuracy of our algorithm. In the case of non-uniform coverage of certain areas in the genome (which has indeed been observed in practice), our algorithm may be less accurate at predicting the copy counts, which may have significant effects in downstream analyses. We believe that these effects can be neutralized if the biases of the sequencing apparatus are known. For example, each read's observed frequency can be adjusted depending on its sequence. The exploration of the exact biases of the NGS platforms and the correction for these is an important avenue for future research.
Footnotes
A loop that is both positive and negative incident to its endpoint is thus represented as a zero column in IG. It is therefore not possible to completely reconstruct G from IG.
Acknowledgments
We are grateful to Allan Borodin, Konstantinos Georgiou, and Gene Myers for helpful comments and careful reading of an earlier version of this manuscript. We would also like to thank Gene Myers and Pavel Pevzner for useful discussion during the development of these algorithms. This work was funded by the NSERC (Discovery Grant), the Canada Foundation for Innovation (an equipment grant), and the NIH (grant RO1-GM81080).
Disclosure Statement
No competing financial interests exist.
References
- Ahuja R.K. Magnanti T.L. Orlin J.B. Network flows: theory, algorithms, and applications. Prentice-Hall; Upper Saddle River, NJ: 1993. [Google Scholar]
- Appa G. Kotnyek B. A bidirected generalization of network matrices. Networks. 2006;47:185–198. [Google Scholar]
- Butler J. Maccallum I. Kleber M., et al. Allpaths: de novo assembly of whole-genome shotgun microreads. Genome Res. 2008;18:810–820. doi: 10.1101/gr.7337908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson M. Pevzner P.A. Tang H. Fragment assembly with short reads. Bioinformatics. 2004;20:2067–2074. doi: 10.1093/bioinformatics/bth205. [DOI] [PubMed] [Google Scholar]
- Chaisson M.J. Pevzner P.A. Short read fragment assembly of bacterial genomes. Genome Res. 2008;18:324–330. doi: 10.1101/gr.7088808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. Skiena S. Assembly for double-ended short-read sequencing technologies, 123–141. In: Mardis E., editor; Kim S., editor; Tang H., editor. Advances in Genome Sequencing Technology and Algorithms. Artech House Publishers; New York: 2007. [Google Scholar]
- Dohm J.C. Lottaz C. Borodina T., et al. SHARCGS, a fast and highly accurate short-read assembly algorithm for de novo genomic sequencing. Genome Res. 2007;17:1697–1706. doi: 10.1101/gr.6435207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmonds J. An introduction to matching [Notes of engineering summer conference] University of Michigan; Ann Arbor: 1967. [Google Scholar]
- Edmonds J. Johnson E.L. Matching, Euler tours, and the Chinese postman. Math. Program. 1973;5:88–124. [Google Scholar]
- Gabow H.N. An efficient reduction technique for degree-constrained subgraph and bidirected network flow problems. STOC. 1983:448–456. [Google Scholar]
- Garey M.R. Johnson D.S. Computers and Intractability: A Guide to the Theory of NP-Completeness. W.H. Freeman; New York: 1979. [Google Scholar]
- Goldberg A.V. An efficient implementation of a scaling minimum-cost flow algorithm. J. Algorithms. 1997;22:1–29. [Google Scholar]
- Hernandez D. Francois P. Farinelli L., et al. De novo bacterial genome sequencing: millions of very short reads assembled on a desktop computer. Genome Res. 2008;18:802–809. doi: 10.1101/gr.072033.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochbaum D.S. Monotonizing linear programs with up to two nonzeroes per column. Oper. Res. Lett. 2004;32:49–58. [Google Scholar]
- Jeck W.R. Reinhardt J.A. Baltrus D.A., et al. Extending assembly of short DNA sequences to handle error. Bioinformatics. 2007;23:2942–2944. doi: 10.1093/bioinformatics/btm451. [DOI] [PubMed] [Google Scholar]
- Kececioglu J.D. Exact and approximation algorithms for DNA sequence reconstruction. University of Arizona; Tucson: 1992. [Ph.D. dissertation]. [Google Scholar]
- Kececioglu J.D. Sankoff D. Exact and approximation algorithms for sorting by reversals, with application to genome rearrangement. Algorithmica. 1995;13:180–210. [Google Scholar]
- Lawler E.L. Combinatorial Optimization: Networks and Matroids. Dover; New York: 2001. [Google Scholar]
- Myers E.W. The fragment assembly string graph. ECCB/JBI 85. 2005. [DOI] [PubMed]
- Myers E.W. Toward simplifying and accurately formulating fragment assembly. J. Comput. Biol. 1995;2:275–290. doi: 10.1089/cmb.1995.2.275. [DOI] [PubMed] [Google Scholar]
- Pevzner P.A. L-Tuple DNA sequencing: computer analysis. J. Biomol. Struct. Dyn. 1989;7:63–73. doi: 10.1080/07391102.1989.10507752. [DOI] [PubMed] [Google Scholar]
- Pevzner P.A. Tang H. Fragment assembly with double-barreled data. ISMB (Suppl. Bioinform.) 2001:225–233. doi: 10.1093/bioinformatics/17.suppl_1.s225. [DOI] [PubMed] [Google Scholar]
- Pevzner P.A. Tang H. Waterman M.S. An Eulerian path approach to DNA fragment assembly. Proc. Natl. Acad. Sci. USA. 2001;98:9748–9753. doi: 10.1073/pnas.171285098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pevzner P.A. Tang H. Tesler G. De novo repeat classification and fragment assembly. Proc. RECOMB 2004. 2004:213–222. doi: 10.1101/gr.2395204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrijver A. Combinatorial Optimization, Volume A. Springer; New York: 2003. [Google Scholar]
- Warren R.L. Sutton G.G. Jones S.J., et al. Assembling millions of short DNA sequences using SSAKE. Bioinformatics. 2007;23:500–501. doi: 10.1093/bioinformatics/btl629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino D. Birney E. Velvet: algorithms for de novo short read assembly using De Bruijn graphs. Genome Res. 2008 doi: 10.1101/gr.074492.107, 18, 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]