

Abstract
The endocannabinoids and their attending CB1 cannabinoid receptors have been implicated in the control of cognition, but their possible roles in dementias are still unclear. In the present study, we used liquid chromatography/mass spectrometry to conduct an endocannabinoid-targeted lipidomic analysis of post mortem brain samples from 38 Alzheimer’s disease (AD) patients and 17 control subjects, matched for age and post mortem interval. The analysis revealed that midfrontal and temporal cortex tissue from AD patients contains, relative to control subjects, significantly lower levels of the endocannabinoid anandamide and its precursor 1-stearoyl, 2-docosahexaenoyl-sn-glycero-phosphoethanolamine-N-arachidonoyl (NArPE). No such difference was observed with the endocannabinoid 2-arachidonoyl-sn-glycerol or 15 additional lipid species. In AD patients, but not in control subjects, statistically detectable positive correlations were found between (a) anandamide content in midfrontal cortex and scores of the Kendrick’s digit copying test (P=0.004, r=0.81; n=10), which measures speed of information processing; and (b) anandamide content in temporal cortex and scores of the Boston naming test (P=0.027, r=0.52; n=18), which assesses language facility. Furthermore, anandamide and NArPE levels in midfrontal cortex of the study subjects inversely correlated with levels of the neurotoxic amyloid peptide, Aβ42, while showing no association with Aβ40 levels, amyloid plaque load or tau protein phosphorylation. Finally, high endogenous levels of Aβ42 in APPSWE/Neuro-2a cells directly reduced anandamide and NArPE concentrations in cells lysates. The results suggest that an Aβ42-dependent impairment in brain anandamide mobilization contributes to cognitive dysfunction in AD.
Keywords: endocannabinoid, anandamide, amyloid β42, cognitive dysfunction, Alzheimer’s disease, human brain, lipidomics
1. Introduction
The endocannabinoids are lipid-derived mediators that participate in the control of neurotransmission. The two major endocannabinoids identified so far, anandamide and 2-arachidonoyl-sn-glycerol (2-AG), are released on demand through activity- or receptor-dependent hydrolysis of membrane phospholipid precursors, and act on presynaptic CB1-type cannabinoid receptors to inhibit neurotransmitter release (Piomelli et al., 2007; Katona and Freund, 2008). They regulate synaptic plasticity in various regions of the brain through both short-term and long-term mechanisms, which include depolarization-induced suppression of inhibition at hippocampal GABAergic synapses and long-term depression at cortical or accumbal glutamatergic synapses (Hashimotodani, 2007; Chevaleyre et al., 2006; Mato et al., 2008).
Anandamide and 2-AG are produced through two separate enzymatic pathways (Astarita and Piomelli, 2009; Piomelli, 2003). Anandamide formation starts with the transfer of arachidonic acid from phosphatidylcholine to phosphatidylethanolamine (PE), which generates a diverse group of N-arachidonoyl-substituted PE species (NArPE) (Piomelli, 2003). Anandamide is released by the hydrolysis of NArPE, which requires either a NAPE-specific phospholipase D (PLD) (Okamoto et al., 2004) or the sequential actions of phospholipase C and PTPN22 phosphatase (Liu et al., 2006). After release, anandamide is internalized by neurons and glia and then degraded by intracellular fatty acid amide hydrolase (FAAH) (Piomelli, 2003).
Evidence indicates that endocannabinoid signaling is involved in the regulation of normal cognition. Pharmacological or genetic blockade of CB1 receptors in mice impairs short- and long-term memory extinction (Lutz, 2007; de Oliveira Alvares et al., 2008; Marsicano et al, 2002) while disruption of FAAH-mediated anandamide hydrolysis enhances extinction in mice (Varvel et al., 2007) and improves memory consolidation in rats (Mazzola et al., 2009). Additionally, mutant mice lacking CB1 receptors display an accelerated cognitive decline with aging (Bilkei-Gorzo et al., 2005), which is accompanied by alterations in spine morphology (Ballesteros-Yáñez et al., 2007). Deficits in endocannabinoid signaling might also contribute to dementias. CB1 and CB2 cannabinoid receptors were found to be associated with amyloid β-protein (Aβ) plaques in post mortem brain tissue from subjects with Alzheimer’s disease (AD), (Ramirez et al., 2005). Moreover, elevated expression levels of CB2 receptors and FAAH were observed in immune cells surrounding senile plaques in subjects with AD or Down’s syndrome (Núñez et al., 2008; Centonze et al., 2007; Benito et al., 2007). Lastly, pharmacological agents that enhance endocannabinoid activity were shown to exert beneficial effects in animal models of Aβ-induced toxicity. Cannabinoid agonists prevented Aβ-triggered microglial activation and neurotoxicity in primary cell cultures, suggesting that cannabinoid receptor activation may reduce neuroinflammation (Ehrhart et al., 2005; Ramirez et al., 2005). Consistent with these results, the anandamide reuptake inhibitor VDM-11 reversed hippocampal damage and loss of memory retention in rodents treated with Aβ42 peptide (van der Stelt et al., 2006). Although CB1 receptor expression was not found to be altered in the post mortem cortex of AD patients (Lee et al., 2010), the majority of available data collectively suggests an involvement of the endocannabinoid system in the cognitive deterioration that accompanies AD. To test this hypothesis, here we utilized liquid chromatography/mass spectrometry (LC/MS) to characterize the endocannabinoid-related lipidome in the cortex and cerebellum of subjects with AD and non-demented control subjects, closely matched for age and post mortem interval.
2. Methods
2.1. Research subjects
We analyzed frozen samples of brain tissue from a total of 17 non-demented control subjects and 38 pathologically confirmed subjects with AD (males/females: control subjects, 10/7; subjects with AD, 20/18), provided by the Institute for Memory Impairments and Neurological Disorders and the Alzheimer’s Disease Research Center at the University of California, Irvine. Three brain areas were selected for analysis: midfrontal cortex (Broadman area 9), temporal cortex (Broadman area 20) and cerebellum. Subjects were matched for age (in years: control subjects, 80.4±2.1; subjects with AD, 80.5±1.2) and post mortem interval (in hours: control subjects, 4.5±0.4; subjects with AD, 4.2±0.3) (Supplementary Table 1). AD cases met the National Institute on Aging-Reagan Institute criteria for intermediate or high likelihood of AD. Mini-Mental State Examination (MMSE) scores, a broad measure of cognitive function (Folstein et al., 1975), were accessible for 8 control subjects (mean score±SEM = 28.4±0.6; assessed 45.9±9.3 months before death) and 20 subjects with AD (mean score±SEM = 11.4±1.6; assessed 10.5±2.2 months before death). Boston Naming test scores (Kaplan et al., 1983) and Kendrick Digit Copy test scores (Kendrick, 1985) were also available (Supplementary Table 1). Medications taken were monitored in part of the patients (n=11) (Supplementary Table 2).
2.2. Cell cultures
We cultured Neuro-2a and APPSWE cells (kindly provided by Drs. Seong-Hun Kim and Sangram S. Sisodia, University of Chicago, IL) at 37°C with 5% CO2 in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum and antibiotics (100 unit/ml Penicillin G and 100 μg/ml Streptomycin; Invitrogen). For the maintenance of APPSWE cells, G418 (0.4 mg/ml, Invitrogen) was included in the media. After 72 hours of plating into 100-mm culture dishes, cells were collected for analyses.
2.3. Lipid extractions from tissues
Lipid extractions were conducted as described (Astarita et al., 2008). Briefly, frozen tissue samples were weighed and homogenized in cold methanol containing the following internal standards: [2H8]-2-AG, [2H8]-arachidonic acid (both from Cayman Chemical, Ann Arbor, MI), [2H4]-anandamide and 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-heptadecanoyl (both synthesized in the lab) (Astarita et al., 2008). Lipids were extracted by adding chloroform and water (2:1, vol:vol) and fractionated through open-bed silica gel columns by progressive elution with chloroform/methanol mixtures. Fractions eluted from the columns were dried under N2, reconstituted in chloroform/methanol (1:4, vol:vol; 0.1 ml) and subjected to LC/MS.
2.4. Lipid extractions from cells in cultures
Cells were washed with ice-cold phosphate-buffered saline (PBS) and scraped into 1 ml of methanol/water (1:1, vol:vol) containing the internal standards listed above. Protein concentrations were measured using the BCA protein assay (Pierce, Rockford, IL). Lipids were extracted with chloroform/methanol (2:1, vol:vol; 1.5 ml). The organic phases were collected, dried under N2 and dissolved in chloroform/methanol (1:3, vol:vol) for LC/MS analyses.
2.5. LC/MS analyses
Monoacylglycerols (MG) and fatty acid ethanolamides (FAE) – We used an Agilent 1100-LC system coupled to a 1946D-MS detector equipped with an electrospray ionization (ESI) interface (Agilent Technologies, Inc., Palo Alto, CA). Lipids were separated on a reversed-phase XDB Eclipse C18 column (50×4.6 mm i.d., 1.8 μm, Zorbax, Agilent Technologies). They were eluted with a gradient of methanol in water (from 85% to 90% methanol in 2.0 min and 90% to 100% in 3.0 min) at a flow rate of 1.5 ml/min. Column temperature was kept at 40°C. MS detection was in the positive ionization mode, capillary voltage was set at 3 kV and fragmentor voltage was 120 V. N2 was used as drying gas at a flow rate of 13 l/min and a temperature of 350°C. Nebulizer pressure was set at 60 PSI. For quantification purposes, we monitored the sodium adducts of the molecular ions [M+Na]+ in the selected ion-monitoring (SIM) mode, using [2H8]-2-AG (mass-to-charge ratio, m/z 409) and [2H4]-anandamide (m/z 370) as internal standards. Non-esterified fatty acids – We used a reversed-phase XDB Eclipse C18 column (50×4.6 mm i.d., 1.8 μm, Zorbax, Agilent Technologies) eluted with a linear gradient from 90% to 100% of A in B for 2.5 min at a flow rate of 1.5 ml/min with column temperature at 40°C. Mobile phase A consisted of methanol containing 0.25% acetic acid and 5 mM ammonium acetate; mobile phase B consisted of water containing 0.25% acetic acid and 5 mM ammonium acetate. ESI was in the negative mode, capillary voltage was set at 4 kV and fragmentor voltage was 100 V. N2 was used as drying gas at a flow rate of 13 l/min and a temperature of 350°C. Nebulizer pressure was set at 60 PSI. We monitored deprotonated molecular ions [M-H]− in the SIM mode and [2H8]-arachidonic acid (m/z 311) was used as an internal standard. N-acyl-phosphatidylethanolamine (NAPE) – NAPE species, including NArPE, were analyzed as previously described (Astarita and Piomelli, 2009). Briefly, they were separated by LC using an 1100 system (Agilent Technologies) equipped with a Poroshell 300 SB C18 column (2.1 × 75 mm inner diameter, 5 μm; Agilent Technologies) maintained at 50°C. A linear gradient of methanol in water containing 5 mM ammonium acetate and 0.25% acetic acid (from 85% to 100% of methanol in 4 min) was applied at a flow rate of 1 ml/min. MS analyses were conducted using an Ion Trap XCT (Agilent Technologies) set in the negative mode. Capillary voltage was 4.5 kV, with skim1 at −40 V and capillary exit at −151 V. N2 was used as drying gas at a flow rate of 12 l/min, with temperature at 350°C and nebulizer pressure at 80 psi. Helium was used as the collision gas. Extracted ion chromatograms were used to quantify each NAPE precursor ion by monitoring the characteristic lyso-NAPE product ions in MS2 using 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-heptadecanoyl (m/z 942.8 > 704.8) as an internal standard.
2.6. Enzyme-linked immunosorbent assay (ELISA)
Aβ peptides were sequentially extracted from ~200 mg of frozen midfrontal cortex tissue in Tris buffer (0.1 M, pH 6.8; 1.33 ml per 200 mg tissue) containing 1% sodium dodecyl sulphate (SDS) and a protease inhibitor cocktail (MP Biochemicals Inc., Solon, OH). The extracts were centrifuged (100,000 × g for 1 hour at 4°C), pellets were resuspended in 70% formic acid and sonicated on ice. After a second centrifugation (100,000 × g for 1 hour, 4°C), the supernatants were collected and stored at −80°C. Samples were assayed in triplicate on ELISA plates coated with a monoclonal anti-Aβ1–16 antibody (kindly provided by Dr. William Van Nostrand, Stony Brook University, NY) and detection was by biotinylated mouse monoclonal anti-Aβ1–40 and anti-Aβ1–42 antibodies, followed by streptavidine-horseradish peroxidase (HRP) conjugate (Pierce). Ultra-TMB ELISA substrate (Pierce) was added to develop the reaction for 15 min. The reaction was stopped by adding sulfuric acid (2 N) and the plates were analyzed on a Synergy HT Spectrophotometer (Bio-Tek Instruments, Winooski, VT) at 450 nm. Aβ1–40 and Aβ1–42 peptides (Chemicon International Inc., Temecula, CA) were used as standards after a pretreatment with 1,1,1,3,3,3-hexafluoro-2-propanol to prevent fibril formation.
2.7. Western blot analyses
We prepared cell lysates in a buffer containing 10 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% Triton X-100, 0.25% Nonidet P-40, and 2 mM EDTA supplemented with a mixture of protease inhibitors (Roche, Indianapolis, IN). Lysates were centrifuged at 14,000 × g for 10 min at 4°C. Proteins (30 μg) from supernatants were separated by 4–20% SDS-polyacrylamide gel electrophoresis, transferred to polyvinylidene difluoride membranes, and subjected to western blotting using a monoclonal anti-amyloid precursor protein (APP) antibody 6E10 (1:1000; Abcam, Cambridge, MA) or a polyclonal anti-FAAH antibody (Millipore, Billerica, MA).
2.8. Quantitative real-time PCR analyses
Total RNA was extracted from frozen tissue with TRIzolTM (Invitrogen) and quantified. First strand cDNA was synthesized from 2 μg of total RNA by using Superscript II RNase H reverse transcriptase (Invitrogen) following the manufacturer’s instructions. Real-time quantitative PCR was performed in an Mx 3000P system (Stratagene, La Jolla, CA). Primers and fluorogenic probes were synthesized by TIB Molbiol (Adelphia, NJ). The primer/probe sequences were as follows: for mouse FAAH: forward, 5′-CCTTATGCCCTGGAGGTCCT-3′; reverse, 5′-GGAGAAAAGAGCAGCCACCA-3′; TaqMan probe, 5′-TCGGCAGGTGGGCTGTTCAGTGT-3′; for mouse NAPE-PLD: forward, 5′-AACGAGCGGTTCGGCA-3′; reverse, 5′-ATCCAGTCAAGAAGGCCCAA-3′; TaqMan probe, 5′-CGAGCTGCGGTGGTTTGTGCC-3′. mRNA levels were calculated from the average of triplicated reactions through absolute quantification and normalized to the levels of glyceraldehyde 3-phosphate dehydrogenase (GAPDH). For the human brain study, primers and fluorogenic probes for human FAAH were purchased from Applied Biosystems (TaqMan(R) Gene Expression Assays, Hs01038678_m1, Foster City, CA). mRNA expression levels were normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and were calculated by comparative quantification using a calibrator (Gutala et al., 2004).
2.9. In vitro FAAH activity assay
Cells were washed and homogenized in ice-cold 50 mM Tris-HCl, pH 7.5. Homogenates were centrifuged at 1000 × g for 10 min at 4 °C and supernatants were collected for in vitro FAAH assays. Protein concentration was determined using a BCA protein assay kit (Pierce). We measured FAAH activity at 37°C for 30 min in 0.5 ml of Tris buffer (50 mM, pH 7.5) containing fatty acid-free bovine serum albumin (0.05%, w/v), the cell homogenates (50 μg), 10 μM anandamide, and anandamide[ethanolamine-3H] (10,000 cpm, specific activity 60 Ci/mmol; American Radiolabeled Chemicals, St. Louis, MO). The reactions were stopped with chloroform/methanol (1:1, 1 ml) and radioactivity was measured in the aqueous layers by liquid scintillation counting.
2.10. Statistical analyses
Results are expressed as means ± SEM. The significance of differences among groups was evaluated using the two-tailed Student’s t-test and differences were considered significant if P<0.05. Pairwise correlation between variables was assessed by Pearson and confirmed by Spearman rank. Analyses were conducted using GraphPad Prism (GraphPad Software, San Diego, CA).
3. Results
3.1. Anandamide mobilization is impaired in AD
Demographic and clinical information on the subjects included in the present study is provided in Supplementary Tables 1 and 2. There was no effect of age, gender, post mortem interval or medication on the levels of anandamide (Supplementary Figs. 1 and 2) or other endocannabinoid-related lipids targeted by our analysis (Table I and data not shown). By contrast, there was a statistically detectable difference between control subjects and AD patients in the levels of anandamide in samples of midfrontal cortex (P=0.047, Fig. 1A) and temporal cortex (P=0.034, Fig. 1B). No such difference was found in cerebellum (Fig. 1C) or with other lipid species, including 2-AG (Fig. 1D), palmitoylethanolamide (Fig. 1E) and arachidonic acid (Fig. 1F) (Table 1 and Supplementary Tables 3 and 4). The results indicate that anandamide content is selectively lowered in midfrontal and temporal cortex of AD patients, relative to control subjects.
Table 1.
Endocannabinoid-related lipids targeted in the present study.
| Names | m/z | Structure |
|---|---|---|
| 16:0 FAE | 322 | |
| 16:0 MG | 353 | |
| 18:0 FAE | 350 | 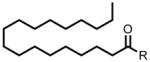 |
| 18:0 MG | 381 | |
| 18:1 Δ9 FAE | 348 | 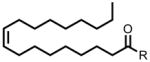 |
| 18:1 Δ9 MG | 379 | |
| 18:2 Δ9,12 FAE | 346 | 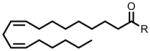 |
| 18:2 Δ9,12 MG | 377 | |
| 18:3 Δ9,12,15 FAE | 344 | 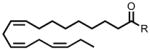 |
| 18:3 Δ9,12,15 MG | 375 | |
| 20:3 Δ8,11,14 FAE | 372 |  |
| 20:3 Δ8,11,14 MG | 403 | |
| 20:4 Δ5,8,11,14 FAE | 370 |  |
| 20:4 Δ5,8,11,14 MG | 401 | |
| 20:4 Δ5,8,11,14 FA | 303 | |
| 22:6 Δ4,7,10,13,16,19 FAE | 394 | |
| 22:6 Δ4,7,10,13,16,19 MG | 425 | |
| 1-stearoyl,2-docosahexaenoyl-sn-glycero-phosphoethanolamine-N-arachidonoyl (NArPE) | 1076.8 > 766.8 | |
| 1-O-octadecenoyl, 2-octadecenoyl-sn-glycero-phosphoethanolamine-N-arachidonoyl (NArPE#) | 1014.8 > 750.8 | |
Fatty acyl ethanolamides (FAE; R=ethanolamine), monoacylglycerols (MG; R=glycerol) and fatty acids (FA; R=OH). Mass-to-charge (m/z) ratios for each lipid species are also indicated.
Figure 1.

Endocannabinoid-related lipids in midfrontal cortex (FC), temporal cortex (TC) and cerebellum (CB) from control subjects (n=16–17) and AD patients (n=36–38). The figure shows levels of anandamide (AEA, A–C), 2-arachidonoyl-sn-glycerol (2-AG, D), palmitoylethanolamide (PEA, E), arachidonic acid (AA, F), and NArPE (G–I). 2-AG levels were slightly increased in FC of AD patients, but this change did not reach statistical significance. *P<0.05, **P<0.01 and ***P<0.001 by two-tailed t-test.
To determine whether this decrease might be due to a defect in anandamide production, we quantified by LC/MS the predominant NArPE species found in the human brain: 1-stearoyl, 2-docosahexaenoyl-sn-glycero-phosphoethanolamine-N-arachidonoyl (Astarita et al., 2008). We found a statistically detectable difference between control subjects and AD patients in NArPE content of midfrontal cortex (P=0.0019, Fig. 1G) and temporal cortex (P=0.0004, Fig. 1H), but not cerebellum (P=0.47, Fig. 1I). Statistical inspection of the data revealed a significant correlation between the levels of NArPE and those of anandamide (P=0.0009 and P<0.0001, r=0.44 and 0.55 in the midfrontal and temporal cortex, respectively, Supplementary Fig. 3), which confirmed the expected precursor-product relationship between the two molecules (Astarita et al., 2008).
The findings reported above suggest that AD is associated with a deficit in brain anandamide production. In addition, we also observed a trend toward elevation of FAAH mRNA levels in temporal cortex of AD patients, but such trend did not reach statistical significance (Supplementary Fig. 4). This observation is consistent with the results of previous studies, which have reported increased FAAH expression in neuritic plaque-associated astrocytes and microglia of AD and Down’s syndrome patients (Benito et al, 2003; Núñez et al, 2008).
3.2. Anandamide levels correlate with cognitive performance
The functional significance of the deficit in anandamide mobilization observed in the cortex of AD patients was explored using correlation analyses. A highly significant positive correlation was found between anandamide content in midfrontal cortex of AD patients and their performance in the Kendrick’s digit copy test (P=0.004 and 0.046, Figs. 2A and 2B), which measures psychomotor speed. By contrast, no correlations were found between anandamide levels in the midfrontal cortex of the patients and scores of either the Boston naming test (Fig. 2C), which assesses the ability to name pictures of objects, or the MMSE test (Supplementary Fig. 5A), a measure of global cognitive function. On the other hand, anandamide levels in temporal cortex of AD patients were positively associated with scores of the Boston naming test (P=0.027, Fig. 2F), but not the Kendrick’s digit copy test (Figs. 2D and 2E). Levels of 2-AG or palmitoylethanolamide displayed no correlation with any of those tests (Supplementary Fig. 6). These correlations do not prove causation, but do suggest that impaired anandamide mobilization in midfrontal and temporal cortex accompanies a deterioration of specific cognitive abilities in subjects with AD.
Figure 2.

Correlation analyses of anandamide levels in midfrontal cortex (FC) and temporal cortex (TC) of AD patients with cognitive test scores. Anandamide (AEA) content in FC correlates with both the number of completed items (P=0.0044) (A) and the time to complete such items (P=0.046) (B) in the Kendrick Digit Copy Test (KDCT) (n=9–10). No correlation was observed with the Boston Naming Test (BNT) scores (n=18, P=0.91) (C). Anandamide levels in TC correlate with BNT scores (n=18, P=0.027) (F), but not with KDCT scores (n=9–10, P=0.14 and 0.36 for D and E, respectively). r, Pearson’s correlation coefficient. *P<0.05 and **P<0.01.
3.3. Aβ42 disrupts anandamide mobilization
To determine whether neurotoxic Aβ peptides can impair anandamide mobilization, we first asked whether cortical levels of Aβ peptides are statistically correlated with those of anandamide in our subject group. As expected, midfrontal cortex tissue from AD patients contained higher concentrations of both SDS-extractable (soluble) and SDS-insoluble forms of Aβ42 and Aβ40, compared to tissue from non-demented control subjects (Figs. 3A and 3D). Importantly, we found a statistically detectable negative correlation between Aβ42 and anandamide content (P=0.0016 and 0.014, Figs. 3B and 3E). Levels of the anandamide precursor, NArPE, were also significantly correlated with insoluble Aβ42 content (P=0.0034, Fig. 3C), while a trend toward correlation, which did not reach statistical significance, was observed with soluble Aβ42 (Fig. 3F). No significant correlations were found between levels of Aβ40 and anandamide or NArPE (Supplementary Fig. 7). A separate analysis for control and AD brains for the correlations also indicated that levels of SDS-insoluble Aβ42 significantly correlate with anandamide and NArPE in AD patients (P=0.03 and 0.04, respectively) (Supplementary Tables 5). In addition, anandamide levels in midfrontal cortex did not correlate with amyloid plaque load, neurofibrillary tangles or ApoE4 genotype (Supplementary Fig. 8), which is suggestive of a specific link between Aβ42 and anandamide mobilization.
Figure 3.

Correlation analyses of Aβ42 and anandamide in midfrontal cortex of control subjects and AD patients. Content of SDS-insoluble (A) and SDS-soluble (D) Aβ40 and Aβ42 in control subjects (n=17) and AD patients (n=38) were measured by ELISA. Levels of anandamide (B) and NArPE (C) showed a highly significant correlation with SDS-insoluble Aβ42 (P=0.0016 and 0.0034 for B and C, respectively). A correlation between anandamide or NArPE and soluble Aβ42 was also observed (n=53–55) (P=0.014 and 0.098 for E and F, respectively). r, Pearson’s correlation coefficient. *P<0.05 and **P<0.01 by two-tailed t-test.
To further explore this link, we stably overexpressed the familial early-onset AD (FAD)-associated Swedish mutant form of amyloid precursor protein (APPSWE) in mouse Neuro-2a cells (Thinakaran et al., 1996). Overexpression of APPSWE and accumulation of cell-associated Aβ40 and Aβ42 were confirmed using western blot and ELISA assays, respectively (Figs. 4A and 4B). These changes were associated with a significant reduction in the levels of anandamide (Fig. 4C) and those of 1-O-octadecenoyl, 2-octadecenoyl-sn-glycero-phosphoethanolamine-N-arachidonoyl (NArPE#), which we have identified by LC/MS as the most abundant NArPE species in Neuro-2a cells (Fig. 4D). Additionally, whereas NAPE-PLD mRNA levels were not affected (Fig. 4E), FAAH mRNA levels were significantly increased in APPSWE cells compared to control cells (Fig. 4F). Such increase was accompanied by elevated expression of FAAH protein (Fig. 4G) as well as in vitro FAAH activity (Fig. 4H). APPSWE overexpression had no effect on cell viability (data not shown). These findings provide direct evidence that abnormally high levels of endogenous Aβ42 alter anandamide mobilization in neuronal cells.
Figure 4.

Impaired anandamide production in Neuro-2a cells stably overexpressing Swedish mutant APP. Neuro-2a cells stably expressing human APPSWE were harvested 72 hours after split, along with control cells. Cell lysates were analyzed for APP protein expression by western blotting (A) and levels of Aβ40 and Aβ42 were measured by ELISA (B). Lipids were extracted and levels of anandamide (AEA) (C) and 1-O-octadecenoyl, 2-octadecenoyl-sn-glycero-phosphoethanolamine-N-arachidonoyl (NArPE#) (D) were analyzed by LC/MS (n=4). Levels of NAPE-PLD (E) and FAAH (F) mRNA were measured by quantitative real-time PCR (n=4). Cellular amount of FAAH protein measured by western blotting (G) as well as in vitro FAAH activity (H) were higher in APPSWE cells compared to native Neuro-2a cells. **P<0.01 and ***P<0.001 by two-tailed t-test.
4. Discussion
The objective of the present study was to investigate the link, suggested by previous reports, between brain endocannabinoid signaling and AD (Ramirez et al., 2005; Núñez et al., 2008; Centonze et al., 2007; Benito et al., 2007). Our targeted lipidomic analyses of human brain tissue provide new evidence in support of such a link by showing that levels of the endocannabinoid anandamide are significantly lower in midfrontal and temporal cortex of subjects with AD than of non-demented subjects closely matched for age and post mortem interval. To identify potential mechanisms contributing for this AD-associated alteration, we quantified the anandamide precursor, NArPE, in the same brain samples (Cadas et al., 1996; Cadas et al., 1997; Astarita et al., 2008). The results indicate that NArPE content is also markedly reduced in the cortex of AD patients, which is suggestive of a deficit in anandamide production. While limited by the use of post mortem tissue, in which endocannabinoid levels are likely to be altered (Schmid et al., 1995), the functional significance of our results is underscored by the identification of highly significant positive correlations between cortical anandamide content and the patient’s performance in cognitive measures of psychomotor speed and language.
Synaptic deterioration is a hallmark of AD pathology (Cotman and Anderson, 2000). It does not appear, however, that the changes in cortical anandamide levels documented here were the consequence of a generalized loss of synaptic lipids. Arguing against this possibility, we found that among the 16 endocannabinoid-related lipid species targeted by our analyses, only anandamide and its precursor NArPE were altered in AD. In particular, we observed no significant changes in the levels of another endocannabinoid, 2-AG, a polyunsaturated fatty acid, arachidonic acid, and a lipid amide, palmitoylethanolamide. These lipid molecules provide a sensitive indicator of synaptic lipid degradation because they are released in substantial amounts during ischemic brain damage (Panikashvili et al., 2001; Schabitz et al., 2002). The results of prior lipid-profile studies of human brain strengthen the idea that AD is associated with a restricted set of lipid abnormalities rather than a generalized loss of neuronal lipids (Soderberg et al., 1991; Skinner et al., 1993; Prasad et al., 1998; Guan et al., 1999; Lukiw et al., 2005; Fraser et al., 2010; Astarita et al., 2010). Also supportive of this conclusion are two additional findings. First, van der Stelt et al have shown that injections of neurotoxic Aβ42 into the rat hippocampus cause a transient increase in 2-AG levels followed by a more persistent decrease in anandamide levels (van der Stelt et al., 2006). Second, we show here that cortical levels of anandamide correlate with the patients’ performance in cognitive tests of psychomotor speed and linguistic ability, but not with amyloid plaque burden or tau-protein hyperphosphorylation, two features of AD neuropathology that are linked to neuronal cell death (Haass and Selkoe, 2007). Notably, animal experiments show that altered endocannabinoid signaling in hippocampus might also contribute to memory loss following Aβ42 administration (van der Stelt et al., 2006; Mazzola et al., 2003).
Neurotoxic amyloid Aβ peptides are thought to play a key role in the pathogenesis of AD (Gandy, 2005). It is significant, therefore, that cortical levels of anandamide and its precursor NArPE were inversely correlated with those of the highly amyloidogenic peptide, Aβ42 (Iwatsubo et al., 1994; McGowan et al., 2005), but not with those of the less toxic species, Aβ40 (Dahlgren et al., 2002). Though further research is needed to determine if this correlation represents cause and effect, our experiments with APPSWE-overexpressing Neuro-2a cells provided direct evidence that pathological accumulation of Aβ42 can disrupt anandamide mobilization, while having no overt effect on cell viability. These studies suggest that excessive Aβ42 can simultaneously impact both anandamide production, by curtailing availability of the anandamide precursor NArPE, and anandamide degradation, by increasing expression of the anandamide-hydrolyzing enzyme FAAH. Similar alterations have been observed in brain tissue of subjects with AD and Down’s syndrome (Benito et al, 2003; Núñez et al, 2008; present study), supporting the possibility that an Aβ42-linked mechanism may impair anandamide mobilization in AD and remove a protective influence of this endocannabinoid messenger against neural toxicity and inflammation (Ehrhart et al., 2005; Ramirez et al., 2005; van der Stelt et al., 2006).
The molecular events engaged by Aβ42 to affect anandamide mobilization were not investigated in the present study. Interesting parallels are offered, however, by prior findings showing that pro-inflammatory stimuli down-regulate lipid amide biosynthesis in innate immune cells (Solorzano et al, 2009) and nerve injury enhances FAAH expression in peripheral sensory neurons (Lever et al., 2009). While additional studies are needed to fill this knowledge gap, it is tempting to speculate that pharmacological agents that strengthen anandamide signaling, such as FAAH inhibitors, might be beneficial to improve cognition in AD.
Supplementary Material
Acknowledgments
This work was supported by grants from National Institute on Drug Abuse (ARRA to D.P.) and the Alzheimer’s Association (IIRG-08-92000 to K.-M.J.). The Alzheimer’s Disease Research Center Neuropathology Core and the Institute for Memory Impairments and Neurological Disorders of the University of California, Irvine, are supported by grants from the National Institute on Aging (P50 AG016573 and P01 AG00538). The contribution of the Agilent Technologies/University of California, Irvine Analytical Discovery Facility, Center for Drug Discovery is gratefully acknowledged.
Footnotes
Disclosure statement
The authors declare no actual or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Astarita G, Ahmed F, Piomelli D. Identification of biosynthetic precursors for the endocannabinoid anandamide in the rat brain. J Lipid Res. 2008;49:48–57. doi: 10.1194/jlr.M700354-JLR200. [DOI] [PubMed] [Google Scholar]
- Astarita G, Piomelli D. Lipidomic analysis of endocannabinoid metabolism in biological samples. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:2755–2767. doi: 10.1016/j.jchromb.2009.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astarita G, Jung KM, Berchtold NC, Nguyen VQ, Gillen DL, Head E, Cotman CW, Piomelli D. Deficient liver biosynthesis of docosahexaenoic acid correlates with cognitive impairment in Alzheimer’s disease. PLoS One. 2010;5:e12538. doi: 10.1371/journal.pone.0012538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros-Yáñez I, Valverde O, Ledent C, Maldonado R, DeFelipe J. Chronic cocaine treatment alters dendritic arborization in the adult motor cortex through a CB1 cannabinoid receptor-dependent mechanism. Neuroscience. 2007;146:1536–1545. doi: 10.1016/j.neuroscience.2007.03.017. [DOI] [PubMed] [Google Scholar]
- Benito C, Núñez E, Pazos MR, Tolón RM, Romero J. The endocannabinoid system and Alzheimer’s disease. Mol Neurobiol. 2007;36:75–81. doi: 10.1007/s12035-007-8006-8. [DOI] [PubMed] [Google Scholar]
- Benito C, Núñez E, Tolón RM, Carrier EJ, Rábano A, Hillard CJ, Romero J. Cannabinoid CB2 receptors and fatty acid amide hydrolase are selectively overexpressed in neuritic plaque-associated glia in Alzheimer’s disease brains. J Neurosci. 2003;23:11136–11141. doi: 10.1523/JNEUROSCI.23-35-11136.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilkei-Gorzo A, Racz I, Valverde O, Otto M, Michel K, Sastre M, Zimmer A. Early age-related cognitive impairment in mice lacking cannabinoid CB1 receptors. Proc Natl Acad Sci USA. 2005;102:15670–15675. doi: 10.1073/pnas.0504640102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadas H, Gaillet S, Beltramo M, Venance L, Piomelli D. Biosynthesis of an endogenous cannabinoid precursor in neurons and its control by calcium and cAMP. J Neurosci. 1996;16:3934–3942. doi: 10.1523/JNEUROSCI.16-12-03934.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadas H, di Tomaso E, Piomelli D. Occurrence and biosynthesis of endogenous cannabinoid precursor, N-arachidonoyl phosphatidylethanolamine, in rat brain. J Neurosci. 1997;17:1226–1242. doi: 10.1523/JNEUROSCI.17-04-01226.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centonze D, Finazzi-Agrò A, Bernardi G, Maccarrone M. The endocannabinoid system in targeting inflammatory neurodegenerative diseases. Trends Pharmacol Sci. 2007;28:180–187. doi: 10.1016/j.tips.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Takahashi KA, Castillo PE. Endocannabinoid-mediated synaptic plasticity in the CNS. Annu Rev Neurosci. 2006;29:37–76. doi: 10.1146/annurev.neuro.29.051605.112834. [DOI] [PubMed] [Google Scholar]
- Cotman CW, Anderson AJ. The brain’s microenvironment, early functional loss, and the conversion to Alzheimer’s disease. Ann NY Acad Sci. 2000;924:112–116. doi: 10.1111/j.1749-6632.2000.tb05569.x. [DOI] [PubMed] [Google Scholar]
- Dahlgren KN, Manelli AM, Stine WB, Jr, Baker LK, Krafft GA, LaDu MJ. Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J Biol Chem. 2002;277:32046–32053. doi: 10.1074/jbc.M201750200. [DOI] [PubMed] [Google Scholar]
- Ehrhart J, Obregon D, Mori T, Hou H, Sun N, Bai Y, Klein T, Fernandez F, Tan J, Shytle RD. Stimulation of cannabinoid receptor 2 (CB2) suppresses microglial activation. J Neuroinflammation. 2005;2:29. doi: 10.1186/1742-2094-2-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- Fraser T, Tayler H, Love S. Fatty acid composition of frontal, temporal and parietal neocortex in the normal human brain and in Alzheimer’s disease. Neurochem Res. 2010;35:503–513. doi: 10.1007/s11064-009-0087-5. [DOI] [PubMed] [Google Scholar]
- Gandy S. The role of cerebral amyloid beta accumulation in common forms of Alzheimer disease. J Clin Invest. 2005;115:1121–1129. doi: 10.1172/JCI25100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Z, Wang Y, Cairns NJ, Lantos PL, Dallner G, Sindelar PJ. Decrease and structural modifications of phosphatidylethanolamine plasmalogen in the brain with Alzheimer disease. J Neuropathol Exp Neurol. 1999;58:740–747. doi: 10.1097/00005072-199907000-00008. [DOI] [PubMed] [Google Scholar]
- Gutala RV, Reddy PH. The use of real-time PCR analysis in a gene expression study of Alzheimer’s disease post-mortem brains. J Neurosci Methods. 2004;132:101–107. doi: 10.1016/j.jneumeth.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Hashimotodani Y, Ohno-Shosaku T, Kano M. Endocannabinoids and synaptic function in the CNS. Neuroscientist. 2007;13:127–137. doi: 10.1177/1073858406296716. [DOI] [PubMed] [Google Scholar]
- Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y. Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta 42(43) Neuron. 1994;13:45–53. doi: 10.1016/0896-6273(94)90458-8. [DOI] [PubMed] [Google Scholar]
- Kaplan E, Goodglass H, Weintraub S. Boston Naming Test. Lee & Febiger; Philadelphia: 1983. [Google Scholar]
- Katona I, Freund TF. Endocannabinoid signaling as a synaptic circuit breaker in neurological disease. Nat Med. 2008;14:923–930. doi: 10.1038/nm.f.1869. [DOI] [PubMed] [Google Scholar]
- Kendrick DC. Kendrick Cognitive Tests for the Elderly. NFER-Nelson; Windsor: 1985. [Google Scholar]
- Lee JH, Agacinski G, Williams JH, Wilcock GK, Esiri MM, Francis PT, Wong PT, Chen CP, Lai MK. Intact cannabinoid CB1 receptors in the Alzheimer’s disease cortex. Neurochem Int. 2010;57:985–989. doi: 10.1016/j.neuint.2010.10.010. [DOI] [PubMed] [Google Scholar]
- Lever IJ, Robinson M, Cibelli M, Paule C, Santha P, Yee L, Hunt SP, Cravatt BF, Elphick MR, Nagy I, Rice AS. Localization of the endocannabinoid-degrading enzyme fatty acid amide hydrolase in rat dorsal root ganglion cells and its regulation after peripheral nerve injury. J Neurosci. 2009;29:3766–3780. doi: 10.1523/JNEUROSCI.4071-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Wang L, Harvey-White J, Osei-Hyiaman D, Razdan R, Gong Q, Chan AC, Zhou Z, Huang BX, Kim HY, Kunos G. A biosynthetic pathway for anandamide. Proc Natl Acad Sci USA. 2006;103:13345–13350. doi: 10.1073/pnas.0601832103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukiw WJ, Cui JG, Marcheselli VL, Bodker M, Botkjaer A, Gotlinger K, Serhan CN, Bazan NG. A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. J Clin Invest. 2005;115:2774–2783. doi: 10.1172/JCI25420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz B. The endocannabinoid system and extinction learning. Mol Neurobiol. 2007;36:92–101. doi: 10.1007/s12035-007-8004-x. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann C, Zieglgänsberger W, Di Marzo V, Lutz B. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- Mato S, Lafourcade M, Robbe D, Bakiri Y, Manzoni OJ. Role of the cyclic-AMP/PKA cascade and of P/Q-type Ca++ channels in endocannabinoid-mediated long-term depression in the nucleus accumbens. Neuropharmacology. 2008;54:87–94. doi: 10.1016/j.neuropharm.2007.04.014. [DOI] [PubMed] [Google Scholar]
- Mazzola C, Micale V, Drago F. Amnesia induced by beta-amyloid fragments is counteracted by cannabinoid CB1 receptor blockade. Eur J Pharmacol. 2003;477:219–225. doi: 10.1016/j.ejphar.2003.08.026. [DOI] [PubMed] [Google Scholar]
- Mazzola C, Medalie J, Scherma M, Panlilio LV, Solinas M, Tanda G, Drago F, Cadet JL, Goldberg SR, Yasar S. Fatty acid amide hydrolase (FAAH) inhibition enhances memory acquisition through activation of PPAR-alpha nuclear receptors. Learn Mem. 2009;16:332–337. doi: 10.1101/lm.1145209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan E, Pickford F, Kim J, Onstead L, Eriksen J, Yu C, Skipper L, Murphy MP, Beard J, Das P, Jansen K, Delucia M, Lin WL, Dolios G, Wang R, Eckman CB, Dickson DW, Hutton M, Hardy J, Golde T. Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron. 2005;47:191–199. doi: 10.1016/j.neuron.2005.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Núñez E, Benito C, Tolón RM, Hillard CJ, Griffin WS, Romero J. Glial expression of cannabinoid CB(2) receptors and fatty acid amide hydrolase are beta amyloid-linked events in Down’s syndrome. Neuroscience. 2008;151:104–110. doi: 10.1016/j.neuroscience.2007.10.029. [DOI] [PubMed] [Google Scholar]
- Okamoto Y, Morishita J, Tsuboi K, Tonai T, Ueda N. Molecular characterization of a phospholipase D generating anandamide and its congeners. J Biol Chem. 2004;279:5298–5305. doi: 10.1074/jbc.M306642200. [DOI] [PubMed] [Google Scholar]
- de Oliveira Alvares L, Pasqualini Genro B, Diehl F, Molina VA, Quillfeldt JA. Opposite action of hippocampal CB1 receptors in memory reconsolidation and extinction. Neuroscience. 2008;154:1648–1655. doi: 10.1016/j.neuroscience.2008.05.005. [DOI] [PubMed] [Google Scholar]
- Panikashvili D, Simeonidou C, Ben-Shabat S, Hanus L, Breuer A, Mechoulam R, Shohami E. An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature. 2001;413:527–531. doi: 10.1038/35097089. [DOI] [PubMed] [Google Scholar]
- Piomelli D, Astarita G, Rapaka R. A neuroscientist’s guide to lipidomics. Nat Rev Neurosci. 2007;8:743–754. doi: 10.1038/nrn2233. [DOI] [PubMed] [Google Scholar]
- Piomelli D. The molecular logic of endocannabinoid signalling. Nat Rev Neurosci. 2003;4:873–884. doi: 10.1038/nrn1247. [DOI] [PubMed] [Google Scholar]
- Prasad KN, Hovland AR, La Rosa FG, Hovland PG. Prostaglandins as putative neurotoxins in Alzheimer’s disease. Proc Soc Exp Biol Med. 1998;219:120–125. doi: 10.3181/00379727-219-44323. [DOI] [PubMed] [Google Scholar]
- Ramírez BG, Blázquez C, Gómez del Pulgar T, Guzmán M, de Ceballos ML. Prevention of Alzheimer’s disease pathology by cannabinoids: neuroprotection mediated by blockade of microglial activation. J Neurosci. 2005;25:1904–1913. doi: 10.1523/JNEUROSCI.4540-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäbitz WR, Giuffrida A, Berger C, Aschoff A, Schwaninger M, Schwab S, Piomelli D. Release of fatty acid amides in a patient with hemispheric stroke: a microdialysis study. Stroke. 2002;33:2112–2114. doi: 10.1161/01.str.0000023491.63693.18. [DOI] [PubMed] [Google Scholar]
- Schmid PC, Krebsbach RJ, Perry SR, Dettmer TM, Maasson JL, Schmid HH. Occurrence and postmortem generation of anandamide and other long-chain N-acylethanolamines in mammalian brain. FEBS Lett. 1995;375:117–120. doi: 10.1016/0014-5793(95)01194-j. [DOI] [PubMed] [Google Scholar]
- Skinner ER, Watt C, Besson JA, Best PV. Differences in the fatty acid composition of the grey and white matter of different regions of the brains of patients with Alzheimer’s disease and control subjects. Brain. 1993;116:717–725. doi: 10.1093/brain/116.3.717. [DOI] [PubMed] [Google Scholar]
- Söderberg M, Edlund C, Kristensson K, Dallner G. Fatty acid composition of brain phospholipids in aging and in Alzheimer’s disease. Lipids. 1991;26:421–425. doi: 10.1007/BF02536067. [DOI] [PubMed] [Google Scholar]
- Solorzano C, Zhu C, Battista N, Astarita G, Lodola A, Rivara S, Mor M, Russo R, Maccarrone M, Antonietti F, Duranti A, Tontini A, Cuzzocrea S, Tarzia G, Piomelli D. Selective N-acylethanolamine-hydrolyzing acid amidase inhibition reveals a key role for endogenous palmitoylethanolamide in inflammation. Proc Natl Acad Sci USA. 2009;106:20966–20971. doi: 10.1073/pnas.0907417106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Stelt M, Mazzola C, Esposito G, Matias I, Petrosino S, De Filippis D, Micale V, Steardo L, Drago F, Iuvone T, Di Marzo V. Endocannabinoids and beta-amyloid-induced neurotoxicity in vivo: effect of pharmacological elevation of endocannabinoid levels. Cell Mol Life Sci. 2006;63:1410–1424. doi: 10.1007/s00018-006-6037-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thinakaran G, Teplow DB, Siman R, Greenberg B, Sisodia SS. Metabolism of the “Swedish” amyloid precursor protein variant in neuro2a (N2a) cells. Evidence that cleavage at the “beta-secretase” site occurs in the golgi apparatus. J Biol Chem. 1996;271:9390–9397. doi: 10.1074/jbc.271.16.9390. [DOI] [PubMed] [Google Scholar]
- Varvel SA, Wise LE, Niyuhire F, Cravatt BF, Lichtman AH. Inhibition of fatty-acid amide hydrolase accelerates acquisition and extinction rates in a spatial memory task. Neuropsychopharmacology. 2007;32:1032–1041. doi: 10.1038/sj.npp.1301224. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.