

Abstract
Plasmepsins (PMs) are pepsin-like aspartic proteases present in different species of parasite Plasmodium. Four Plasmodium species (P. vivax, P. ovale, P. malariae, and the most lethal P. falciparum) are mainly responsible for causing human malaria that affects millions worldwide. Due to the complexity and rate of parasite mutation coupled with regional variations, and the emergence of P. falciparum strains which are resistant to antimalarial agents such as chloroquine and sulfadoxine/pyrimethamine, there is constant pressure to find new and lasting chemotherapeutic drug therapies. Since many proteases represent therapeutic targets and PMs have been shown to play an important role in the survival of parasite, these enzymes have recently been identified as promising targets for the development of novel antimalarial drugs. The genome of P. falciparum encodes ten PMs (PMI, PMII, PMIV-X and histo-aspartic protease (HAP)), four of which (PMI, PMII, PMIV and HAP) reside within the food vacuole, are directly involved in degradation of human hemoglobin, and share 50-79% amino acid sequence identity. This review focuses on structural studies of only these four enzymes, including their orthologs in other Plasmodium species. Almost all original crystallographic studies were performed with PMII, but more recent work on PMIV, PMI, and HAP resulted in a more complete picture of the structure-function relationship of vacuolar PMs. Many structures of inhibitor complexes of vacuolar plasmepsins, as well as their zymogens, have been reported in the last 15 years. Information gained by such studies will be helpful for the development of better inhibitors that could become a new class of potent antimalarial drugs.
Keywords: Aspartic protease, Malaria, Crystal structure, Plasmepsin, Inhibitors
1. Introduction
Malaria is the most prevalent human disease caused by infection by a parasite. It is estimated that 300-500 million people become ill every year, and 1-3 million of them die, mostly pregnant women and children [1,2]. The causative agents of malaria are various species of Plasmodium, with P. falciparum, P. vivax, P. ovale, and P. malariae being principally responsible for malaria in humans. The deadliest form of malaria is caused by P. falciparum. In recent years, some human cases of malaria have also been reported to result from infection by P. knowlesi – a parasite that infects monkeys in certain forested areas of South-East Asia [3]. The parasites spread to people through the bites of female Anopheles mosquitoes. Several drugs are available for treating malaria [4], with sulfadoxine-pyrimethamine and artemisinin-based combinations [5] most commonly used in current medical practice. However, recent reports show that the number of deaths of malaria patients has increased because of development of drug resistance of P. falciparum and P. vivax [4]; multidrug-resistant strains of P. falciparum are now emerging in several parts of the world. Because of the rapid development of resistance to the current antimalarial drugs, discovery of their new, potent, and long-lasting replacements has become essential.
During its erythrocytic growth phase, the parasite degrades most of the host cell hemoglobin [4,6,7] and utilizes the amino acids obtained through this mechanism for biosynthesis of its own proteins [8], also reducing the colloid-osmotic pressure within the host cell to prevent its premature lysis [9]. The degradation process that takes place in the food vacuole of the parasite [6] involves a number of plasmepsins (PMs), enzymes belonging to the pepsin family of aspartic proteases [2,10]. These enzymes were initially called hemoglobinases [11], but the current name has been in common use since 1994 [12]. The total number of plasmepsins varies between different Plasmodium strains, with ten PMs identified in the genome of P. falciparum [10]. Only four of them, PMI, PII, PMIV and histo-aspartic protease (HAP), reside in the acidic food vacuole and are presumed to be involved in hemoglobin degradation [2], whereas the other plasmepsins most likely play different roles [13,14]. In this review, the name “plasmepsin” will refer to only the vacuolar enzymes, unless specifically stated otherwise. Vacuolar PMs are highly homologous, sharing 50-79% amino acid sequence identity [15]. Due to their important role in providing nutrients for the rapidly growing parasites, these enzymes have been identified as promising targets for the development of novel antimalarial drugs [4]. Indeed, inhibitors of aspartic proteases have been shown to exhibit potent antiparasitic activity [11,16-19]. Nevertheless, it is still controversial whether inhibition of vacuolar plasmepsins is responsible for the biological effects of such inhibitors, since knock-out studies showed that these four plasmepsins have overlapping roles in hemoglobin degradation [7]. Additionally, it has been shown that even deletion of all vacuolar PMs does not fully remove the sensitivity of the parasites to inhibitors of pepsin-like enzymes [20]. Some of these questions might only be answered if more structural and biological data for different PMs would become available.
As mentioned above, plasmepsins are pepsin-like aspartic proteases [21-24]. A molecule of a typical pepsin-like aspartic protease usually consists of a single polypeptide chain folded into two structurally similar domains. The active site is located in the cleft formed by these two domains [21], with each domain contributing a single catalytic aspartic acid residue (Asp32 and Asp215; pepsin numbering will be used consistently throughout this review) [25]. The side chains of the two aspartates and a water molecule found in the apoenzymes in their vicinity are generally coplanar and their inner carboxyl oxygens are located within hydrogen bond distance from each other. Another characteristic structural feature of this family of aspartic proteases is the presence in the N-terminal domain of a β-hairpin loop, known as “flap” [21,22]. The flap covers the active site [22] and plays an important role during catalysis. A variety of biochemical and structural studies have been done in order to elucidate the catalytic mechanism of these enzymes [22]. Although some details of the mechanism are still debatable, it is generally agreed that one aspartic acid acts as a catalytic base and the other one as a catalytic acid, activating the water molecule located between the aspartates [21,22,25,26]. It is likely that Asp215 is responsible for the initial activation of the water molecule, generating the nucleophile which attacks the amide carbon of the substrate. The tetrahedral intermediate thus generated accepts a proton from Asp32 and forms the products [21,27]. PMI, PII, and PMIV all contain these two catalytic aspartic acid residues and utilize the same catalytic mechanism [28-30]. In contrast, Asp32 is replaced by His32 in HAP, indicating that the catalytic mechanism of this enzyme must differ [2,15,31,32], but its details are not yet clarified. Recently solved crystal structures of HAP [15] disproved the previously proposed hypothesis that HAP is serine protease with aspartic protease fold [32], whereas they neither confirmed nor disproved the results of computational studies that postulated Asp215 to be the sole residue directly involved in catalytic mechanism of HAP, with His32 playing only a supporting role [31].
PMII has been the subject of by far the largest number of structural studies, most likely because it was the easiest one to crystallize (Table 1). Several structures of PMIV have also been reported in the last decade, whereas structures of PMI and HAP became available only relatively recently. Taken together, the available structural data provide insights into similarities and differences found among vacuolar plasmepsins and may provide guidance for creation of potent new inhibitors of this family of enzymes.
Table 1.
Crystal structures of vacuolar plasmepsins (P. falciparum unless noted otherwise) that are have been deposited in the Protein Data Bank by March 2011.
| Protein | PDB code | Resol (Å) | Ligand | Year deposited | Reference | Remarks |
|---|---|---|---|---|---|---|
| PMI | 3QRV | 2.4 | - | 2011 | [44] | |
| 3QS1 | 3.1 | KNI-10006 | 2011 | [44] | ||
| PMII | 1SME | 2.7 | Pepstatin A | 1997 | [39] | |
| 1PFZ | 1.85 | - | 1999 | [35] | zymogen | |
| 1LF2 | 1.8 | RS370 | 2002 | [28] | ||
| 1LF3 | 2.7 | EH58 | 2002 | [29] | ||
| 1LF4 | 1.9 | - | 2002 | [29] | ||
| 1LEE | 1.9 | RS367 | 2002 | [28] | ||
| 1M43 | 2.4 | Pepstatin A | 2002 | - | ||
| 1ME6 | 2.7 | Statine-based inhibitor (C25H47N3O6) | 2004 | [41] | ||
| 1XDH | 1.7 | Pepstatin A | 2005 | [81] | ||
| 1XE5 | 2.4 | Pepstatin analogue (C29H47F6N5O9) | 2005 | [81] | ||
| 1XE6 | 2.8 | Pepstatin analogue (C28H45F6N5O9) | 2005 | [81] | ||
| 2BJU | 1.56 | Achiral inhibitor (C37H44N4O3) | 2005 | [78] | ||
| 2IGX | 1.70 | Achiral inhibitor (C41H49N5O2) | 2006 | [79] | ||
| 2IGY | 2.60 | Achiral inhibitor (C35H48N4O) | 2006 | [79] | ||
| 1W6H | 2.2 | Inhibitor (C32H45BrN6O7), bulky P1 side chain | 2006 | [82] | ||
| 1W6I | 2.7 | Pepstatin A | 2006 | [82] | ||
| 2R9B | 2.8 | Reduced peptide inhibitor | 2007 | [58] | ||
| 3F9Q | 1.9 | - | 2009 | [64] | Re-refinement of 1LF4 | |
| HAP | 3FNS | 2.5 | - | 2009 | [15] | |
| 3FNT | 3.3 | Pepstatin A | 2009 | [15] | ||
| 3FNU | 3.0 | KNI-10006 | 2009 | [15] | ||
| 3QVI | 2.2 | KNI-10395 | 2011 | - | ||
| 3QVC | 1.9 | - | 2011 | - | zymogen | |
| PMIV | 1QS8 | 2.5 | Pepstatin A | 1999 | [40] | (P. vivax) |
| 1MIQ | 2.5 | - | 2002 | [40] | zymogen (P. vivax) | |
| 1LS5 | 2.8 | Pepstatin A | 2003 | [29] | ||
| 2ANL | 3.3 | KNI-764 | 2006 | [38] | P. malariae |
2. Primary structure of plasmepsins
Although four vacuolar plasmepsins (PMI, PMII, HAP, and PMIV) have been identified in P. falciparum, other infectious strains of the parasite contain only a single plasmepsin in their food vacuole, an ortholog of PMIV [33]. Similarly to many other proteases, plasmepsins are synthesized as inactive zymogens (proplasmepsins), which contain N-terminal prosegments that are removed during maturation [34]. The zymogen forms of PMI, PMII, HAP, and PMIV contain 452, 453, 451, and 449 amino acid residues, respectively (Fig. 1) [33,35-38]. The prosegments are generally longer in vacuolar proplasmepsins than in other eukaryotic aspartic proteases, in which they are only up to ~50 amino acids long [21,34]. By contrast, prosegments of PMI, PMII, HAP, and PMIV consist of 123, 124, 123, and 121 amino acid residues, respectively. These four plasmepsins exhibit ~63% sequence identity, but are only ~35% homologous to mammalian enzymes renin and cathepsin D [36]. Sequence similarity among pepsin-like proteases does not extend to their prosegments (Fig. 1) [35], which are homologous within subfamilies such as vacuolar plasmepsins, but not throughout the whole family. Prosegments of vacuolar plasmepsins contain 21 amino acids (39p to 59p) which form a transmembrane helix [35] characteristic of type II membrane proteins; this helix provides an anchor to the membrane [36]. Prosegments of other plasmepsins (V-X) differ in their lengths and primary structures.
Figure 1.

Structure-based sequence alignment of proplasmepsins from different human malaria parasites with porcine pepsinogen. The Plasmodium species in this alignment are P. falciparum (pf), P. vivax (pv), P. ovale (po), P. malariae (pm), and P. knowlesi (pk). The alignment was performed with ClustalW [83] and the secondary structure was plotted using ESPript [84]. Similar residues identified by ESPript (global score=0.5) are shown in red letters and identical residues are highlighted by red background. The secondary structural elements as seen in the structure of the zymogen form of pfPMII (1PFZ) are drawn above the sequences. The catalytic residues are marked by stars. The disulfide links are identified as green numbers below the corresponding cysteine residues. The in vivo cleavage site of the prosegment is shown by a red triangle.
Cleavage of prosegements from the zymogens produces soluble active plamepsins with molecular weight of around 37kDa [7,36]. The mature forms of plasmepsins are slightly longer than other eukaryotic aspartic proteases which are ~327 amino acids long. Similarly to other pepsin-like aspartic proteases, signature sequences Asp32-Thr33-Gly34-Ser35 and Asp215-Ser216-Gly217-Thr218 are present in the N- and C-terminal domains, respectively, of PMI, PMII, and PMIV (Fig. 1). However, HAP is a unique plasmepsin since the catalytic Asp32 in the N-terminal domain is replaced by histidine, and Gly34 and Gly217 are replaced by alanines. In addition, other substitutions are also found in the functionally important flap (residues 70-83), with the most important one being the replacement of the catalytically important Tyr75 by Ser (Fig. 1).
3. Structural features of plasmepsins
3.1. The fold of plasmepsin molecules
The first crystal structures of the members of the plasmepsin family were those of PMII, and the number of structures available for this enzyme still surpasses the combined number of structures of the three other vacuolar plasmepsins. The first crystal structure of PMII was published in 1996 [39], followed by a series of high resolution structures of the apoenzyme as well as of complexes with various inhibitors (Table 1). The first structure of PMIV ortholog from P. vivax was determined in 1999 [40], and of P. falciparum PMIV several years later [29]. Although the biochemical properties of HAP [2,41] and of PMI [2,42,43] have been subject to extensive investigations, crystal structures of these two enzymes were solved only very recently [15,44], due to significant difficulties in preparing sufficient amounts of recombinant forms of these proteins. These problems were finally overcome through introduction of unique expression constructs that yielded soluble and active PMI [45] and HAP [37], suitable for crystallographic studies. Table 1 provides a summary of all structures of vacuolar plasmepsins that are currently (April 2011) deposited in the Protein Data Bank (PDB).
A comparison of the structures of these four plasmepsins, in the apo- and complexed forms, shows them to be very similar. The overall fold of PMs is the same as of other eukaryotic aspartic proteases [21], examples of which include porcine pepsin [46], endothiapepsin [47], chymosin [48], renin [49], and cathepsin D [50]. Fig. 1 compares the sequences of proplasmepsins from different Plasmodium species with the sequence of porcine pepsinogen. The secondary structure elements as seen in the structure of proPMII [35] are also marked as an example, since the secondary structure of all vacuolar plasmepsins is very similar, consisting of mainly β sheets and only a few short α helices. A schematic diagram showing the three-dimensional structure of the mature form of PMII is shown in Fig. 2A. The molecule is bilobal, with two topologically similar N- and C-terminal domains related by a pseudo-twofold rotation axis [15,29,38,44]. The substrate-binding cleft which is located between the two domains contains the catalytic residues, Asp32 and Asp215. The amino and carboxyl ends of the polypeptide chain of each PMII domain are assembled into a characteristic six-stranded -sheet which serves to suture the domains together [22]. The N-terminal domain contains a β-hairpin flexible loop, known as flap, which covers the substrate-binding cleft. The flap is usually found in an open conformation in apoenzymes and in closed conformation in the complexes with inhibitors, similarly to the conformations assumed in other pepsin-like aspartic proteases [21,22]. The only exceptions among vacuolar plasmepsins are provided by the structures of PMII complexed with an achiral inhibitor [51] and by HAP complexed with KNI-10006 [15]. In these two structures the flaps assume an open conformation despite the presence of inhibitor molecule(s) bound to the enzymes. Because of its flexibility, a complete flap or its part are not visible in some of the reported crystal structures of plasmepsins [35,44].
Figure 2.

Structures of plasmepsins. (A) Three-dimensional structure of apo-PMII. The secondary structural elements are shown in different colors (magenta for helices, green for strands, and orange for loops and irregular structural elements). Selected residues important for the catalytic mechanism are shown in stick representation. (B) The structure of the HAP dimer (green and orange) in its apo form, with the side chains in one of the active sites shown in stick representation. A Zn2+ ion bound in the active site is shown as a sphere. (C) A unique domain swapped dimer of the HAP-KNI-10395 complex viewed down the 2-fold axis. One protomer is shown in green and the other one in blue.
Vacuolar plasmepsins contain two disulfide bonds, linking together Cys45 with Cys50, and Cys249 with Cys282. The C-terminal disulfide linkage is conserved not only among plasmepsins, but also among all other eukaryotic aspartic proteases [21,39]. The N-terminal disulfide linkage, on the other hand, is also found in mammalian enzymes [22], but not in enzymes from some other organisms, such as fungi. The two loops in the C-terminal domain consisting of residues 238-245 and 276-283 are flexible and assume a variety of different conformations in the plasmepsin structures [15,29,38].
3.2. Plasmepsins create dimers and higher oligomers
Several biochemical and crystallographic studies have attempted to analyze the oligomeric forms of plasmepsins [52,53], but the significance of oligomerization of these enzymes, if any, has not been fully established as yet. It has been shown that monomeric forms of the enzymes are responsible for their activity [52]. Since creation of crystal lattices always involves intermolecular interactions, it is not always easy to determine whether the oligomeric states observed in the crystals are meaningful or not. With that kept in mind, the existence of both crystallographic as well as noncrystallographic dimers of all four vacuolar plasmepsins has been reported [15,29,38,44]. The buried surface area in these dimers ranges from as low as only 609 Å2 (PMIV, 2ANL) to as much as 3789 Å2 (HAP, 3QVI).
The dimeric structures of apo-HAP and HAP-KNI-10395 complex are unique among all plasmepsins. The apoenzyme of HAP was found to make in the crystal a tight dimer (Fig. 2B) involving very close contacts of the C-terminal domains, whereas the N-terminal domains are pointing away from each other [15]. Because of formation of the dimer, the C-terminal helix (residues 225-235) and the loop (residues 238-245) are displaced from their position usually seen in pepsin-like aspartic proteases. As a result of formation of this tight dimer the loop consisting of residues 276-283 of the second molecule is inserted into the putative active site of the first molecule. A zinc ion present in the active site is tetrahedrally coordinated by His32 and Asp215 from one molecule, Glu278A located in the intruding loop of the other molecule, and by a water molecule [15]. Two hydrophobic residues from the same loop, Ile279A and Phe279B, are packed inside a hydrophobic pocket by Phe109A, Ile80, Met104, Ile107, and Val120 of the other molecule [15]. Although HAP is not a metalloprotease, the coordination of the Zn2+ ion in the active site is similar to that observed in some metalloproteases, such as DppA (D-aminopeptidase, 1HI9) [54]. The flap is found in an open conformation.
Crystals of the HAP-KNI-10395 complex contain two tight domain-swapped dimers (A-B and C-D), related by a local 2-fold axis (Fig. 2C) (PDB ID 3QVI; Bhaumik et al., unpublished). As in the apoenzyme, the helix containing residues 225-235 and the following loop composed of residues 238-245 are displaced and the C-terminal loop consisting of residues 276-283 of one molecule is packed in the active site of the other molecule. The most interesting feature of this HAP dimer is swapping of the N-terminal β-strand (residues 0-9) of the enzyme (Fig. 2C). The first β-strand of one monomer forms a part of an antiparallel β-sheet in the other one, forming a number of hydrogen bonds with residues 164-167. The surface area buried upon formation of the domain swapped dimer in this complex is 3789 Å2, calculated for each monomer. Domain swapping has not been reported for other plasmepsins (or, for that matter, any other aspartic proteases).
3.3. Proplasmepsin structures
Similarly to other eukaryotic aspartic proteases, plasmepsins are synthesized as inactive zymogens that must be enzymatically processed in order to remove their prosegment fragments, thus generating active enzymes [55]. The mechanisms of activation of proplasmepsins in vitro and in vivo are different. In vivo, the processing occurs within the conserved sequence: (Y/H)LG*(S/N)XXD [56]. Initially it was proposed that inside the acidic food vacuole of the parasite, proPMI and proPMII are activated by a maturase, likely a cysteine protease, in a process that requires acidic pH [36]. Later Banerjee et al. [56] proposed that, in vivo, proplasmepsins are processed in the acidic condition with similar kinetics by a novel convertase which is not inhibited by a general cysteine protease inhibitor. In vitro, the autoactivation of recombinant proPMII takes place at pH 4.7 by autolysis at the Phe112p-Leu113p bond, 12 residues upstream of the wild type N terminus [35]. It is also important to note that the location of the cleavage site of the recombinant PMII varies depending on the conditions used [42]. It has been reported that in vitro autoactivation of recombinant HAP takes place at Lys119p-Ser120p, four residues upstream of the native cleavage site (Gly123p-Ser-1) [57]. Xiao et al. [45] have also reported that the autoactivation of recombinant PMI takes place in the acidic conditions (pH=4.5-5.5) at Leu116p-Thr117p, seven residues upstream from the native cleavage site, whereas Liu et al. reported an additional cleavage between Phe111p and Phe112p [58] . P. vivax PM (which exhibits the highest sequence identity with P. falciparum PMIV) undergoes autocatalytic activation under acidic conditions at Tyr121p-Leu122p, two residues upstream from the native cleavage site.
Crystal structures of truncated zymogens of PMII, HAP, and pvPMIV have been determined [35,40]. Since part of the prosegment is normally attached to the membrane and thus making zymogens not amenable to crystallization as soluble proteins, only the last 48 residues of the prosegment were present in the expressed constructs. These studies enabled unambiguous interpretation of the structural features responsible for the lack of enzymatic activity of the proplasmepsins, as well as elucidated their mechanism of activation. The three available structures of proplasmepsins show almost identical mode of interactions between the prosegment and the fragments corresponding to the mature enzyme. Interactions with the prosegment introduce large shifts in the position of the N- and C-terminal domains of the enzyme compared to its mature form. The prosegments of plasmepsin zymogens have a clearly defined secondary structure (Fig. 3A), consisting of a β-strand (80p-87p) followed by an α-helix (89p-99p), a helical turn, a second α-helix (103p-113p), and a coil connection to the mature segment. The pro-mature junction containing residues 120p to 1 is not visible in the structure of HAP zymogen because of lack of the corresponding electron density. The prosegment forms a number of hydrogen bonds and hydrophobic interactions with the parts of the molecule that corresponds to mature plasmepsin [35,40]. The pro-mature junction (Gly-Ser/Asn), is present in a “Tyr-Asp” tight β-loop which is positioned in a constrained conformation by several hydrogen bonded interactions (Fig. 3B) [35,40]. A sequence comparison (Fig. 1) shows that both residues responsible for forming the “Tyr-Asp” loop are conserved in all plasmepsin zymogens except for proPMI, where tyrosine is replaced by histidine. Asp2, which is present at the center of the hydrogen bonding network, maintains the structure of “Tyr-Asp” loop. The first 13-14 residues of the mature plasmepsin polypeptide fold mainly in a random coil, with the exception of a single 310 helical turn. This one turn helix is present at one side of the active site cleft. Two important hydrogen bonded interactions conserved in all three zymogen structures are present in this first segment of the mature enzyme. The prosegment is rich in positively charged residues, which help to stabilize proplasmepsins at neutral pH [35].
Figure 3.

The structure of the zymogen of PMII. (A) A schematic chain tracing with the prosegment colored pink, the first 13 residues of the mature PMII polypeptide chain blue, and the remaining portion of the mature PMII green. The two catalytic aspartates are shown in stick representation. (B) Stereoview of the junction between the propeptide and mature PMII. Residues N121p- N3 and I238-F242 are shown with the carbon atoms in gray and green, respectively. (C) The “immature” active site in proPMII. Two active site loops are shown with gray carbons and residues 9-15 are shown with carbons in green. Water molecules are shown as red spheres and hydrogen bonds are marked with dashed lines.
A comparison of the structures of proplasmepsins with those of gastric protease zymogens, porcine pepsinogen A [59] and human progastricsin [60], indicates a significant difference in the mode of inhibition of the catalytic activity [35]. In the zymogens of gastric proteases the active site cleft is inaccessible to the substrates, being blocked by the prosegment [61], but no similar blockage is seen in proplasmepsins. In all three structures the mechanism of inhibition involves physical separation of the domains, preventing creation of the fully formed active sites [40]. It has been shown that the prosegment of proplasmepsins associates with the C-terminal domain of the protein, and together with the mature N terminus forms a “harness” that keeps the two active site aspartates apart (Fig. 3C) [35].
An autoactivation mechanism of plasmepsins under acidic conditions has been proposed based on the results of structural studies [35,40]. Several key salt bridges and a hydrogen bonding network involving Asp or Glu residues are disrupted at low pH due to protonation of Asp2 of the conserved “Tyr-Asp” loop, resulting in disruption of the hydrogen bonds made by its side chain [35]. As a result, the “Tyr-Asp” loop opens up and the length of the prosegment increases by five residues, preventing it from keeping the two domains separated. The loss of several key hydrogen bonds and salt bridges leads to dissociation of the prosegment helices from the C-terminal domain and thus allows the molecule to assume the final active conformation [35].
3.4. Catalytic residues and their environment
Since vacuolar plasmepsins are typical pepsin-like aspartic proteases [21], their active sites resemble those of other members of the family (except for HAP with its unique active site). Structures of plasmepsins with bound peptidic inhibitors delineate the substrate-binding sites located in the large cleft formed between the N- and C-terminal domains of the protein [29,38,44]. Residues directly responsible for the catalytic activity of plasmepsins are Asp32 and Asp215 (Fig. 1) [2]. These two residues are located on the two quasi-symmetric ψ loops [28,40,62]. The main chains of the aspartates are involved in the “fireman's grip” [21,63] hydrogen bonding pattern (Fig. 4A). The side chains of these two active site residues also interact with the adjacent Ser35, Gly34, Gly217, and Thr218. With a single exception, the carboxylate groups of the two catalytic aspartates are nearly exactly coplanar in all published structures [40]. However, re-refinement of the originally deposited structure of the uncomplexed PMII (1LF4) in which the carboxylates were originally reported as coplanar resulted in a model (3F9Q) in which the plane of the carboxylate group of Asp215 is rotated by 66° from its original position [64]. The significance of this observation is still not clear.
Figure 4.

The environment of the active sites of plasmepsins. (A) The active site of apo-PMII. Important water molecules are shown as red spheres. (B) One of the active sites of the apo-HAP dimer. The residues from one protomer are shown with carbons colored gray and Glu278A’ (from the other protomer, marked with a prime) is in magenta. Important water molecules are shown as red spheres. The bound Zn2+ ion is shown as a gray sphere.
The active sites of the apoenzymes contain a water molecule which is bound in between the two aspartates [29,44]. This water molecule is activated by the aspartates during the catalytic process and serves as a nucleophile. Sequence alignment shows (Fig. 1) that vacuolar plasmepsins have a serine in position 216, whereas in other eukaryotic aspartic proteases the equivalent residue is often threonine [21]. Crystal structures of PMI, II and IV show that this substitution does not affect the architecture of the active site and that the same hydrogen bonding pattern is maintained. The other important residues in the active site region are Tyr75 and Trp39. In a majority of the structures of PMI, II and IV complexed with inhibitors the flap is in a closed conformation and the side chain of Tyr75 forms a hydrogen bond with the side chain of Trp39 [29,38,44]. This hydrogen bonded interaction is important for the catalytic activity of PM I, II and IV, the enzymes that utilize the catalytic mechanism common to pepsin-like aspartic proteases [21,22,63,65].
In HAP, the catalytic Asp32 in the N-terminal domain is replaced by His32 [15]; several other substitutions of the functionally important residues are found in the flap area. Importantly, the conserved Tyr75 and Val/Gly76 have been replaced in HAP by Ser and Lys, respectively [15]. Some features of the active sites of the dimeric apoenzyme form of HAP are unique, with each of them containing a tightly bound Zn2+ cation. The ion is tetrahedrally coordinated by the side chains of His32 and Asp215 from one monomer, Glu278A from the other monomer, and a water molecule [15] (Fig. 4B). The presence of Zn2+ cation in the HAP active site has disrupted the conserved catalytically important hydrogen bond between the side chains of Asp215 and Thr218. His32 is hydrogen bonded to the side chain of Ser35 which interacts with Trp39 via a water molecule [15]. The flap is in an open conformation and Ser75 and Lys76 are far away from the active site. However, although the coordination of Zn2+ ion in the HAP active site resembles the coordination of one of the Zn2+ ions in a metalloprotease such as DppA [54], it is not likely that HAP is a metalloprotease [15].
4. Inhibitors of plasmepsins
4.1. General features
The inhibitors used for structural studies of plasmepsins are mechanism based [4], thus a brief summary of the catalytic mechanism of plasmepsins (other than HAP) is in order. Crystals structures of plasmepsins complexed with peptidic inhibitors indicate that a peptide substrate binds to the active site in an extended conformation [15,29]. Similarly to the convention utilized for other proteases [66], the substrate residues (P1-Pn/P1 -Pn ) and the corresponding binding sites (S1-Sn/S1 -Sn ) in the plasmepsin active sites are denoted based on their positions relative to the scissile amide linkage. From the biochemical [2] and structural studies [29,38,44,67] it is clear that PMs utilize the same catalytic mechanism as other aspartic proteases. The water molecule that is activated by the active site aspartates in order to attack the peptide bond of the substrate is seen in the structures of the apoenzymes of PMI and PMII [29,44]. Nucleophilic attack by the activated water molecule creates a tetrahedral intermediate which is further protonated, leading to the break of the peptide bond and creation of the products. Despite both structural [15] and computational [31] studies of HAP, its catalytic mechanism is not yet sufficiently clear.
Most inhibitors of plasmepsins are transition-state analogs containing noncleavable inserts such as reduced amides, statines, hydroxyethylamines, norstatines, dihydroxyethylenes, phosphinates, or difluoroketones [4]. Several approaches have been used to identify potent inhibitors of plasmepsins. Inhibitors of cathepsin D, renin, and HIV-1 PR have been tested for their ability to inhibit plasmepsins, and some of them were also specifically modified for that purpose [39,42]. In particular, the KNI compounds which were initially created in order to inhibit HIV-1 PR, but later shown to be active also against HTLV-1 PR [68], have also been shown to inhibit PMII [69]. The core of many KNI compounds is made of an α-hydroxy-β-amino acid derivative, allophenylnorstatine, which contains a hydroxymethylcarbonyl isostere [41]. One of these inhibitors, KNI-10006, was shown to have broad specificity for plasmepsins, inhibiting PMI, II, HAP and IV [41]. Chemical structures of the inhibitors reported to date in crystallographic studies of plasmepsins are shown in Table 2. The chemical nature and other properties of plasmepsin inhibitors have been previously reviewed in detail [4] and will not be repeated here.
Table 2.
Chemical formulas of the inhibitors bound to plasmepsins in the crystal structures of their complexes deposited in the PDB.
| No | Structure | Name | PDB |
|---|---|---|---|
| 1 |
|
Pepstatin A | 1XDH, 1W6I, 1M43, 1SME, 1QS8, 3FNT, 1LS5 |
| 2 |
|
Statine-based inhibitor | 1ME6 |
| 3 |
|
Pepstatin analogue | 1XE5 |
| 4 |
|
Pepstatin analogue | 1XE6 |
| 5 |

|
Inhibitor with bulky P1 side chain | 1W6H |
| 6 |
|
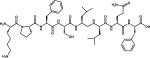
Peptide-based inhibitor | 2R9B |
| 7 |
|
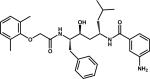
RS367 | 1LEE |
| 8 |
|
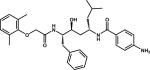
RS370 | 1LF2 |
| 9 |
|
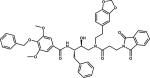
EH58 | 1LF3 |
| 10 |
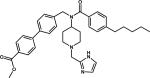
|
Achiral inhibitor | 2BJU |
| 11 |
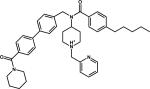
|
Achiral Inhibitor | 2IGX |
| 12 |
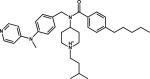
|
Achiral Inhibitor | 2IGY |
| 13 |

|
KNI-764 | 2ANL |
| 14 |
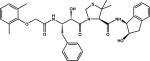
|
KNI-10006 | 3FNU, 3QS1 |
| 15 |
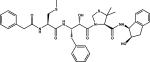
|
KNI-10395 | 3QVI |
4.2. Inhibitor binding to PMII
Binding of inhibitors to the PMII active site has been reviewed previously [4], with the exception of a recently published structure (2R9B) of a complex with a peptidomimetic inhibitor [58]. Some of the structures are available only in the form of PDB deposits and have not been analyzed in detail in primary literature (Table 1). Although several more specific inhibitors of plasmepsins have been utilized, pepstatin A has been used in the majority of the structural studies (Table 1). Pepstatin A, a universal aspartic protease inhibitor, was shown to inhibit hemoglobin degradation by the extract of digestive vacuole of P. falciparum [70]. Since inhibition studies on the recombinant PMII showed that pepstatin A is a picomolar (Ki=0.006 nM) inhibitor for this enzyme [39], this inhibitor was used in the initial crystallographic studies [39]. Several crystal structures of this one and other plasmepsins complexed with pepstatin A and its analogs have been subsequently determined (Table 1). Pepstatin A binds in the active site of PMII in an extended conformation [29,39]. The central hydroxyl group of the inhibitor is inserted in between the carboxylate groups of Asp32 and Asp215. The side chains of Ser77, Tyr189, and Ser219, as well as the main chains of Gly34, Asn74, Val76, Ser77, Gly217, and Ser219 form several hydrogen bonded interactions with pepstatin A (Fig. 5A and Table 3). The binding modes of pepstatin A analogues (Table 1; structures 1XE5, 1XE6, 1W6I, 1ME6) in the PMII active site are similar to the mode seen in the PMII-pepstatin A complexes.
Figure 5.

Inhibitor binding in the active site of PMII. (A) Interactions of pepstatin A with PMII (PDB 1XDH). Pepstatin A is shown in ball-and-stick representation with the carbons colored green. Protein residues are shown as thinner sticks with carbons in gray and hydrogen bonds are marked with dashed lines. (B) Binding mode of one of the two achiral inhibitors found in the crystal structure 2BJU to the “flap pocket” of PMII. The inhibitor is shown in ball-and-stick model with the carbons colored green. Protein residues are shown as thinner sticks with carbons in gray.
Table 3.
Substrate/inhibitor binding pockets of plasmepsins defined by the structures of their complexes with pepstatin A
| Binding pocket | PMI | PMII | PMIV | HAP | Pepsin |
|---|---|---|---|---|---|
| S1 | Ile30 | Ile30 | Ile30 | Leu30 | Ile30 |
| Tyr75 | Tyr75 | Tyr75 | Ser75 | Tyr75 | |
| Phe117 | Phe117 | Phe117 | Val117 | Phe117 | |
| Phe109A | Phe109A | Leu109A | Phe109A | - | |
| Ile120 | Ile120 | Ile120 | Val120 | Ile120 | |
| Gly217 | Gly217 | Gly217 | Ala217 | Gly217 | |
| S2 | Thr218 | Thr218 | Thr218 | Thr218 | Thr218 |
| Thr222 | Thr222 | Thr222 | Thr222 | Thr222 | |
| Ile300 | Ile300 | Ile300 | Val300 | Ile300 | |
| Val289 | Leu289 | Val289 | Ile289 | Met289 | |
| Ile287 | Ile287 | Leu287 | Val287 | Glu287 | |
| S3 | Met13 | Met13 | Met13 | Leu13 | Glu13 |
| Ala111 | Thr111 | Leu111 | Phe111 | Phe111 | |
| S4 | Ile287 | Ile287 | Leu287 | Val287 | Glu287 |
| Ser219 | Ser219 | Ser219 | Ser219 | Ser219 | |
| Ser220 | Ala220 | Thr220 | Val220 | Leu220 | |
| S1′ | Tyr75 | Tyr75 | Tyr75 | Ser75 | Tyr75 |
| Val76 | Val76 | Gly76 | Lys76 | Gly76 | |
| S2′ | Ser35 | Ser35 | Ser35 | Ser35 | Ser35 |
| Met73 | Met73 | Ile73 | Leu73 | Ile73 | |
| Tyr75 | Tyr75 | Tyr75 | Ser75 | Tyr75 | |
| Leu128 | Leu128 | Leu128 | Leu128 | Ile128 | |
| Tyr189 | Tyr189 | Phe189 | Met189 | Tyr189 | |
| S3′ | Val76 | Val76 | Gly76 | Lys76 | Gly76 |
| S4′ | Leu128 | Leu128 | Leu128 | Leu128 | Ile128 |
| Ile130 | Ile130 | Ile130 | Ile130 | Ala130 | |
| Tyr189 | Tyr189 | Phe189 | Met189 | Tyr189 |
The structure of PMII complexed with a statine-based inhibitor containing bulky substituents at positions P1 (p-bromobenzoyloxy) and P3 (Pyridyl) (1W6H) has been described [4]. In this complex, the P1 group is bound in the S1-S3 pocket, making hydrophobic contacts with the side chains of Phe109A and Thr111. The side chain of Phe109A has changed its conformation to provide the stacking interaction to pbromobenzoyloxy group of the inhibitor. Crystal structures of PMII complexed with other inhibitors with bulky substitutions at different substrate binding pockets have also been determined (Table 1; structures 2R9B, 1LF2, 1LF3, 1LEE) [28,29,58]. In the complex 2R9B, the catalytic water molecule is visible because of the absence of the central hydroxyl group in the inhibitor [58]. The N-terminal part of the inhibitor is solvent exposed and the C-terminal phenylalanine group makes hydrophobic interactions with Pro292 from other subunit. In the PMII complexes 2R9B, 1LF2, 1LF3, and 1LEE the active site is much wider compared to the PMII-pepstatin A/pepstatin A analogue complex structures. In these structures the flap and the loop Leu292-Pro297 were displaced from their positions to create more space in the active site. In the PMII-EH58 complex structure, the loop Ile238-Tyr245 assumes a different conformation and Phe242 is involved in hydrophobic interactions with the inhibitor in another subunit [29].
A high resolution crystal structure of PMII complexed with a potent achiral inhibitor containing a 4-aminopiperidine group (2BJU) has been determined [51]. The structure shows a unique mode of binding of two inhibitor molecules accompanied by major conformational changes in the active site area (Fig. 5B). Even though the inhibitor binds in the active site, the flap assumed an open conformation, with its tip lifted by as much as 6 Å (Val80) to 9 Å (Ser81), compared to the pepstatin A bound structure (1SME) [51]. Rotation of Trp39 side chain created a large hydrophobic “flap pocket” [15,71] that is oriented towards the core of the protein. The existence of a similar pocket was noted before in the structure of human renin [72]. The hydrogen bond between the side chains of Trp39 and Tyr75 is disrupted in the structure with the achiral inhibitor and two inhibitor molecules could be unambiguously placed in the active site area. One molecule is deeply buried in the newly formed hydrophobic pocket formed by the side chains of Trp39, Pro41, Val80 Phe109A, Val104, Ile120, and Tyr112 and was proposed to be solely responsible for the high inhibitory activity. The n-pentyl chain of the inhibitor has ideal length and volume to fit optimally in the flap pocket. The second inhibitor molecule makes significantly fewer interactions with the protein molecule and also contacts the loop Ile238-Tyr245 of a neighboring molecule [51]. The tightly bound inner inhibitor molecule occupies the pocket S1 and parts of the S1, S3 and S5 pockets, whereas the loosely bound outer inhibitor molecule occupies the S2, S4, and S6 pockets, as well as the space filled by the backbone of peptidomimetic inhibitors [51]. The catalytic water molecule is also visible in between the two catalytic aspartic acid side chains.
A comparison of the apoenzyme and complexed structures of PMII shows that the enzyme exhibits significant structural flexibility in order to accommodate bulky groups of the inhibitors in the substrate binding cleft. The hydrophobic flap pocket is unique and is utilized for creating an unconventional binding mode of the inhibitor. Among all the complexed structures, the two loop regions composed of residues Ile238-Tyr245 and Tyr274-Asn285 are observed to be in different conformations.
4.3. Inhibitor binding to PMIV
The first crystal structure of P. vivax ortholog of PMIV was solved in 1999 but analyzed only later [40]. It was followed by the structure of PMIV from P. falciparum which was mentioned only in passing [29], and by the subsequently determined structure of P. malariae ortholog [38]. pfPMIV exhibits 69% overall sequence identity and 68% active site identity with pfPMII [73]. The mode of binding of pepstatin A in the PMIV active site (structure 1LS5) is similar to the one observed in the PMII-pepstatin A complexes, despite the differences between some amino acids located in the binding site pockets. Met73 and Val76 of the flap in PMII are replaced in PMIV by the less bulky Ile73 and Gly76, respectively. Other significant differences include Phe109A, Thr111, Ile287, Leu289 and Phe291 in PMII being substituted by Leu109A, Ile111, Leu287, Val289 and Ile291 in PMIV, respectively. Because of these substitutions, the binding pocket of PMIV is more open compared to its counterpart in PMII. A difference between Plasmodium species is the replacement of Tyr189 in pfPMIV by Phe189 in pmPMIV, whereas other important active site residues in the substrate binding pockets of PMIV from these two species are identical.
Crystal structure of pmPMIV complexed with KNI-764 [38] shows an unexpected orientation of the compound in the active site, different from the previous models [74,75]. Although KNI-764 is a peptidomimetic inhibitor, its chain direction is opposite to the direction of a natural substrate or of any other peptidomimetic inhibitors, such as pepstatin A [39]. Because of the inversion of the orientation of the binding of KNI-764, the P1 allophenylnorstatine group of the inhibitor is bound to the S1 pocket of the enzyme, and, conversely, the P1′ dimethylthioproline group is bound to the S1 pocket (Fig. 6). The S2 pocket is occupied by the 2-methylbenzoyl group and the S2 pocket is occupied by the 3-hydroxy-2-methylbenzoyl group. The inhibitor is bound in the active site by several hydrogen bonded interactions which include the hydrogen bonds between the central hydroxyl group of the inhibitor and the two catalytic aspartic acid residues. The allophenylnorstatine moiety makes hydrophobic interactions with Phe189, Ile291 and Ile300. The dimethylthioproline group has hydrophobic contact with the side chains of Tyr75 and Ser77. The 3-hydroxy-2-methylbenzoyl group is making hydrophobic contacts with the side chains of the residues Ile73, Tyr75, Leu128 and Phe189. A water mediated hydrogen bond is present between O2 of 3-hydroxy-2-methylbenzoyl group and the main chain of Thr74. The 2-methylbenzoyl group is bound between the tip of the flap and the loop composed of residues Met283-Asp292 making hydrophobic contacts with the side chains of Thr218, Thr222 and Val289. Computational studies using this complexed structure (2ANL) proposed a different orientation of the 2-methylbenzoyl group [76] because of flipping of the amide bond between P1′-P2′. A flipped amide bond between P1′-P2′ in KNI-10006 inhibitor was observed in the PMI-KNI-10006 complex structure [44] which is discussed below.
Figure 6.

A stereoview showing the binding mode of KNI-764 in the PMIV active site. The inhibitor is shown as ball-and-stick with the carbons colored green. The protein residues are shown as thinner sticks with carbons in gray. A water molecule is shown as a red sphere and hydrogen bonds are dashed.
4.4. Inhibitor binding to PMI
The first crystal structures of PMI, for the apoenzyme and for a complex KNI-10006, have been determined only very recently [44]. PMI shares overall 73% sequence identity and 84% active site identity with PMII [4]. A comparison of the crystal structure of the PMI-KNI-10395 complex with PMII complexed with pepstatin A (1XDH) shows that there are only four variations among the active site residues which could affect inhibitor binding. The differences between PMII and PMI are limited to the substitution of Thr111, Ser115, Leu289, and Phe291 in the former by Ala111, Gly115, Val289 and Leu291 in the latter, respectively (Table 3). The mode of binding of KNI-10006 in the active site of PMI is similar to the one observed in PMIV-KNI-764 complex structure [38], with the peptide chain direction opposite to the putative direction of the substrate. As in other similar structures, the hydroxyl group of the allophenylnorstatine moiety of the inhibitor in the PMI-KNI-10006 complex is placed in between the two catalytic aspartates (Fig. 7). The phenyl moiety of allophenylnorstatine makes hydrophobic contacts with the side chains of Leu291 and Ile300, as well as with Val76 from the flap. The 2,6-dimethylphenyloxymethyl group of the inhibitor is placed in a hydrophobic pocket of the active site, making apolar contacts with Met73, Tyr75, Leu128, Ile130, and Tyr189. The dimethylthioproline moiety of the inhibitor is stacked in a hydrophobic pocket formed by Ile30, Tyr75, Phe109A, and Ile120. The 2-aminoindanol moiety is positioned by forming a hydrogen bond between its hydroxyl group and the main chain NH group of Ser219. It is important to note that the 2-aminoindanol group is also involved in hydrophobic interactions with the side chain of Phe242 from another molecule. Similar hydrophobic interactions were reported in the PMII-EH58 complex [29].
Figure 7.

Stereoview showing the binding mode of KNI-10006 in the PMI active site. The inhibitor is shown as ball-and-stick model with the carbons colored green. Protein residues are shown as thinner sticks, with carbons colored gray for one protomer and magenta for the other protomer. The prime mark on Phe242 indicates that this residue is from a different protomer than Asp32 and Asp215. Hydrogen bonds are marked with dashed lines.
4.5. Inhibitor binding to HAP
P. falciparum HAP is the most divergent vacuolar plasmepsin [2], with no counterpart in other characterized species of Plasmodium. The mature enzyme exhibits 60% overall sequence identity compared to PMII, but only 39% identity in the active site region [41]. Crystal structures of HAP complexed with pepstatin A, KNI-10006, and KNI-10395 have been determined [15].
The overall mode of binding of pepstatin A to HAP (3FNT) resembles the mode of binding of this inhibitor that was previously reported in the structures of PMII (1XDH) [39] and PMIV (1LS5) [29]. The inhibitor is bound in an extended conformation, with the statine hydroxyl positioned between Asp215 and His32. However, orientation of the C-terminal half of the inhibitor is distinctly different from that found in complexes with other pepsin-like proteases, including plasmepsins. Instead of wrapping around the flap, as observed with the other enzymes, this part of the molecule is oriented towards loop 287-292, making extensive interactions with the residues comprising this fragment [15]. The flap is closed in the structure of the complex and Lys76, located at its tip, interacts with the inhibitor via hydrophobic contacts with the side chain of the P3’ Sta residue. The ω-amino group of Lys76 is linked via a hydrogen bond to the carbonyl oxygen of Ala at the P2’ subsite. The shift of the C-terminal half of pepstatin A may be due to the presence in HAP of an unusually large residue at the tip of the flap and its resulting interactions. Only two hydrogen bonds between pepstatin A and HAP are clearly identified, fewer than in complexes with other plasmepsins [29], due to a different orientation of the C-terminal half of the inhibitor, which prevents formation of hydrogen bonds with the flap residues. The side chains of pepstatin A at both termini of the molecule are also involved in extensive hydrophobic interactions with the protein. The isovaleryl group at the N terminus of the inhibitor and the side chain of Val at P3 interact with Val12, Leu13, and Phe111 in HAP. Phe111 corresponds to Thr111 and Ile111 in PMII and IV, respectively (Fig. 1). The P2 Val interacts with the methyl group of the side chain of Thr218, as well as with the side chains of Val287 and Ile289. The side chain of P1 Sta interacts with Phe109A, while Ala at P2’ is involved in hydrophobic interactions with Met189 and Leu291. Finally, the side chain of Glu292 helps to stabilize the conformation of the C terminus of pepstatin A.
The binding mode of KNI-10006 to HAP is significantly different from that of pepstatin A (Fig. 8A), as well as from other KNI inhibitors bound to various aspartic proteases [15]. The hydroxyl group in the central part of the inhibitor points away from the catalytic residues, in contrast to its orientation in the structures of either HIV-1 PR [77] or PMIV [29], in which it is positioned between the active site aspartates. The predominant interactions of KNI-10006 are with the flap and this inhibitor does not make any contacts with either the loop 283-292 or with several other hydrophobic residues conserved in plasmepsins and in other pepsin-like enzymes. However, there is striking similarity in the binding mode of KNI-10006 to HAP and the deeply buried molecule of an achiral inhibitor bound to PMII (2BJU) [78] (Fig. 8B). In the latter structure, two inhibitor molecules are bound to a single PMII molecule, with the second inhibitor molecule oriented in a way that is reminiscent of the binding mode of pepstatin A to HAP. Both the n-pentyl chain of the achiral inhibitor and the 2,6-dimethylphenyloxymethyl (DMP) moiety of KNI-10006 occupy the flap pocket [15]. In the HAP complex with KNI-10006, this pocket is open and the conformation of the flap is similar to its conformation in the apoenzyme. Similarly to the previously described flap pocket of PMII, its counterpart in HAP is predominantly hydrophobic (Fig. 8C). An insertion of Phe109A in HAP and PMII, or Leu109A in PMIV changes the architecture of this pocket compared to other pepsin-like enzymes and makes it even more hydrophobic in plasmepsins. It should be noted, however, that the side chain of Leu112 in pepsin is oriented in such a way that it occupies some of the space taken by the residue 109A in plasmepsins, thus contributing to the interactions with the ligand and partially compensating for the absence of the extra residue in the “flap pocket”. Another important residue, located at the entrance to the flap pocket in HAP is Phe111, substituted by threonine in PMII and by leucine in PMIV. These differences between plasmepsins may influence their preferences for specific ligands.
Figure 8.

Binding of inhibitors in the active site of HAP and a comparison with other plasmepsins. (A) Different binding modes of KNI inhibitors in HAP (green) and PMIV (purple). The flaps are shown in a ribbon representation. (B) Overlay of the inhibitors based on the superposition of the corresponding proteins in the structures of the complexes: pepstatin A (cyan) from HAP complex (3FNT); KNI-10006 (green) from HAP complex (3FNU); and two achiral inhibitor molecules (violet and yellow) from PMII complex (2BJU). (C) Surface representation of the HAP flap pocket. KNI-10006 is shown as ball and stick with carbons in cyan. The surfaces of protein carbon, nitrogen and oxygen atoms are shown in wheat-yellow, blue and red color, respectively.
A surprising feature of the recently determined structure of the HAP-KNI-10395 complex is the presence of a domain swapped dimer and a unique mode of inhibitor binding. The conformation of KNI-10395 is considerably deformed, with the inhibitor chain turning back on itself, thus creating a U-shaped structure. Although the central hydroxyl group of the inhibitor is bound close to the catalytic His32 and Asp215, it is not positioned directly in between these two residues, but is hydrogen bonded to the OD2 of Asp215 via water molecule. The inhibitor forms two intramolecular hydrogen bonds. Because of formation of a tight domain-swapped dimer, the flap pocket is filled by the residues from the loop 276’-283’ from the other subunit. The flap is in an open conformation and the side chain of Trp39 is flipped away from the flap pocket, forming a hydrogen bond with Ser35 through Wat377. The NE2 atom of His32 is hydrogen bonded to the main chain carbonyl oxygen of Ile279A’ and to the side chain carboxyl oxygen (OE2) of Glu278A’ through Wat38.
All three currently available structures of the inhibitor complexes of HAP stress the unique nature of this plasmepsin and its differences from the other vacuolar enzymes. The mechanism-based inhibitors of plasmepsins are effective against this enzyme, although its mechanism is likely not the same as for typical pepsin-like proteases..However, the fact that at least same inhibitors retain activity against all vacuolar plasmepsins bodes well for the possibility of creating universal compounds which could inhibit all these enzymes simultaneously.
4.6. Structure-assisted development of high affinity plasmepsin inhibitors
Vacuolar plasmepsins have been identified as targets for the development of new antimalarial drugs as these enzymes play a key role in the lifecycle of the Plasmodium parasites [4]. Recent characterization and knockout studies of four vacuolar plasmepsins have indicated that these enzymes have overlapping roles [7], thus the most effective inhibitors should be able to cross-react with all four of them. When designed for a primary target, they should be capable of maintaining their high affinity against other vacuolar plasmepsins by adjusting their conformations. Molecules with such properties have been named “adaptive inhibitors” [73] and their development relies on full understating of the differences among the active sites of plasmepsins. Despite high overall sequence identity between vacuolar plasmepsins (50-79%) [15], there are some important substitutions in the active sites (Table 3). Because of the paucity of structural data, most of the initial inhibitor design was done using PMII as a target [73]. A number of potent achiral inhibitors active against PMI, PMII, and PMIV have been developed, but they have not been tested against HAP [79].
The development of high affinity inhibitors of vacuolar plasmepsins has been aided by detailed analysis of their active sites [73,80]. It has been proposed that the KNI compounds containing flexible and asymmetric functional groups could adjust well to the different active site environment [41,73]. Biochemical studies have shown that KNI-10006 inhibits well all four vacuolar plasmepsins [41]. Structural studies of the KNI compounds have also progressed, with the determination of the crystal structure of PMIV-KNI-764 complex [38]. Recent crystal structures of PMI and HAP complexed with KNI-10006 elucidated different modes of binding of the same compound in the active sites of these two enzymes [15,44]. These crystal structures clearly emphasized the flexibility of KNI-10006, a compound that might serve as a lead molecule to develop high affinity inhibitors for all vacuolar plasmepsins. Based on the crystal structures of HAP-KNI-10006 and PMIV-KNI-764 complexes, several new KNI compounds have already been developed [19]. Among those new compounds, KNI-10743 and KNI-10742 show extremely potent inhibitory activity against PMII, whereas KNI-10740 exhibited the most potent antimalarial activity in the series [19]. From the structural analysis presented in this review, it is likely that KNI-10743 and KNI-10742 should also inhibit PMI, PMIV, and HAP with very high affinity.
5. Conclusion
Crystal structures of the zymogens, apoenzymes, and inhibited forms of vacuolar plasmepsins have provided extensive information about these closely related enzymes with redundant activity. It has been shown that plasmepsin zymogens differ in the way that they protect the active site from the zymogens of other aspartic proteases. Detailed studies of the inhibitor complexes of plasmepsins, sometimes using the same inhibitor for multiple enzymes, have elucidated both the similarities and the differences between them, helping in the design of either very specific or less specific compounds. Whereas the mode of activity of PMI, PMII, and PMIV is well understood, HAP presents still something of a puzzle and much more work will be needed before its mode of action will be clear. The success of the future development of antimalarial drugs active against these enzymes will depend on better understanding whether it is their inhibition that leads to anti-parasitic properties of the inhibitors of aspartic proteases, or whether such inhibitors are active against enzymes other than vacuolar plasmepsins. There is still hope, however, that research on structural properties of vacuolar plasmepsins may lead to practical results.
Acknowledgement
We are grateful to Professor Ben Dunn for constructive criticism of a draft of this manuscript. This project was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Greenwood BM, Bojang K, Whitty CJ, Targett GA. Malaria. Lancet. 2005;365:1487–1498. doi: 10.1016/S0140-6736(05)66420-3. [DOI] [PubMed] [Google Scholar]
- 2.Banerjee R, Liu J, Beatty W, Pelosof L, Klemba M, Goldberg DE. Four plasmepsins are active in the Plasmodium falciparum food vacuole, including a protease with an active-site histidine. Proc. Natl. Acad. Sci. USA. 2002;99:990–995. doi: 10.1073/pnas.022630099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fong YL, Cadigan FC, Coatney GR. A presumptive case of naturally occurring Plasmodium knowlesi malaria in man in Malaysia. Trans. R. Soc. Trop. Med. Hyg. 1971;65:839–840. doi: 10.1016/0035-9203(71)90103-9. [DOI] [PubMed] [Google Scholar]
- 4.Ersmark K, Samuelsson B, Hallberg A. Plasmepsins as potential targets for new antimalarial therapy. Med. Res. Rev. 2006;26:626–666. doi: 10.1002/med.20082. [DOI] [PubMed] [Google Scholar]
- 5.Kremsner PG, Krishna S. Antimalarial combinations. Lancet. 2004;364:285–294. doi: 10.1016/S0140-6736(04)16680-4. [DOI] [PubMed] [Google Scholar]
- 6.Francis SE, Sullivan DJ, Jr., Goldberg DE. Hemoglobin metabolism in the malaria parasite Plasmodium falciparum. Annu. Rev. Microbiol. 1997;51:97–123. doi: 10.1146/annurev.micro.51.1.97. [DOI] [PubMed] [Google Scholar]
- 7.Liu J, Gluzman IY, Drew ME, Goldberg DE. The role of Plasmodium falciparum food vacuole plasmepsins. J. Biol. Chem. 2005;280:1432–1437. doi: 10.1074/jbc.M409740200. [DOI] [PubMed] [Google Scholar]
- 8.Sherman IW, Tanigoshi L. Incorporation of 14C-amino-acids by malaria (plasmodium lophurae) IV. In vivo utilization of host cell haemoglobin. Int. J. Biochem. 1970;1:635–637. [Google Scholar]
- 9.Esposito A, Tiffert T, Mauritz JM, Schlachter S, Bannister LH, Kaminski CF, Lew VL. FRET imaging of hemoglobin concentration in Plasmodium falciparum-infected red cells. PLoS ONE. 2008;3:e3780. doi: 10.1371/journal.pone.0003780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coombs GH, Goldberg DE, Klemba M, Berry C, Kay J, Mottram JC. Aspartic proteases of Plasmodium falciparum and other parasitic protozoa as drug targets. Trends Parasitol. 2001;17:532–537. doi: 10.1016/s1471-4922(01)02037-2. [DOI] [PubMed] [Google Scholar]
- 11.Francis SE, Gluzman IY, Oksman A, Knickerbocker A, Mueller R, Bryant ML, Sherman DR, Russell DG, Goldberg DE. Molecular characterization and inhibition of a Plasmodium falciparum aspartic hemoglobinase. EMBO J. 1994;13:306–317. doi: 10.1002/j.1460-2075.1994.tb06263.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hill J, Tyas L, Phylip LH, Kay J, Dunn BM, Berry C. High level expression and characterisation of Plasmepsin II, an aspartic proteinase from Plasmodium falciparum. FEBS Lett. 1994;352:155–158. doi: 10.1016/0014-5793(94)00940-6. [DOI] [PubMed] [Google Scholar]
- 13.Russo I, Babbitt S, Muralidharan V, Butler T, Oksman A, Goldberg DE. Plasmepsin V licenses Plasmodium proteins for export into the host erythrocyte. Nature. 2010;463:632–636. doi: 10.1038/nature08726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boddey JA, Hodder AN, Gunther S, Gilson PR, Patsiouras H, Kapp EA, Pearce JA, Koning-Ward TF, Simpson RJ, Crabb BS, Cowman AF. An aspartyl protease directs malaria effector proteins to the host cell. Nature. 2010;463:627–631. doi: 10.1038/nature08728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bhaumik P, Xiao H, Parr CL, Kiso Y, Gustchina A, Yada RY, Wlodawer A. Crystal structures of the histo-aspartic protease (HAP) from Plasmodium falciparum. J. Mol. Biol. 2009;388:520–540. doi: 10.1016/j.jmb.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carroll CD, Patel H, Johnson TO, Guo T, Orlowski M, He ZM, Cavallaro CL, Guo J, Oksman A, Gluzman IY, Connelly J, Chelsky D, Goldberg DE, Dolle RE. Identification of potent inhibitors of Plasmodium falciparum plasmepsin II from an encoded statine combinatorial library. Bioorg. Med. Chem. Lett. 1998;8:2315–2320. doi: 10.1016/s0960-894x(98)00419-3. [DOI] [PubMed] [Google Scholar]
- 17.Johansson PO, Chen Y, Belfrage AK, Blackman MJ, Kvarnstrom I, Jansson K, Vrang L, Hamelink E, Hallberg A, Rosenquist A, Samuelsson B. Design and synthesis of potent inhibitors of the malaria aspartyl proteases plasmepsin I and II. Use of solid-phase synthesis to explore novel statine motifs. J. Med. Chem. 2004;47:3353–3366. doi: 10.1021/jm031106i. [DOI] [PubMed] [Google Scholar]
- 18.Hidaka K, Kimura T, Ruben AJ, Uemura T, Kamiya M, Kiso A, Okamoto T, Tsuchiya Y, Hayashi Y, Freire E, Kiso Y. Antimalarial activity enhancement in hydroxymethylcarbonyl (HMC) isostere-based dipeptidomimetics targeting malarial aspartic protease plasmepsin. Bioorg. Med. Chem. 2008;16:10049–10060. doi: 10.1016/j.bmc.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miura T, Hidaka K, Uemura T, Kashimoto K, Hori Y, Kawasaki Y, Ruben AJ, Freire E, Kimura T, Kiso Y. Improvement of both plasmepsin inhibitory activity and antimalarial activity by 2-aminoethylamino substitution. Bioorg. Med. Chem. Lett. 2010;20:4836–4839. doi: 10.1016/j.bmcl.2010.06.099. [DOI] [PubMed] [Google Scholar]
- 20.Moura PA, Dame JB, Fidock DA. Role of Plasmodium falciparum digestive vacuole plasmepsins in the specificity and antimalarial mode of action of cysteine and aspartic protease inhibitors. Antimicrob. Agents Chemother. 2009;53:4968–4978. doi: 10.1128/AAC.00882-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davies DR. The structure and function of the aspartic proteinases. Annu. Rev. Biophys. Biophys. Chem. 1990;19:189–215. doi: 10.1146/annurev.bb.19.060190.001201. [DOI] [PubMed] [Google Scholar]
- 22.Dunn BM. Structure and mechanism of the pepsin-like family of aspartic peptidases. Chem. Rev. 2002;102:4431–4458. doi: 10.1021/cr010167q. [DOI] [PubMed] [Google Scholar]
- 23.Andreeva NS, Rumsh LD. Analysis of crystal structures of aspartic proteinases: On the role of amino acid residues adjacent to the catalytic site of pepsin-like enzymes. Protein Sci. 2001;10:2439–2450. doi: 10.1110/ps.25801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blundell TL. The Aspartic Proteinases - an Historical Overview. In: James MNG, editor. The Aspartic Proteinases. Plenum Press; 1998. pp. 1–13. [PubMed] [Google Scholar]
- 25.Suguna K, Bott R, Padlan E, Subramanian E, Sheriff S, Cohen G, Davies D. Structure and refinement at 1.8 Å resolution of the aspartic proteinase from Rhizopus chinensis. J. Mol. Biol. 1987;196:877–900. doi: 10.1016/0022-2836(87)90411-6. [DOI] [PubMed] [Google Scholar]
- 26.Coates L, Tuan HF, Tomanicek S, Kovalevsky A, Mustyakimov M, Erskine P, Cooper J. The catalytic mechanism of an aspartic proteinase explored with neutron and X-ray diffraction. J. Am. Chem. Soc. 2008;130:7235–7237. doi: 10.1021/ja801269x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suguna K, Padlan EA, Smith CW, Carlson WD, Davies DR. Binding of a reduced peptide inhibitor to the aspartic proteinase from Rhizopus chinensis: Implications for a mechanism of action. Proc. Natl. Acad. Sci. USA. 1987;84:7009–7013. doi: 10.1073/pnas.84.20.7009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Asojo OA, Afonina E, Gulnik SV, Yu B, Erickson JW, Randad R, Medjahed D, Silva AM. Structures of Ser205 mutant plasmepsin II from Plasmodium falciparum at 1.8 Å in complex with the inhibitors rs367 and rs370. Acta Crystallogr. 2002;D58:2001–2008. doi: 10.1107/s0907444902014695. [DOI] [PubMed] [Google Scholar]
- 29.Asojo OA, Gulnik SV, Afonina E, Yu B, Ellman JA, Haque TS, Silva AM. Novel uncomplexed and complexed structures of plasmepsin II, an aspartic protease from Plasmodium falciparum. J. Mol. Biol. 2003;327:173–181. doi: 10.1016/s0022-2836(03)00036-6. [DOI] [PubMed] [Google Scholar]
- 30.Gupta D, Yedidi RS, Varghese S, Kovari LC, Woster PM. Mechanism-based inhibitors of the aspartyl protease plasmepsin II as potential antimalarial agents. J. Med. Chem. 2010;53:4234–4247. doi: 10.1021/jm100233b. [DOI] [PubMed] [Google Scholar]
- 31.Bjelic S, Aqvist J. Computational prediction of structure, substrate binding mode, mechanism, and rate for a malaria protease with a novel type of active site. Biochemistry. 2004;43:14521–14528. doi: 10.1021/bi048252q. [DOI] [PubMed] [Google Scholar]
- 32.Andreeva N, Bogdanovich P, Kashparov I, Popov M, Stengach M. Is histoaspartic protease a serine protease with a pepsin-like fold? Proteins. 2004;55:705–710. doi: 10.1002/prot.20078. [DOI] [PubMed] [Google Scholar]
- 33.Dame JB, Yowell CA, Omara-Opyene L, Carlton JM, Cooper RA, Li T. Plasmepsin 4, the food vacuole aspartic proteinase found in all Plasmodium spp. infecting man. Mol. Biochem. Parasitol. 2003;130:1–12. doi: 10.1016/s0166-6851(03)00137-3. [DOI] [PubMed] [Google Scholar]
- 34.Khan AR, James MNG. Molecular mechanisms for the conversion of zymogens to active proteolytic enzymes. Protein Sci. 1998;7:815–836. doi: 10.1002/pro.5560070401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bernstein NK, Cherney MM, Loetscher H, Ridley RG, James MN. Crystal structure of the novel aspartic proteinase zymogen proplasmepsin II from Plasmodium falciparum. Nature Struct. Biol. 1999;6:32–37. doi: 10.1038/4905. [DOI] [PubMed] [Google Scholar]
- 36.Francis SE, Banerjee R, Goldberg DE. Biosynthesis and maturation of the malaria aspartic hemoglobinases plasmepsins I and II. J. Biol. Chem. 1997;272:14961–14968. doi: 10.1074/jbc.272.23.14961. [DOI] [PubMed] [Google Scholar]
- 37.Xiao H, Sinkovits AF, Bryksa BC, Ogawa M, Yada RY. Recombinant expression and partial characterization of an active soluble histo-aspartic protease from Plasmodium falciparum. Protein Expr. Purif. 2006;49:88–94. doi: 10.1016/j.pep.2006.02.022. [DOI] [PubMed] [Google Scholar]
- 38.Clemente JC, Govindasamy L, Madabushi A, Fisher SZ, Moose RE, Yowell CA, Hidaka K, Kimura T, Hayashi Y, Kiso Y, Agbandje-McKenna M, Dame JB, Dunn BM, McKenna R. Structure of the aspartic protease plasmepsin 4 from the malarial parasite Plasmodium malariae bound to an allophenylnorstatine-based inhibitor. Acta Crystallogr. 2006;D62:246–252. doi: 10.1107/S0907444905041260. [DOI] [PubMed] [Google Scholar]
- 39.Silva AM, Lee AY, Gulnik SV, Maier P, Collins J, Bhat TN, Collins PJ, Cachau RE, Luker KE, Gluzman IY, Francis SE, Oksman A, Goldberg DE, Erickson JW. Structure and inhibition of plasmepsin II, a hemoglobin- degrading enzyme from Plasmodium falciparum. Proc. Natl. Acad. Sci. USA. 1996;93:10034–10039. doi: 10.1073/pnas.93.19.10034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bernstein NK, Cherney MM, Yowell CA, Dame JB, James MN. Structural insights into the activation of P. vivax plasmepsin. J. Mol. Biol. 2003;329:505–524. doi: 10.1016/s0022-2836(03)00444-3. [DOI] [PubMed] [Google Scholar]
- 41.Nezami A, Kimura T, Hidaka K, Kiso A, Liu J, Kiso Y, Goldberg DE, Freire E. High-affinity inhibition of a family of Plasmodium falciparum proteases by a designed adaptive inhibitor. Biochemistry. 2003;42:8459–8464. doi: 10.1021/bi034131z. [DOI] [PubMed] [Google Scholar]
- 42.Moon RP, Tyas L, Certa U, Rupp K, Bur D, Jacquet C, Matile H, Loetscher H, Grueninger-Leitch F, Kay J, Dunn BM, Berry C, Ridley RG. Expression and characterisation of plasmepsin I from Plasmodium falciparum. Eur. J. Biochem. 1997;244:552–560. doi: 10.1111/j.1432-1033.1997.00552.x. [DOI] [PubMed] [Google Scholar]
- 43.Luker KE, Francis SE, Gluzman IY, Goldberg DE. Kinetic analysis of plasmepsins I and II aspartic proteases of the Plasmodium falciparum digestive vacuole. Mol. Biochem. Parasitol. 1996;79:71–78. doi: 10.1016/0166-6851(96)02651-5. [DOI] [PubMed] [Google Scholar]
- 44.Bhaumik P, Horimoto Y, Xiao H, Miura T, Hidaka K, Kiso Y, Wlodawer A, Yada RY, Gustchina A. Crystal structures of the free and inhibited forms of plasmepsin I (PMI) from Plasmodium falciparum. J. Struct. Biol. 2011 doi: 10.1016/j.jsb.2011.04.009. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xiao H, Tanaka T, Ogawa M, Yada RY. Expression and enzymatic characterization of the soluble recombinant plasmepsin I from Plasmodium falciparum. Protein Eng. Des. Sel. 2007;20:625–633. doi: 10.1093/protein/gzm066. [DOI] [PubMed] [Google Scholar]
- 46.Andreeva NS, Zdanov A, Gustchina AE, Fedorov AA. Structure of ethanol-inhibited porcine pepsin at 2 Å resolution and binding of the methyl ester of phenylalanyl-diiodotyrosine to the enzyme. J. Biol. Chem. 1984;259:11353–11366. [PubMed] [Google Scholar]
- 47.Erskine PT, Coates L, Mall S, Gill RS, Wood SP, Myles DA, Cooper JB. Atomic resolution analysis of the catalytic site of an aspartic proteinase and an unexpected mode of binding by short peptides. Protein Sci. 2003;12:1741–1749. doi: 10.1110/ps.0305203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gilliland GL, Winborne EL, Nachman J, Wlodawer A. The three-dimensional structure of recombinant bovine chymosin at 2.3 Å resolution. Proteins. 1990;8:82–101. doi: 10.1002/prot.340080110. [DOI] [PubMed] [Google Scholar]
- 49.Foundling SI, Cooper J, Watson FE, Cleasby A, Pearl LH, Sibanda BL, Hemmings A, Wood SP, Blundell TL, Valler MJ. High resolution X-ray analyses of renin inhibitor-aspartic proteinase complexes. Nature. 1987;327:349–352. doi: 10.1038/327349a0. [DOI] [PubMed] [Google Scholar]
- 50.Baldwin ET, Bhat TN, Gulnik S, Hosur MV, Sowder RC, II, Cachau RE, Collins J, Silva AM, Erickson JW. Crystal structures of native and inhibited forms of human cathepsin D: implications for lysosomal targeting and drug design. Proc. Natl. Acad. Sci. USA. 1993;90:6796–6800. doi: 10.1073/pnas.90.14.6796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Prade L, Jones AF, Boss C, Richard-Bildstein S, Meyer S, Binkert C, Bur D. X-ray structure of plasmepsin II complexed with a potent achiral inhibitor. J Biol. Chem. 2005;280:23837–23843. doi: 10.1074/jbc.M501519200. [DOI] [PubMed] [Google Scholar]
- 52.Liu J, Istvan ES, Goldberg DE. Hemoglobin-degrading plasmepsin II is active as a monomer. J. Biol. Chem. 2006;281:38682–38688. doi: 10.1074/jbc.M608535200. [DOI] [PubMed] [Google Scholar]
- 53.Xiao H, Briere LA, Dunn SD, Yada RY. Characterization of the monomerdimer equilibrium of recombinant histo-aspartic protease from Plasmodium falciparum. Mol. Biochem. Parasitol. 2010;173:17–24. doi: 10.1016/j.molbiopara.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 54.Remaut H, Bompard-Gilles C, Goffin C, Frere JM, Van Beeumen J. Structure of the Bacillus subtilis D-aminopeptidase DppA reveals a novel self-compartmentalizing protease. Nature Struct. Biol. 2001;8:674–678. doi: 10.1038/90380. [DOI] [PubMed] [Google Scholar]
- 55.Koelsch G, Mares M, Metcalf P, Fusek M. Multiple functions of pro-parts of aspartic proteinase zymogens. FEBS Lett. 1994;343:6–10. doi: 10.1016/0014-5793(94)80596-2. [DOI] [PubMed] [Google Scholar]
- 56.Banerjee R, Francis SE, Goldberg DE. Food vacuole plasmepsins are processed at a conserved site by an acidic convertase activity in Plasmodium falciparum. Mol. Biochem. Parasitol. 2003;129:157–165. doi: 10.1016/s0166-6851(03)00119-1. [DOI] [PubMed] [Google Scholar]
- 57.Parr CL, Tanaka T, Xiao H, Yada RY. The catalytic significance of the proposed active site residues in Plasmodium falciparum histoaspartic protease. FEBS J. 2008;275:1698–1707. doi: 10.1111/j.1742-4658.2008.06325.x. [DOI] [PubMed] [Google Scholar]
- 58.Liu P, Marzahn MR, Robbins AH, Gutierrez-de-Teran H, Rodriguez D, McClung SH, Stevens SM, Jr., Yowell CA, Dame JB, McKenna R, Dunn BM. Recombinant plasmepsin 1 from the human malaria parasite Plasmodium falciparum: enzymatic characterization, active site inhibitor design, and structural analysis. Biochemistry. 2009;48:4086–4099. doi: 10.1021/bi802059r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.James MNG, Sielecki AR. Molecular structure of an aspartic proteinase zymogen, porcine pepsinogen, at 1.8 Å resolution. Nature. 1986;319:33–38. doi: 10.1038/319033a0. [DOI] [PubMed] [Google Scholar]
- 60.Moore SA, Sielecki AR, Chernaia MM, Tarasova NI, James MNG. Crystal and molecular structures of human progastricsin at 1.62 Å resolution. J. Mol. Biol. 1995;247:466–485. doi: 10.1006/jmbi.1994.0154. [DOI] [PubMed] [Google Scholar]
- 61.Sielecki AR, Fujinaga M, Read RJ, James MN. Refined structure of porcine pepsinogen at 1.8 Å resolution. J. Mol. Biol. 1991;219:671–692. doi: 10.1016/0022-2836(91)90664-r. [DOI] [PubMed] [Google Scholar]
- 62.Andreeva NS, Gustchina AE. On the supersecondary structure of acid proteases. Biochem. Biophys. Res. Commun. 1979;87:32–42. doi: 10.1016/0006-291x(79)91643-7. [DOI] [PubMed] [Google Scholar]
- 63.Pearl L, Blundell T. The active site of aspartic proteinases. FEBS Lett. 1984;174:96–101. doi: 10.1016/0014-5793(84)81085-6. [DOI] [PubMed] [Google Scholar]
- 64.Robbins AH, Dunn BM, Agbandje-McKenna M, McKenna R. Crystallographic evidence for noncoplanar catalytic aspartic acids in plasmepsin II resides in the Protein Data Bank. Acta Crystallogr. 2009;D65:294–296. doi: 10.1107/S0907444908041632. [DOI] [PubMed] [Google Scholar]
- 65.Suguna K, Padlan EA, Bott R, Boger J, Parris KD, Davies DR. Structures of complexes of rhizopuspepsin with pepstatin and other statine-containing inhibitors. Proteins. 1992;13:195–205. doi: 10.1002/prot.340130303. [DOI] [PubMed] [Google Scholar]
- 66.Schechter I, Berger A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967;27:157–162. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
- 67.Friedman R, Caflisch A. The protonation state of the catalytic aspartates in plasmepsin II. FEBS Lett. 2007;581:4120–4124. doi: 10.1016/j.febslet.2007.07.033. [DOI] [PubMed] [Google Scholar]
- 68.Maegawa H, Kimura T, Arii Y, Matsui Y, Kasai S, Hayashi Y, Kiso Y. Identification of peptidomimetic HTLV-I protease inhibitors containing hydroxymethylcarbonyl (HMC) isostere as the transition-state mimic. Bioorg. Med. Chem. Lett. 2004;14:5925–5929. doi: 10.1016/j.bmcl.2004.09.034. [DOI] [PubMed] [Google Scholar]
- 69.Nezami A, Luque I, Kimura T, Kiso Y, Freire E. Identification and characterization of allophenylnorstatine-based inhibitors of plasmepsin II, an antimalarial target. Biochemistry. 2002;41:2273–2280. doi: 10.1021/bi0117549. [DOI] [PubMed] [Google Scholar]
- 70.Gluzman IY, Francis SE, Oksman A, Smith CE, Duffin KL, Goldberg DE. Order and specificity of the Plasmodium falciparum hemoglobin degradation pathway. J. Clin. Invest. 1994;93:1602–1608. doi: 10.1172/JCI117140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zurcher M, Gottschalk T, Meyer S, Bur D, Diederich F. Exploring the flap pocket of the antimalarial target plasmepsin II: the “55 % rule” applied to enzymes. ChemMedChem. 2008;3:237–240. doi: 10.1002/cmdc.200700236. [DOI] [PubMed] [Google Scholar]
- 72.Oefner C, Binggeli A, Breu V, Bur D, Clozel JP, D'Arcy A, Dorn A, Fischli W, Gruninger F, Guller R, Hirth G, Marki H, Mathews S, ller M, Ridley RG, Stadler H, Vieira E, Wilhelm M, Winkler F, Wostl W. Renin inhibition by substituted piperidines: a novel paradigm for the inhibition of monomeric aspartic proteinases? Chem. Biol. 1999;6:127–131. doi: 10.1016/s1074-5521(99)89004-8. [DOI] [PubMed] [Google Scholar]
- 73.Nezami A, Freire E. The integration of genomic and structural information in the development of high affinity plasmepsin inhibitors. Int. J. Parasitol. 2002;32:1669–1676. doi: 10.1016/s0020-7519(02)00196-0. [DOI] [PubMed] [Google Scholar]
- 74.Abdel-Rahman HM, Kimura T, Hidaka K, Kiso A, Nezami A, Freire E, Hayashi Y, Kiso Y. Design of inhibitors against HIV, HTLV-I, and Plasmodium falciparum aspartic proteases. Biol. Chem. 2004;385:1035–1039. doi: 10.1515/BC.2004.134. [DOI] [PubMed] [Google Scholar]
- 75.Kiso A, Hidaka K, Kimura T, Hayashi Y, Nezami A, Freire E, Kiso Y. Search for substrate-based inhibitors fitting the S2' space of malarial aspartic protease plasmepsin II. J. Pept. Sci. 2004;10:641–647. doi: 10.1002/psc.609. [DOI] [PubMed] [Google Scholar]
- 76.Gutierrez-de-Teran H, Nervall M, Ersmark K, Liu P, Janka LK, Dunn B, Hallberg A, Aqvist J. Inhibitor binding to the plasmepsin IV aspartic protease from Plasmodium falciparum. Biochemistry. 2006;45:10529–10541. doi: 10.1021/bi0609669. [DOI] [PubMed] [Google Scholar]
- 77.Fitzgerald PMD, McKeever BM, VanMiddlesworth JF, Springer JP, Heimbach JC, Leu C-T, Herber WK, Dixon RAF, Darke PL. Crystallographic analysis of a complex between human immunodeficiency virus type 1 protease and acetyl-pepstatin at 2.0 Å resolution. J. Biol. Chem. 1990;265:14209–14219. [PubMed] [Google Scholar]
- 78.Prade L, Jones AF, Boss C, Richard-Bildstein S, Meyer S, Binkert C, Bur D. X-ray structure of plasmepsin II complexed with a potent achiral inhibitor. J. Biol. Chem. 2005;280:23837–23843. doi: 10.1074/jbc.M501519200. [DOI] [PubMed] [Google Scholar]
- 79.Boss C, Corminboeuf O, Grisostomi C, Meyer S, Jones AF, Prade L, Binkert C, Fischli W, Weller T, Bur D. Achiral, cheap, and potent inhibitors of Plasmepsins I, II, and IV. ChemMedChem. 2006;1:1341–1345. doi: 10.1002/cmdc.200600223. [DOI] [PubMed] [Google Scholar]
- 80.Bhargavi R, Sastry GM, Murty US, Sastry GN. Structural and active site analysis of plasmepsins of Plasmodium falciparum: potential anti-malarial targets. Int. J. Biol. Macromol. 2005;37:73–84. doi: 10.1016/j.ijbiomac.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 81.Binkert C, Frigerio M, Jones A, Meyer S, Pesenti C, Prade L, Viani F, Zanda M. Replacement of isobutyl by trifluoromethyl in pepstatin A selectively affects inhibition of aspartic proteinases. Chembiochem. 2006;7:181–186. doi: 10.1002/cbic.200500180. [DOI] [PubMed] [Google Scholar]
- 82.Johansson PO, Lindberg J, Blackman MJ, Kvarnstrom I, Vrang L, Hamelink E, Hallberg A, Rosenquist A, Samuelsson B. Design and synthesis of potent inhibitors of plasmepsin I and II: X-ray crystal structure of inhibitor in complex with plasmepsin II. J Med. Chem. 2005;48:4400–4409. doi: 10.1021/jm040884n. [DOI] [PubMed] [Google Scholar]
- 83.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gouet P, Courcelle E, Stuart DI, Metoz F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]