

Abstract
Methods suggested for reconstructing regulatory networks can be divided into two sets based on how the activity level of transcription factors (TFs) is inferred. The first group of methods relies on the expression levels of TFs, assuming that the activity of a TF is highly correlated with its mRNA abundance. The second treats the activity level as unobserved and infers it from the expression of the genes that the TF regulates. While both types of methods were successfully applied, each suffers from drawbacks that limit their accuracy. For the first set, the assumption that mRNA levels are correlated with activity is violated for many TFs due to post-transcriptional modifications. For the second, the expression level of a TF which might be informative is completely ignored. Here we present the post-transcriptional modification model (PTMM) that, unlike previous methods, utilizes both sources of data concurrently. Our method uses a switching model to determine whether a TF is transcriptionally or post-transcriptionally regulated. This model is combined with a factorial HMM to reconstruct the interactions in a dynamic regulatory network. Using simulated and real data, we show that PTMM outperforms the other two approaches discussed above. Using real data, we also show that PTMM can recover meaningful TF activity levels and identify post-transcriptionally modified TFs, many of which are supported by other sources.
Supporting website: www.sb.cs.cmu.edu/PTMM/PTMM.html
Key words: dynamic regulatory networks, machine learning, post-transcriptional modification
1. Introduction
Transcriptional gene regulation is a dynamic process that utilizes a network of interactions. This process is primarily controlled by transcription factors (TFs) that bind DNA, and activate or repress sets of genes. Regulatory networks activate hundreds of genes as part of a biological system such as the cell cycle (Spellman et al., 1998; Rustici et al., 2004) and circadian rhythm (Panda et al., 2002), in response to internal and external stimuli (Gasch et al., 2000; Nau et al., 2002) and during development (Arbeitman et al., 2002). Proper functioning of these networks is essential for all living organisms. For example, several diseases are associated with partial or complete loss of appropriate transcriptional regulation (Theuns and Van Broeckhoven, 2000). Determining accurate models for these regulatory networks is thus an important challenge.
A major source of information regarding these networks is gene expression data, which measures the effects that TFs have on their regulated targets. Many methods have been suggested for using this data and other data sources for reconstructing regulatory networks. One of the key challenges faced by methods aimed at reconstructing such networks is to infer the activity levels of the factors regulating the network. While for some TFs the activity levels can be determined by looking at their expression levels, many of the master TFs are post-transcriptionally regulated and can be active even if their expression levels do not change (Beer and Tavazoie, 2004).
So far, methods suggested for reconstructing regulatory networks can be divided into two major groups based on how they infer the activity levels of the TFs. The first set of methods (Zou and Conzen, 2005; Tanay and Shamir, 2001; D'haeseleer et al., 2000; Segal et al., 2003) relies on the mRNA levels measured for TFs and uses these to represent the activity levels of TFs. The second group of methods (Beer and Tavazoie, 2004; Sabatti and James, 2006; Ernst et al., 2007; Rogers et al., 2007) treats the activity levels of TFs as completely unobserved values and infers them from the mRNA levels of their regulated genes.
While both methods have proven useful for many different reconstruction efforts, each suffers from drawbacks that can limit their ability to accurately reconstruct the networks. The first set of methods is less appropriate in cases where TFs are post-transcriptionally modified, which may lead to activity levels that are not reflected in the mRNA levels measured for these TFs (Ideker et al., 2001). The second group of methods overcomes this problem, but does not take advantage of the information from the mRNA levels of the TFs. There are many cases in which TFs' activities are reflected in their mRNA levels (Segal et al., 2003), and ignoring these levels may reduce the ability to correctly model the activity levels of these TFs.
A possible way to combine the two approaches is to measure protein levels in addition to gene expression levels (Kannan et al., 2007). However, this data cannot account for other post-transcriptional events, including phosphorylation and nuclear exclusion (Bose et al., 2005). In addition, it requires proteomics measurements that are not always available and have a limited ability to identify low abundance proteins (Washburn et al., 2003). A number of methods have been proposed for projecting expression data on known interaction networks to identify active edges. However, these methods do not attempt to reconstruct a network but rather combine expression data with an existing network to determine functional links in a specific condition (Sohler and Zimmer, 2005).
Another approach was proposed by Nachman et al. (2004), which infers regulatory networks from expression data using a dynamic Bayesian network. They model the unobserved TF activity levels by hidden variables and then use a post-processing step to link these levels to known TFs based on their expression levels. Thus, this model is a variant of the second set of methods discussed above, since the measured expression levels for TFs are not used when reconstructing the network.
Here we present an algorithm combining the two types of methods mentioned above during the reconstruction phase to get the “best of both worlds”: For transcriptionally regulated TFs, we infer their activity levels from their mRNA levels, and for post-transcriptionally regulated TFs, we rely on the mRNA levels of their target genes. The key insight we make is that, when using time series expression data, we can compare the mRNA levels of a TF with the expression levels of genes regulated by the TF in consecutive time points in order to determine whether the mRNA levels correlate with the activity levels for that TF. This allows us to determine which TFs are post-transcriptionally modified and which are not. For this, we developed the post-transcriptional modification model (PTMM), a variant of factorial hidden Markov model (Ghahramani and Jordan, 1997) that accounts for the factor-specific correlation between TF's mRNA levels and activity levels. For each TF, we maintain a binary indicator representing whether or not this TF is post-transcriptionally modified. If a TF is post-transcriptionally modified, we treat its activity levels as unobserved variables whose values can be inferred from the observed expression levels of genes regulated by this TF using a Kalman filter-based model. If a TF is not post-transcriptionally modified, we use its expression levels as prior and combine them with the expression levels of its target genes to infer its posterior activity. This posterior accounts for the noisy measurement of the TF's expression levels, which might lead to slightly different protein activity levels.
We tested our method and compared it to methods that rely only on target genes or on TF's mRNA levels. Using simulated and real expression data, we show that our method has higher accuracy in detecting the post-transcriptional modification events, in inferring the hidden activity levels of TFs, and in predicting the regulatory relationships between TFs and genes. We also discovered some candidate post-transcriptionally modified TFs, which are validated by other sources.
2. Methods
We start this section with the introduction of the PTMM, which combines time series expression data from multiple experimental conditions and static TF-gene interaction data to reconstruct temporal regulatory networks and to infer whether a TF is post-transcriptionally modified. We also introduce an associated EM algorithm to (i) learn the parameters of a PTMM, (ii) infer whether or not each TF is post-transcriptionally modified, and (iii) infer the hidden activity levels of each TF. In addition to inferring TF activity levels, PTMM can also determine new TF-gene interactions. For each known or inferred interaction, the learned model assigns a condition independent weight between a TF and each of the genes it regulates, representing the strength of this interaction. We conclude this section with the description of our experimental procedure.
2.1. Post-transcriptional modification model
Let m be the number of a set of genes whose expression level is measured at a series of time points under a variety of experimental conditions (datasets). Let n represent the number of a subset of these m genes that are TFs. A PTMM defines a joint probability distribution over an observed time series of gene expression levels, unobserved time series of TF activity levels, and the unobserved post-transcriptional status for each TF (modified or unmodified). We use PTMM to estimate which TFs are post-transcriptionally modified, to infer the hidden activity levels of TFs over time, to determine which genes are regulated by each TFs, and to assign a weight to these regulatory interactions.
Let Gi,d,t represent the expression level of gene i (1 ≤ i ≤ m) in dataset d at time t. Without loss of generality, we assume that the first n (n < m) genes encode for TFs. Let Tj,d,t denote the (hidden) activity level of TF j (the protein product of gene j) in dataset d at time t. Each gene may be regulated by zero, one, or several TFs. Let wi,j denote the weight with which gene i is regulated by TF j. A positive weight means that TF j is an activator of gene i, a negative weight implies that TF j represses gene i, and a weight of zero indicates that gene i is not regulated by TF j. Similar to other methods (Wang et al., 2007; Geier et al., 2007), PTMM models the observed expression level for gene i at each time point t as the linear superposition of contributions from each of the TFs that regulates this gene. More precisely:
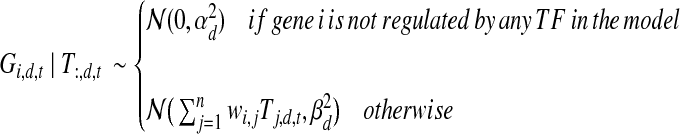 |
(1) |
where 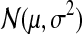 represents a Gaussian distribution with mean μ and variance σ2. In Equation 1, the expression profile of a gene over time is a noisy realization of the weighted sum of activity profiles of the TFs which regulate this gene. At each time point, the expression level is modeled using a Gaussian distribution whose variance is either
represents a Gaussian distribution with mean μ and variance σ2. In Equation 1, the expression profile of a gene over time is a noisy realization of the weighted sum of activity profiles of the TFs which regulate this gene. At each time point, the expression level is modeled using a Gaussian distribution whose variance is either 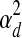 or
or 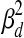 , depending on whether the gene is believed to be regulated by at least one TF. If it is regulated by at least one TF, then the variance
, depending on whether the gene is believed to be regulated by at least one TF. If it is regulated by at least one TF, then the variance 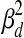 is used to represent experimental measurement noise. If PTMM cannot assign a regulator to a gene, it may be the case that the gene is regulated by TFs that are not included in the model. These genes are assumed to have a higher variance since some of their variance can be attributed to deficiencies in the model. Thus, we use a different variance,
is used to represent experimental measurement noise. If PTMM cannot assign a regulator to a gene, it may be the case that the gene is regulated by TFs that are not included in the model. These genes are assumed to have a higher variance since some of their variance can be attributed to deficiencies in the model. Thus, we use a different variance, 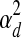 , for these genes.
, for these genes.
For each TF j, we maintain a global binary indicator Zj independent of experimental conditions and constant over time, indicating whether this TF is post-transcriptionally modified. Zj is a random variable following a Bernoulli distribution with parameter ρ. We treat ρ as a pre-specified constant representing the proportion of TFs that are post-transcriptionally modified. Based on this indicator, we assume that each TF follows one of these two models: (i) If TF j is not post-transcriptionally modified, i.e., Zj = 0, we model the activity profile of TF j as a noisy realization of its gene's expression profile with one time point lag (Fig. 1a; i.e., 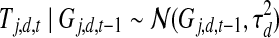 ).
). 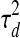 represents the possible experimental noise that may lead to slight differences between TF activity levels and mRNA levels. The one time point lag accounts for the time of translation from mRNA to protein. It also makes the model computationally sound, preventing possible loops in the time slice model (allowing, for example, self-regulation by TFs). The first time point in each dataset is modeled by a Gaussian distribution with zero mean and variance
represents the possible experimental noise that may lead to slight differences between TF activity levels and mRNA levels. The one time point lag accounts for the time of translation from mRNA to protein. It also makes the model computationally sound, preventing possible loops in the time slice model (allowing, for example, self-regulation by TFs). The first time point in each dataset is modeled by a Gaussian distribution with zero mean and variance 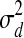 . (ii) The second option is that the TF j is post-transcriptionally modified (i.e., Zj = 1). For these TFs, the change in activity levels over time is modeled as a hidden Markov chain (Fig. 1b). The activity level of the TF at time point t (i.e., Tj,d,t) is dependent on the activity level of this same TF at time point t − 1 (i.e., Tj,d,t − 1). This dependency is modeled as a Gaussian random walk (i.e.,
. (ii) The second option is that the TF j is post-transcriptionally modified (i.e., Zj = 1). For these TFs, the change in activity levels over time is modeled as a hidden Markov chain (Fig. 1b). The activity level of the TF at time point t (i.e., Tj,d,t) is dependent on the activity level of this same TF at time point t − 1 (i.e., Tj,d,t − 1). This dependency is modeled as a Gaussian random walk (i.e., 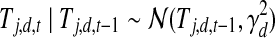 ). The variance
). The variance 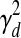 determines the likely amount of change in the TF's activity level between consecutive time points. The activity level of each TF at the very first time point in dataset d is modeled by a Gaussian distribution with mean 0 and variance
determines the likely amount of change in the TF's activity level between consecutive time points. The activity level of each TF at the very first time point in dataset d is modeled by a Gaussian distribution with mean 0 and variance 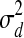 . This dataset-specific variance allows integrating multiple datasets in which the activity levels at the first time point for some TFs may differ from 0 (e.g., cell cycle experiments). Figure 1c presents the full graphical model of a PTMM, using indicator variables Zj to select between the two cases.
. This dataset-specific variance allows integrating multiple datasets in which the activity levels at the first time point for some TFs may differ from 0 (e.g., cell cycle experiments). Figure 1c presents the full graphical model of a PTMM, using indicator variables Zj to select between the two cases.
FIG. 1.

Graphical model representations for the following: (a) TFs with no post-transcriptional modification (Zj = 0), (b) post-transcriptionally modified TFs (Zj = 1), and (c) the general case (complete PTMM model). Observed variables are shaded. Tj,d,t is the (hidden) activity level of TF j at time point t in dataset d. Gi,d,t is the observed expression level for gene i at time point t in dataset d. The edge from TF j to gene i exists if and only if gene i is regulated by TF j, i.e., wi,j ≠ 0, where wi,j represents the weight of each edge. The edge from gene j to its protein product, TF j, exists when there is no post-transcriptional modification for TF j. Each TF j has a global binary indicator variable Zj, indicating whether the TF is post-transcriptionally modified. D plates correspond to the D datasets.
Note that within a dataset, the expression noise parameters 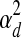 ,
, 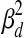 and
and 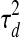 are shared across genes/TFs, and the TF activity level smoothness term
are shared across genes/TFs, and the TF activity level smoothness term 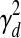 is shared across TFs. We estimate different noise parameters for each dataset d, to allow for the possibility that noise levels may differ across datasets from different labs using different array platforms. On the other hand, we assume the regulation between gene i and TF j is independent of experimental conditions. That is, the weight parameters wi,j are shared across all datasets.
is shared across TFs. We estimate different noise parameters for each dataset d, to allow for the possibility that noise levels may differ across datasets from different labs using different array platforms. On the other hand, we assume the regulation between gene i and TF j is independent of experimental conditions. That is, the weight parameters wi,j are shared across all datasets.
2.2. Penalized likelihood score
Given a set of TFs, a set of genes, and a collection of gene expression datasets, we train the PTMM by inferring which TFs are post-transcriptionally modified, the TFs activity levels, which genes are regulated by each TF, and by estimating the various PTMM parameters wi,j, αd, βd, τd, γd, and σd. These estimates are chosen to maximize a penalized complete log-likelihood score subject to the constraint that any gene be regulated by at most C TFs (i.e., it has at most C incoming edges). This constraint is motivated by the fact that recent high throughput studies find that most genes are regulated by only a few TFs (Harbison et al., 2004). The constrained penalized log-likelihood score is Score(o, h, z : W, θ), where o, h, z represent observed gene expression levels, hidden TF activity levels, and unobserved post-transcriptional modification (PTM) indicators, respectively, and θ includes all model parameters other than the regulation weights W.
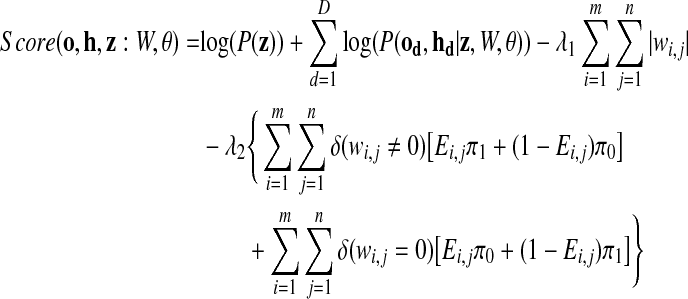 |
subject to:
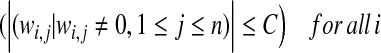 |
(2) |
Here od and hd are the observed expression levels for genes and the unobserved activity levels for TFs in dataset d, respectively. The score contains two regularization terms. The first imposes an L1 penalty on the weights, thereby encouraging most TF-gene regulation weights to be zero (Tibshirani, 1996). The second term incorporates prior knowledge obtained from earlier binding experiments. Ei,j is a binary indicator which is 1 if gene i is thought a priori to be bound by TF j and 0 otherwise. δ(·) is 1 if · is true, and 0 otherwise. π0 is a penalty term paid when the model selects a TF-gene edge weight that is inconsistent with prior assumptions (i.e., including an edge that is not assumed a priori, or excluding one that is). π1 is a smaller penalty term for using edges that are supported by prior assumptions from binding experiments, and for dropping edges that are not supported by binding experiments. Since we use highly trusted binding data to form our prior assumptions, we set 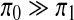 . Thus, the learned model is encouraged to assign wi,j weights consistent with prior knowledge, though it may depart from these priors if the incurred penalty is offset by improved data likelihood. Such departures might result from incompleteness or noise in prior binding datasets, or from the fact that only a subset of bound TFs may affect a target gene's expression (Hu et al., 2007). π1 and π0 are user defined and indicate confidence in the prior assumptions regarding the static binding data. λ1 and λ2 are constants representing the tradeoff between likelihood term and regularization terms, which can be used to control the tradeoff between precision and recall in predicting TF-gene regulatory relationships.
. Thus, the learned model is encouraged to assign wi,j weights consistent with prior knowledge, though it may depart from these priors if the incurred penalty is offset by improved data likelihood. Such departures might result from incompleteness or noise in prior binding datasets, or from the fact that only a subset of bound TFs may affect a target gene's expression (Hu et al., 2007). π1 and π0 are user defined and indicate confidence in the prior assumptions regarding the static binding data. λ1 and λ2 are constants representing the tradeoff between likelihood term and regularization terms, which can be used to control the tradeoff between precision and recall in predicting TF-gene regulatory relationships.
2.3. Inference and learning for PTMM
To learn the PTMM, we use an approximate EM algorithm to attempt to maximize Score(o, h, z : W, θ). The algorithm iteratively performs an E step in which the current model parameters W and θ are used to calculate the expected values of the hidden TF activity levels h and the most likely values of the unobserved PTM indicators z, followed by an M step in which these activity levels h and indicators z are used to re-estimate the model parameters W and θ. These two steps are iterated until convergence.
E step: Given all model parameters, we employ a generalized mean field algorithm (Xing et al., 2003) to iteratively infer the PTM indicators z and the hidden activity levels h of TFs. This variational inference method is based on non-overlapping clusters of random variables. Specifically in the PTMM, the hidden chain of activity levels for each TF forms a cluster and each PTM indicator forms an additional cluster. The E step iterates until convergence by inferring values for one cluster assuming the current values for all other clusters.
To infer the hidden activity levels for TF j given the most likely assignment of the PTM indicators z and expected activity levels for all other TFs, we first examine the current assignment of Zj: (i) If Zj = 0, i.e., TF j is not post-transcriptionally modified, we can compute the posterior distribution of Tj,d,t independently for each time point t in each dataset d. In this case, the prior of Tj,d,t is a Gaussian distribution whose mean is the expression level of its corresponding gene at time point t − 1 in dataset d, (i.e., Gj,d,t−1) and whose variance is 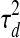 . The posterior of Tj,d,t depends on the observed expression levels of genes regulated by TF j as well as the hidden activity levels of other TFs, which have overlapping target genes due to the v-structure in directed graphical model (Pearl, 1988). Since we have fixed the activity levels for other TFs while inferring the level for TF j, we subtract out their assumed contributions to the observed expression levels of the target genes for TF j, in order to estimate the contribution due solely to TF j:
. The posterior of Tj,d,t depends on the observed expression levels of genes regulated by TF j as well as the hidden activity levels of other TFs, which have overlapping target genes due to the v-structure in directed graphical model (Pearl, 1988). Since we have fixed the activity levels for other TFs while inferring the level for TF j, we subtract out their assumed contributions to the observed expression levels of the target genes for TF j, in order to estimate the contribution due solely to TF j:
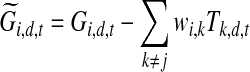 |
(3) |
where 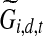 is the adjusted expression level which represents the inferred contribution of TF j to the expression level for gene i at time point t in dataset d. The posterior of Tj,d,t depends only on these adjusted expression levels. This posterior is a Gaussian distribution due to the conjugacy of Gaussian distribution to itself and it is straightforward to obtain the posterior mean and variance. (ii) If Zj = 1, i.e., TF j is post-transcriptionally modified, we first calculate the adjusted expression levels for the genes regulated by TF j. Given these adjusted expression levels the posterior for the activity level of TF j is no longer dependent on the activity levels of other TFs and the resulting model is equivalent to a single hidden Markov chain for TF j regulating multiple genes with their adjusted expression levels. Let
is the adjusted expression level which represents the inferred contribution of TF j to the expression level for gene i at time point t in dataset d. The posterior of Tj,d,t depends only on these adjusted expression levels. This posterior is a Gaussian distribution due to the conjugacy of Gaussian distribution to itself and it is straightforward to obtain the posterior mean and variance. (ii) If Zj = 1, i.e., TF j is post-transcriptionally modified, we first calculate the adjusted expression levels for the genes regulated by TF j. Given these adjusted expression levels the posterior for the activity level of TF j is no longer dependent on the activity levels of other TFs and the resulting model is equivalent to a single hidden Markov chain for TF j regulating multiple genes with their adjusted expression levels. Let 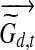 denote the m-dimensional column vector for adjusted expression levels of all genes in dataset d at time point t. We can then write for TF j:
denote the m-dimensional column vector for adjusted expression levels of all genes in dataset d at time point t. We can then write for TF j:
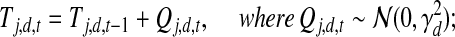 |
(4) |
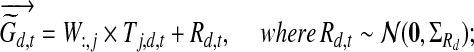 |
(5) |
where W is the m-by-n regulation weight matrix (0 indicates no edge) and W:,j represents the jth column of this matrix corresponding to the regulation weights associated with TF j. Here 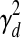 determines the probable rate of change of the TF activities over time (i.e., Qj,d,t). ΣRd is a m-by-m diagonal matrix where the ith diagonal element is
determines the probable rate of change of the TF activities over time (i.e., Qj,d,t). ΣRd is a m-by-m diagonal matrix where the ith diagonal element is 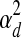 if wi,j = 0 for all j and
if wi,j = 0 for all j and 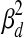 otherwise. It determines the variance in the noise in the observed expression levels (i.e., Rd,t).
otherwise. It determines the variance in the noise in the observed expression levels (i.e., Rd,t).
As Equations 4 and 5 show, for post-transcriptionally modified TFs, when the parameters are known, the model reduces to a special case of Kalman filter (Murphy, 2002) model with one-dimensional hidden chain. Inference on it can be done efficiently by computing the posterior of hidden variables Tj,d,t. The probabilities are all Gaussian distributed, and the computation is tractable because of the conjugacy of the Gaussian distribution.
Inferring the unobserved assignment of the PTM indicator Zj of TF j is in fact a model selection process. Given the inferred expected values of activity levels of TF j, we examine which model explains this TF better. That is, if the no PTM model (Fig. 1a) has a higher likelihood than the PTM model (Fig. 1b) we assign Zj as 0. Otherwise, we assign Zj as 1. This likelihood can be easily computed as the product of the local conditional probabilities of all nodes associated with TF j. Along with the assignment of each indicator, we can also output a confidence score for this assignment which we define as the log ratio between the likelihood scores of two different models.
Before the iterations in the E step, we need to initialize the hidden variables in the PTMM. The activity levels of TFs are initialized by a standard Kalman filter model assuming all TFs are post-transcriptionally modified, and the PTM indicators for TFs are initialized by computing the correlation between the initialized TFs' activity profiles and their corresponding genes' expression profiles and setting the less correlated TFs to be post-transcriptionally modified.
M step: Given the expected activity levels of TFs and the most likely assignment of PTM indicators inferred in the E step, we learn new parameters by attempting to maximize the Score function subject to the constraint discussed above (number of TFs for each gene). We can calculate exact solutions for the variance terms γ, σ and τ by zeroing the partial derivatives of the penalized complete log-likelihood of data defined in Equation 2. We also calculate maximum likelihood estimates for α and β by fixing the regulation weights W.
Unlike the noise parameters the weight parameters W cannot be computed in closed form because of the limit on the number of incoming edges for each gene. Instead, we first conduct a greedy search to associate TFs with each gene, and then solve an optimization problem to obtain estimates for W. To find the optimal set of regulating TFs for each gene i, the algorithm first computes the penalized likelihood score for the case that gene i is not regulated by any TF (wi,j = 0 for all j). It next adds one TF at a time up to C. Assume we have selected a set c of TFs for gene i (|c| < C). To determine whether we should add to the set c, we loop over all TFs j where j ∉ c. For each such TF, we create the set cj = c ∪{j} and solve the following optimization problem which is equivalent to maximizing the penalized likelihood score using TFs in cj:
for gene i, minimize:
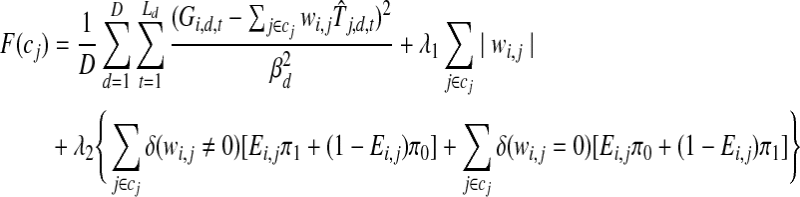 |
where Ld is the number of time points in dataset d and 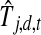 is the inferred (from the E step) expected activity level of TF j at time t in dataset d. All other notations are adopted from Equation 2. This optimization problem is solved using a subspace trust region method (Coleman and Li, 1996). (We use the fminunc method in MATLAB.)
is the inferred (from the E step) expected activity level of TF j at time t in dataset d. All other notations are adopted from Equation 2. This optimization problem is solved using a subspace trust region method (Coleman and Li, 1996). (We use the fminunc method in MATLAB.)
We choose the TF j that minimizes F(cj) among all TFs not in c. If F(cj) < F(c), we set c = c ∪{j} and repeat the above search. Otherwise we assign to gene i all TFs in c using weights computed from the solution to the optimization problem for c. All other weights are set to 0. If we reach a set c containing C TFs, we stop and assign these to the gene. This algorithm searches for the optimal TFs for each gene independently and can be easily parallelized.
2.4. ChIP-chip experiments
A Yeast strain harboring a 9myc tagged version of Gcn4 (Lee et al., 2002) was grown to early logarithmic phase and transferred to heat shock at 37°C for 20 min in a manner similar to that of Gasch et al. (2000). Chromatin was cross-linked by Formaldehyde, and ChIP was performed in a manner similar to that of Lee et al. (2002). Products were cleaned by the PCR cleanup system (Promega) and then amplified using the WGA system (Sigma). 100 ng was taken, randomly primed, and incubated with Klenow enzyme and amino-allyl-dUTP over night. Products were cleaned by the microcon system (Millipore). Products were purified after coupling on Qiaquick PCR purification kit (Qiagen), and hybridized to a spotted glass microarray, which was printed with all intergenic regions of Saccharomyces cerevisiae as polymerase chain reaction (PCR) products (the array design is similar to the one used by Lee et al. [2002]). The arrays were scanned by an Axon-4000B scanner and analyzed using the Axon-pro software. The hybridization values were put into the error model designed by Lee et al. (2000) and normalized using the Lowess function (Matlab software). Complete results are available to download from the supporting website: www.sb.cs.cmu.edu/PTMM/PTMM.html.
3. Experiments and Results
We tested PTMM's performance on both simulated and real gene expression time series data. Using simulated data we show that our algorithm can indeed recover the hidden activity levels of TFs and determine whether a TF is post-transcriptionally modified. Using real data, we show that by PTMM we can reconstruct meaningful TF activity profiles, detect known post-transcriptionally regulated TFs and improve the ability to determine TF-gene regulatory relationships. We also compared PTMM on both simulated and real expression data with two methods representing the two approaches mentioned in the introduction:
Kalman Filter model (KF): This model corresponds to methods that assume that TF activity levels can only be inferred from the expression levels of its regulated genes. For this we set all PTM indicators in PTMM to 1. Thus, PTMM reduces to a Kalman filter model. We can infer the hidden activity levels of TFs efficiently by standard inference method for Kalman filter (Murphy, 2002).
No Post-transcriptional modification model (NP): This model corresponds to methods that use a TF's expression levels to infer its activity levels, i.e., all PTM indicators are fixed to 0 in PTMM.
For comparison of predicting PTM indictors we use a post-processing step for both methods in which we compute the Pearson correlation between the inferred activity profile for each TF and the expression profile of its corresponding gene. A cutoff is applied to turn this correlation score to binary PTM indicators.
The maximum number C of regulating TFs for one gene was set to three in all experiments below.
3.1. Results on simulated data
We first synthesized n TF activation profiles using a random walk model and used a noisy version of these profiles as the mRNA levels for the TFs. Next we randomly selected P percent of TFs and set them to be post-transcriptionally modified. For these TFs, we replaced their mRNA level with Gaussian noise with mean 0 (though we kept their activity levels and used it to generate the profiles for the regulated genes). Finally we randomly generated a TF-gene regulation weight matrix. We used the TF activity levels and the weight matrix to generate the observed expression values for all genes and added i.i.d. random noise to each time point for each gene.
We varied the percentage of TFs that are post-transcriptionally modified. For all cases we sampled n = 100 TFs and m = 1000 genes. For noise parameters we used the values learned from real data to make the simulation realistic. The prior constants on evidences were set to π0 = 0.7 and π1 = 0.3 by cross validation.
Figure 2a presents the mean squared error between the true and inferred TF activity profiles for each of the methods. As can be seen, PTMM consistently outperformed all other methods. In all three cases, NP cannot capture the underlying activity profiles as accurately as the two other models, because it tends to predict activity levels of post-transcriptionally modified TFs as their mRNA expression levels which significantly compromises its performance. KF ignores all information from a TF's expression levels and infers its activity levels solely from its regulated genes. It has better performance than NP because of the way we constructed the expression levels (linear combination of the activity levels of TFs). However, since it ignores useful data for many TFs (mRNA levels), the reconstructed profiles are still not as good as PTMM.
FIG. 2.

Simulated data results. (a) Mean squared error (MSE) between actual and inferred hidden activity levels of TFs. The three plots correspond to different percentages of post-transcriptionally modified TFs (5%, 10%, and 20%). Gray line is the median. Black box indicates upper and lower quantiles. The black bars are the range of the MSE. Outliers are plotted by “+.” (b) Comparison of precision-recall curves for predicting the PTM indicators. (c) Comparison of precision-recall curves for predicting TF-gene regulatory relationships.
Figure 2b shows precision-recall curves of the prediction of PTM indicators. The precision-recall curves were drawn by increasing the cutoff for the prediction confidence scores (for PTMM) or correlation coefficients (for KF and NP). Again, PTMM outperformed other two methods.
We also tested the ability of all methods to predict the regulatory relationships between TFs and genes by eightfold cross-validation. Since the regulation weights outputted by all methods are continuous values, we applied a cutoff to turn the regulation weights into binary predictions of regulation. Figure 2c shows the precision-recall curves of regulatory relationship prediction by all three methods. Interestingly, the curve for NP starts higher indicating that this model is very powerful at identifying TF-gene interactions when mRNA levels correspond to activity levels. However, the KF model is more general and applies to both transcriptionally and post-transcriptionally regulated TF. Thus the KF curve crosses the NP curve at a recall of 35%. Our method that utilize both sources of information acts as the best of both methods. It starts out very strong (similar to NP method) but unlike NP method it remains strong for higher recall rates as well.
3.2. Yeast expression data
We applied PTMM to Saccharomyces cerevisiae microarray time series data collected under 17 experimental conditions, including various stresses, cell cycle, and DNA damage (see supporting website for complete list: www.sb.cs.cmu.edu/PTMM/PTMM.html). The number of time points in these datasets was 8–24. To construct the prior binding evidence matrix E, we used protein-DNA binding data from MacIsaac et al. (2006). Their binding data offers a list of ORFs with binding sites for each transcription factor at various binding and conservation thresholds. We only used the most confident data where binding p-value is less than 0.001 and motifs are conserved in at least two additional species. We removed TFs that had less than five known targets and TFs that had more than 50% missing expression values in at least one dataset. The remaining 72 TFs were used for the analysis we discuss below. We set the prior knowledge π0 = 0.7, π1 = 0.3 indicating our belief in the high quality binding data. We tested our approach using cross-validation. Thus we also removed all genes that are not known to be bound by any of the 72 modeled TFs. This leaves 1069 genes.
3.2.1. Predicting TF-gene regulatory relationships
We first tested the ability of PTMM to predict TF-gene regulatory relationships by performing eightfold cross-validation. In each fold, we removed the associations of 1/8 of the 1069 genes (i.e., set the corresponding entries in the evidence matrix E to zero) and used all three methods to predict the regulatory relationships for these genes. Again, by varying a cutoff w0 for all edges between genes and TFs, we can generate precision-recall curves for all methods.
Figure 3a presents these curves of all three methods. The results are qualitatively similar to the simulated data results. The NP method starts strong but drops rapidly. The KF method that does not rely on TF expression level is more robust and holds for longer recall rates. Nicely, our method dominates both other methods indicating that it is indeed possible to combine both approaches for modeling regulatory networks. Note that since each gene can only be assigned to up to three out of 72 TFs, a precision rate of close to 50% is quite impressive. Also, it is important to remember that most ChIP-chip experiments were carried out in YPD, whereas the expression data we used is primarily from stress conditions. Thus, some of our false positive might be actually correct prediction and the reason they were not identified before is due to the condition under which the experiments were carried out. This also affects the recall rate, which is low for all methods. Another possible reason for the low recall rate is the protein-DNA binding data combined with strict conservation standard, which makes our prior knowledge arguably incomplete. In fact, we can use the constants λ1 and λ2 to control the level of tradeoff between the precision and recall. Figure 3b shows the precision-recall curves in predicting the TF-gene regulatory relationships when setting both λ1 and λ2 to 0.2. As can be seen, using these values the recall rate substantially improved though the precision drops. Again, the curve of PTMM outperforms the other two methods. In both plots (Fig. 3a,b), the fact that higher weight correlates well with correct TF-gene associations indicates that the recovered TF activity profiles are a good representation of the underlying profiles.
FIG. 3.

Results for yeast expression data. Precision and recall curves for recovering TF-gene interactions in cross-validation studies. The tradeoff constants used are λ1 = 0.3, λ2 = 0.3 (a) and λ1 = 0.2, λ2 = 0.2 (b).
To test whether more data can improve the performance of our algorithm, we measured precision-recall curves using different numbers of datasets. Figure 4a shows four curves corresponding to the performance of PTMM with 1, 4, 8, and 16 datasets. Indeed, more datasets improved both precision and recall. Figure 4b shows the penalized likelihood scores versus the number of iterations. As can be seen, the score converges quickly, reaching a (local) maximum after only a few iterations. Note that, while this convergence may seem fast, it is a direct result of the fact that we are initializing our model with known TF-gene binding data rather than random initializations, which are common in many EM applications. Figure 4c presents the precision-recall curves for setting the maximum number C of associated TFs for each gene to be 3 and 4. As can be seen, setting C bigger does not help in improving the coverage of PTMM.
FIG. 4.

Results for yeast expression data. (a) The precision-recall curves as a function of the number of expression datasets used for PTMM, ranging from 1 to 16 datasets (DS). (b) The penalized likelihood score curve for PTMM versus the number of iterations. (c) Comparison of the precision-recall curves when setting maximum number C of associated TFs for each gene to be 3 and 4.
3.2.2. Insights into post-transcriptional modifications
Of the 72 TFs, PTMM determined that seven are post-transcriptionally modified in at least some of the conditions we looked at. We found strong indications for differences between the transcript level and the activity level of five factors (Gcn4, Msn4, Swi5, Fkh2, Rap1). Most of these factors are known to be regulated post-transcriptionally. For example, the master regulator of amino acid starvation, Gcn4, is regulated at the translational level (Ernst et al., 2007). The stress response TF, Msn4, is regulated at the post-transcriptional level by phosphorylation (Garreau et al., 2000) and by nucleus localization (Gorner et al., 1998; Beck and Hall, 1999). Interestingly, we found that the Msn4 activity follows its mRNA level in stress conditions but differs from them during the cell cycle (Fig. 5a). This observation suggests that the main role of the PTMs is to prevent undesired activation of the stress response pathway. During stress when Msn4 activity is desired, it is induced and activated, whereas in other conditions such as the cell cycle, when its activity may be harmful to cell, it is kept in a silent form and fluctuations in its mRNA level seem to be not important. The cell cycle regulators, Swi5 and Fkh2, are regulated by phosphorylation which is important for the nucleus localization of Swi5 (Moll et al., 1991) and for the activity of Fkh2 (Pic-Taylor et al., 2004). Finally, it has been shown that the phosphorylation of Rap1 (Fig. 5b) affects its binding to DNA (Tsang et al., 1990). The other two factors are Met32 and Mbp1. For Met32, we could not confirm, nor reject the prediction. The last TF predicted to be post-transcriptionally regulated is Mbp1, which is known to be transcriptionally regulated in at least some conditions (Spellman et al., 1998) and may be a false positive result. See supporting website for more figures: www.sb.cs.cmu.edu/PTMM/PTMM.html.
FIG. 5.

The observed mRNA expression levels (black) versus TF activity levels (gray) inferred by PTMM for all 17 experimental conditions (separated by dashed lines) for Msn4 (a) and Rap1 (b).
3.2.3. Validating predictions regarding TF activity levels
To further analyze the predictions regarding TF activity levels, we have looked at a specific condition, yeast response to methyl-methanesulfonate stress (MMS). For this condition, we have both time series expression data (by which we use to make predictions), as well as new ChIP-chip data (Workman et al 2006), which we did not use to learn the model. In their article, Workman et al., (2006) classified each TF they tested as expanding in MMS (regulating more genes when compared to general growth conditions), contracting, or not changing. Thus, we can use this new interaction data to determine whether the predictions made by our method agree with the activity observed by the ChIP-chip experiment, and by the mRNA levels of the TF.
We present results for 24 TFs in Table 1. Overall, we see a good agreement between the predicted activity levels and the observed binding profiles. For example, Pdr1, Uga3, and Dal81 are expanded in MMS, suggesting that they are active. This is accurately predicted by PTMM. Interestingly, PTMM predicts several TFs to have activities that differ from their mRNA level. For example, even though Yap5 is not identified as post-transcriptionally modified, its activity level for this condition is accurately predicted to be lower than its normal level. In contrast, its expression level is actually higher than baseline. PTMM's prediction for Yap5 is validated by the MMS ChIP-chip data, which shows that Yap5 is contracting. Similarly, Swi5 is also predicted to be inactive by PTMM, which is validated by its contracting status. Another example is Gcn4. While the expression of Gcn4 is slightly lower than baseline, PTMM predicts that this factor is post-transcriptionally modified and its activity increases in MMS. While Workman et al. (2006) did not find Gcn4 to be expanding, a more recent study (Ernst et al., 2007) experimentally tested Gcn4's binding in a more appropriate time point (15 min following MMS treatment) and found that Gcn4 greatly expands in MMS as predicted by PTMM. While PTMM was successful in several cases, there are cases where the predictions differed from the MMS ChIP-chip data, including Ino4 and Rim101, which were both expanding in ChIP-chip experiments, though PTMM predicted that they remain unchanged.
Table 1.
Summary of Agreement between Predicted and Observed TF Activity Level in MMS
| TF | Confa | ChIp-chipb | Postc | Activityd | Expressione |
|---|---|---|---|---|---|
| Pdr1 | 213 | e | 0 | ∧ | ∧ |
| Ino4 | 1439 | e | 0 | – | – |
| Rim101 | 1649 | e | 0 | – | – |
| Uga3 | 7065 | e | 0 | ∧ | ∧ |
| Dal81 | 0 | e | 0 | ∧ | ∧ |
| Mcm1 | 1665 | n | 0 | ∧ | ∧ |
| Swi4 | 798 | n | 0 | – | – |
| Cin5 | 1516 | n | 0 | – | – |
| Sok2 | 1289 | n | 0 | – | – |
| Fkh2 | 275 | n | 1 | – | ∨ |
| Yap5 | 1396 | n | 0 | ∧ | ∧ |
| Ixr1 | 1763 | n | 0 | – | – |
| Ndd1 | 1302 | n | 0 | ∨ | ∨ |
| Ace2 | 42 | n | 0 | ∨ | ∨ |
| Adr1 | 1706 | n | 0 | ∨ | ∨ |
| Gcn4 | 488 | n | 1 | ∧ | ∨ |
| Sko1 | 993 | n | 0 | ∨ | ∨ |
| Msn4 | 326 | c | 1 | ∧ | ∧ |
| Ash1 | 1975 | c | 0 | ∨ | ∨ |
| Swi5 | 494 | c | 1 | ∨ | – |
| Hsf1 | 336 | c | 0 | – | – |
| Rtg3 | 1501 | c | 0 | ∧ | ∧ |
| Yap5 | 1396 | c | 0 | ∨ | ∧ |
Confidence score given by PTMM.
(e)xpanding, (c)ontracting, or (n)either, according to ChIp-chip experiment in Workman et al.
PTMM result. 0 for factors determined to be transcriptionally regulated, 1 for post-transcriptionally modified.
Activity levels inferred by PTMM.
Observed gene expression level.
For d and e, up (∧), down (∨) or no obvious tendency (–).
TF, transcription factor; MMS, methyl-methanesulfonate stress; PTMM, Post-Transcriptional Modification Model.
3.3. Chip-ChIp experimental validation
As can be seen in Figure 6a, while the observed expression of Gcn4 under heat shock condition is repressed, its predicted activity levels are induced, suggesting that Gcn4 tends to bind more targets under heat shock condition. To validate this hypothesis, we conducted new Chip-ChIp experiments as described before. Figure 6b shows the numbers of bound genes under YPD condition and under two repeats of heat shock condition in our experiments using a p-value of 0.005 as cutoff. As shown in Figure 6b, the number of bound genes under heat shock condition is significantly larger than under YPD condition, supporting the prediction result by PTMM. The overlap between the two heat shock (HS) repeats is 67 (out of 150 bound genes in each repeat), which is significant with a p-value of 0. To test how this overlap compares to the overlap between HS and YPD, we looked at the average overlap between HS and YPD at two different p-value cutoffs: 0.005 and 0.001. For 0.005, we see an average overlap of 64 (less than the 67 genes found for the two HS repeats). The difference is even more pronounced when we use a stricter p-value, which leads to more accurate (though lower coverage) results. At a p-value of 0.001, the overlap between the two HS repeats is 39 (again, a p-value of 0). This is 50% higher than the average overlap between the HS repeats and YPD (26). Thus, both the p-value analysis and the overlap analysis support our claim of a higher activation level for GCN4 in HS. See supporting website for complete ChIP-chip results: www.sb.cs.cmu.edu/PTMM/PTMM.html.
FIG. 6.

(a) Expression levels (black) and predicted activity levels (gray) of Gcn4 under heat shock condition. (b) Number of bound genes of Gcn4 under two repeats of heat shock condition (HS1, HS2) and YPD condition.
4. Discussion
To date, researchers chose one of two ways to determine the activity levels of TFs when modeling regulatory networks. The first used TF expression levels and the second relied on the levels of genes it regulates. In this article, we have developed a method that utilizes both sources of data for this task. Our method uses a switching model to determine whether a TF is transcriptionally or post-transcriptionally regulated. This model is combined with a factorial HMM to fully model interactions in a dynamic regulatory network.
Factorial HMMs and variants of dynamic Bayesian networks have been suggested in the past for modeling regulatory networks (Nachman et al., 2004), for modeling the activity of neurons in the brain (Mitchell et al., 2006), and for determining functional GO annotations in time series expression experiments (Shi et al., 2007). However, the ability to use both interaction and expression data to model the activity of the hidden layer in these models is a novel aspect of PTMM. As we show using simulated and real expression data, this allows our method to combine the best of both worlds. PTMM outperforms other methods when comparing their ability to predict new TF-gene interactions. Many of the factors predicted to be post-transcriptionally regulated are validated by prior knowledge. Our method is also successful in predicting TF activity in a new condition.
In the future, we would like to further extend this model so that it can utilize additional biological data sources. For example, we would like to use protein-protein interaction to try to explain the activity levels of post-transcriptionally modified TFs. We would also like to apply this method to other species in order to predict new interactions and the mode of activation of TFs.
Acknowledgments
This work was supported in part by the NIH (grant 1RO1 GM085022) and the NSF (Career award 0448453 to Z.B.J.).
Disclosure Statement
No conflicting financial interests exist.
References
- Arbeitman M. Furlong E. Imam F., et al. Gene expression during the life cycle of drosophila melanogaster. Science. 2002;298:2270–2275. doi: 10.1126/science.1072152. [DOI] [PubMed] [Google Scholar]
- Beck T. Hall M. The TOR signalling pathway controls nuclear localization of nutrient-regulated transcription factors. Nature. 1999;402:689–692. doi: 10.1038/45287. [DOI] [PubMed] [Google Scholar]
- Beer M. Tavazoie S. Predicting gene expression from sequence. Cell. 2004;117:185–198. doi: 10.1016/s0092-8674(04)00304-6. [DOI] [PubMed] [Google Scholar]
- Bose S. Dutko J. Zitomer R. Genetic factors that regulate the attenuation of the general stress response of yeast. Genetics. 2005;169:1215–1226. doi: 10.1534/genetics.104.034603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman T.F. Li Y. An interior trust region approach for nonlinear minimization subject to bounds. SIAM J. Optim. 1996;6:418–445. [Google Scholar]
- D'haeseleer P. Liang S. Somogyi R. Genetic network inference: from co-expression clustering to reverse engineering. Bioinformatics. 2000;16:707–726. doi: 10.1093/bioinformatics/16.8.707. [DOI] [PubMed] [Google Scholar]
- Ernst J. Vainas O. Harbison C., et al. Reconstructing dynamic regulatory maps. Nat. EMBO Mol. Syst. Biol. 2007;3:74. doi: 10.1038/msb4100115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garreau H. Hasan R.N. Renault G., et al. Hyperphosphorylation of Msn2p and Msn4p in response to heat shock and the diauxic shift is inhibited by cAMP in Saccharomyces cerevisiae. Microbiology. 2000;146:2113–2120. doi: 10.1099/00221287-146-9-2113. [DOI] [PubMed] [Google Scholar]
- Gasch A. Spellman P. Kao C., et al. Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geier F. Timmer J. Fleck C. Reconstructing gene-regulatory networks from time series, knock-out data, and prior knowledge. BMC Syst. Biol. 2007;1:11. doi: 10.1186/1752-0509-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghahramani Z. Jordan M. Factorial hidden Markov models. Mach. Learn. 1997;29:245–273. [Google Scholar]
- Gorner W. Durchschlag E. Martinez-Pastor M. T., et al. Nuclear localization of the C2H2 zinc finger protein Msn2p is regulated by stress and protein kinase A activity. Genes Dev. 1998;12:586–597. doi: 10.1101/gad.12.4.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbison C.T. Gordon B.D. Lee T.I., et al. Transcriptional regulatory code of a eukaryotic genome. Nature. 2004;431:99–104. doi: 10.1038/nature02800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z. Killion P. Iyer V. Genetic reconstruction of a functional transcriptional regulatory network. Nat. Genet. 2007;39:683–687. doi: 10.1038/ng2012. [DOI] [PubMed] [Google Scholar]
- Ideker T. Thorsson V. Ranish J.A., et al. Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science. 2001;292:929–934. doi: 10.1126/science.292.5518.929. [DOI] [PubMed] [Google Scholar]
- Kannan A. Emili A. Frey B. A Bayesian model that links microarray mRNA measurements to mass spectrometry protein measurements. Proc. RECOMB. 20072007:325–338. [Google Scholar]
- Lee T. Rinaldi N. Robert F., et al. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002;298:799–804. doi: 10.1126/science.1075090. [DOI] [PubMed] [Google Scholar]
- MacIsaac K. Wang T. Gordon D., et al. An improved map of conserved regulatory sites for Saccharomyces cerevisiae. BMC Bioinform. 2006;7:113. doi: 10.1186/1471-2105-7-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell T. Hutchinson R. Rustandi I. Hidden process models. Proc. ICML. 20062006:433–440. [Google Scholar]
- Moll T. Tebb G. Surana U., et al. The role of phosphorylation and the CDC28 protein kinase in cell cycle-regulated nuclear import of the S. cerevisiae transcription factor SWI5. Cell. 1991;66:743–758. doi: 10.1016/0092-8674(91)90118-i. [DOI] [PubMed] [Google Scholar]
- Murphy K. Dynamic bayesian networks: representation, inference and learning. [Ph.D. dissertation] University of California, Berkeley. 2002.
- Nachman I. Regev A. Friedman N. Inferring quantitative models of regulatory networks from expression data. Bioinformatics. 2004;20(Suppl 1):I248–I256. doi: 10.1093/bioinformatics/bth941. [DOI] [PubMed] [Google Scholar]
- Nau G. Richmond J. Schlesinger A., et al. Human macrophage activation programs induced by bacterial pathogens. Proc. Natl. Acad. Sci. USA. 2002;99:1503–1508. doi: 10.1073/pnas.022649799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda S. Antoch M. Miller B., et al. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell. 2002;109:307–320. doi: 10.1016/s0092-8674(02)00722-5. [DOI] [PubMed] [Google Scholar]
- Pearl J. Probabilistic Reasoning in Intelligent Systems: Networks of Plausible Inference. Morgan Kaufmann; New York: 1988. [Google Scholar]
- Pic-Taylor A. Darieva Z. Morgan B.A., et al. Regulation of cell cycle-specific gene expression through cyclin-dependent kinase-mediated phosphorylation of the forkhead transcription factor Fkh2p. Mol. Cell. Biol. 2004;24:10036–10046. doi: 10.1128/MCB.24.22.10036-10046.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers S. Khanin R. Girolami M. Bayesian model-based inference of transcription factor activity. BMC Bioinform. 2007;8:S2. doi: 10.1186/1471-2105-8-S2-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustici G. Mata J. Kivinen K., et al. Periodic gene expression program of the fission yeast cell cycle. Nat. Genet. 2004;36:809–817. doi: 10.1038/ng1377. [DOI] [PubMed] [Google Scholar]
- Sabatti C. James G.M. Bayesian sparse hidden components analysis for transcription regulation networks. Bioinformatics. 2006;22:739–746. doi: 10.1093/bioinformatics/btk017. [DOI] [PubMed] [Google Scholar]
- Segal E. Shapira M. Regev A., et al. Module networks: identifying regulatory modules and their condition-specific regulators from gene expression data. Nat. Genet. 2003;34:166–176. doi: 10.1038/ng1165. [DOI] [PubMed] [Google Scholar]
- Shi Y. Klustein M. Simon I., et al. Continuous hidden process model for time series expression experiments. Bioinformatics. 2007;23:i459–i467. doi: 10.1093/bioinformatics/btm218. [DOI] [PubMed] [Google Scholar]
- Sohler F. Zimmer R. Identifying active transcription factors and kinases from expression data using pathway queries. Bioinformatics. 2005;21:ii115–ii122. doi: 10.1093/bioinformatics/bti1120. [DOI] [PubMed] [Google Scholar]
- Spellman P.T. Sherlock G. Zhang M.Q., et al. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol. Biol. Cell. 1998;9:3273–3297. doi: 10.1091/mbc.9.12.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanay A. Shamir R. Computational expansion of genetic networks. Bioinformatics. 2001;17:S270–S278. doi: 10.1093/bioinformatics/17.suppl_1.s270. [DOI] [PubMed] [Google Scholar]
- Theuns J. Van Broeckhoven C. Transcriptional regulation of Alzheimer's disease genes: implications for susceptibility. Hum. Mol. Genet. 2000;9:2383–2394. doi: 10.1093/hmg/9.16.2383. [DOI] [PubMed] [Google Scholar]
- Tibshirani R. Regression shrinkage and selection via the lasso. J. R. Statist. Soc B. 1996;58:267–288. [Google Scholar]
- Tsang J.S. Henry Y.A., et al. Phosphorylation influences the binding of the yeast RAP1 protein to the upstream activating sequence of the PGK gene. Nucleic Acids Res. 1990;18:7331–7337. doi: 10.1093/nar/18.24.7331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L. Chen G. Li H. Group SCAD regression analysis for microarray time course gene expression data. Bioinformatics. 2007;23:1486–1494. doi: 10.1093/bioinformatics/btm125. [DOI] [PubMed] [Google Scholar]
- Washburn M.P. Koller A. Oshiro G., et al. Protein pathway and complex clustering of correlated mRNA and protein expression analyses in Saccharomycs cerevisiae. Proc. Natl. Acad. Sci. USA. 2003;100:3107–3112. doi: 10.1073/pnas.0634629100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman C.T. Mak H.C. McCuine S., et al. A systems approach to mapping DNA damage response pathways. Science. 2006;312:1054–1059. doi: 10.1126/science.1122088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing E. Jordan M. Russell S. A generalized mean field algorithm for variational inference in exponential families. Proc. UAI. 20032003:583–591. [Google Scholar]
- Zou M. Conzen S.D. A new dynamic Bayesian network (DBN) approach for identifying gene regulatory networks from time course microarray data. Bioinformatics. 2005;21:71–79. doi: 10.1093/bioinformatics/bth463. [DOI] [PubMed] [Google Scholar]