

Abstract
Endosomal sorting complexes required for transport (ESCRT) recognize ubiquitinated cargo and catalyze diverse budding processes including multivesicular body biogenesis, enveloped virus egress and cytokinesis. We present the crystal structure of an N-terminal fragment of the deubiquitinating enzyme AMSH (AMSHΔC) in complex with the C-terminal region of ESCRT-III CHMP3 (CHMP3ΔN). AMSHΔC folds into an elongated 90 Å long helical assembly that includes an unusual MIT domain. CHMP3ΔN is unstructured in solution and helical in complex with AMSHΔC, revealing a novel MIT domain interacting motif (MIM) that does not overlap with the CHMP1-AMSH binding site. ITC and SPR measurements demonstrate an unusual high affinity MIM-MIT interaction. Structural analysis suggests a regulatory role for the N-terminal helical segment of AMSHΔC and its destabilization leads to a loss of function during HIV-1 budding. Our results indicate a tight coupling of ESCRT-III CHMP3 and AMSH functions and provide novel insight into the regulation of ESCRT-III.
Keywords: AMSH, CHMP3, CHMP1A, CHMP1B, ESCRT-III, ubiquitin, HIV-1
Introduction
Internalization of plasma membrane receptors by endocytosis is regulated by ubiquitination and leads to receptor sorting into intralumenal vesicles at the endosome giving rise to multivesicular bodies [1–3]. The sorting process is catalyzed by ESCRTs (Endosomal sorting complexes required for transport) and their associated molecules [2, 4, 5]. Part of the same machinery is recruited by some enveloped viruses to facilitate budding [6–9] and during cytokinesis of eukaryotes [10–12] and Crenarchaea [13, 14].
The ESCRT pathway is linked to the activity of deubiquitinating enzymes (deubiquitinase, DUB, or ubiquin hydrolase or deubiquitylating enzyme) in order to maintain a constant pool of ubiquitin [15]. DUB Doa4 has an essential recycling role in the yeast MVB pathway [16] and controls the function of ESCRTs [17]. The mammalian MVB pathway employs several DUBs including AMSH (associated molecule with the Src homology 3 domain of STAM, also known as STAMBP) [18], AMSH-LP (AMSH-like protein) [19] and UBPY (Ub-specific protease Y) [20]. Both AMSH and UBPY are recruited early in the pathway via interaction with the ESCRT-0 subunit STAM [21–23] and their localization to early endosomes is supported by interaction with the clathrin heavy chain [24].
Deubiquitination by AMSH is thought to precede cargo incorporation into MVB vesicles and lysosomal degradation [25, 26]. Depletion of AMSH was reported to increase degradation of epidermal growth factor receptor (EGFR) [18]. In contrast a dominant negative form of CHMP3 prevents AMSH localization to the endosome, which in turn inhibits EGFR degradation [27]. Other studies implicate UBPY in the control of EGFR ubiquitination [20, 22, 28]. Besides EGFR, AMSH is involved in downregulation of the calcium sensing receptor [29], G protein-coupled receptors [30], the protease-activated receptor 2 [31] and the CXCR4 chemokine receptor [32]. The latter study suggests that AMSH not only regulates cargo ubiquitination but also ubiquitination of the endocytic machinery [32]. Furthermore, depletion of AMSH leads to defects in cytokinesis [33] and inactive AMSH exerts a dominant negative effect on retrovirus budding [34, 35].
The N-terminus of AMSH is predicted to contain a MIT domain [36]. This is followed by a central proline rich region providing the framework for STAM SH3 domain binding [37], which was suggested to control the isopeptidase activity of the C-terminal JAMM domain, a metalloprotease with specificity for K63-linked polyubiquitin chains [19, 21]. AMSH interacts with ESCRT-III CHMP1 [35, 36], CHMP2A and CHMP3 [21, 27, 34-36, 38] as well as Ist1 [39]. ESCRT-III proteins are auto-inhibited in the cytosol [34, 40, 41]. Activation leads to membrane interaction [42, 43] and polymerization in vitro [41, 44–46] and in vivo inducing membrane deformation [47]. Current models implicate ESCRT-I and II in vesicle formation [48] while the ordered assembly of ESCRT-III catalyzes membrane fission [49–52]. ESCRT-III proteins contain MIT domain interacting motifs (MIMs) within their C-termini which bind to MIT domains of VPS4 and spastin [53–56].
In order to understand the structural basis of the AMSH-CHMP3 interaction we defined an N-terminal AMSH domain (AMSHΔC) by proteolysis that allowed the isolation of a soluble complex with a C-terminal fragment of CHMP3. The crystal structure of AMSHΔC-CHMP3ΔN reveals an unconventional MIT domain within AMSHΔC. The CHMP3 C-terminus adopts a helical conformation upon binding and the molecular details of the interaction provide a novel MIM sequence motif. Although CHMP3 binds with high affinity to AMSH, ESCRT-III CHMP1A and B show only micromolar interactions. Mutagensis studies suggest further that AMSH employs a different molecular surface for CHMP1 interaction. Structural analyses indicate that the N-terminal helix preceding the MIT domain is mobile consistent with data demonstrating that this region might control the activity of AMSH during HIV-1 budding. Although the CHMP3-AMSH interaction site overlaps with the VPS4 binding site the high KD renders it unlikely to be displaced by VPS4.
Results
Determination of a minimal AMSH-CHMP3 complex
Trypsin proteolysis of the AMSH(1–206)-CHMP3 complex described previously [40] was used to identify AMSH residues 1 to 146 (AMSHΔC) as a stable core fragment. N-terminally His-tagged AMSHΔC showed limited solubility on its own but produced a monodisperse complex in association with a C-terminal fragment of CHMP3 containing residues 183 to 222 (CHMP3ΔN). The AMSHΔC-CHMP3ΔN complex was purified by size exclusion chromatography and complex formation was confirmed by detection of the CHMP3ΔN peptide in the elution peak by mass spectrometry.
In order to compare the affinity of AMSH to CHMP3ΔN with previous measurements employing full length CHMP3 [40], MBP-AMSH(1–206) was used for ITC analysis with CHMP3ΔN, which produced an equilibrium dissociation constant (KD) of 63± nM (enthalpy ΔH=−15.1±0.09 kcal/mol) (Figure S1A and B). MBP-AMSH(1–146) was constructed to determine whether a shorter form of AMSH influences binding to CHMP3ΔN. SPR measurements with His-MBP-CHMP3ΔN coupled to the chip and MBP-AMSHΔC as analyte revealed a KD of 113 nM based on the association constant (ka) of 6.79 103 M−1 s−1 and the dissociation constant (kd) of 7.64 10−4 s−1 confirming the high affinity interaction observed with the longer AMSH(1–206) construct (Figure S1C).
Structure of the AMSHΔC-CHMP3ΔN complex
The structure of AMSHΔC in complex with CHMP3ΔN was determined from a selenomethionine-containing crystal using the single wavelength anomalous dispersion method and diffraction data to 1.75 Å (Table 1). This resulted in a readily interpretable electron density map (Figure 1A) revealing two complexes in the asymmetric crystal unit. AMSHΔC folds into a 90 Å long elongated helical assembly (Figure 1B). The N-terminal α-helical segment 1 folds back onto α-helices 2 and 5 which are followed by an extended conformation. Helices 2, 3 and 4–5 form a three helical bundle structure reminiscent of a MIT domain although a break in helix 4 at Ile 98 disrupts the helical continuity of the third helix. The latter extends far beyond the core of the MIT domain and spans a distance of 90 Å (Figure 1B). However, the conserved features of a MIT domain are evident from superpositioning of the Cα atoms with the structure of the VPS4 MIT domain revealing a r.m.s.d. of 2.6 Å (Figure 1C).
Table I.
Data collection and refinement statistics
| Native | SeMet | |
|---|---|---|
| Data collection | ||
| Space group | P41 | P41 |
| Cell dimensions | ||
| a = b, c (Å) | 45.97, 206.91 | 45.92, 206.90 |
| Peak | ||
| Wavelength (Å) | 0.9760 | 0.9795 |
| Resolution (Å) | 44.86 -1.75(1.84 - 1.75) | 69.84 - 2.90(3.06-2.90) |
| Rmerge | 0.08 (0.64) | 0.14 (0.52) |
| I/σI | 5.1 (1.2) | 5.1 (1.4) |
| Completeness (%) | 99.8 (100.0) | 99.9 (100.0) |
| Redundancy | 3.3 (3.3) | 4.1 (4.2) |
| Refinement | ||
| Resolution (Å) | 1.75 | |
| No. reflections | 141,404(20,743) | |
| Rwork/Rfree | 0.19/0.23 | |
| No. atoms | ||
| Protein | 2698 | |
| Water | 386 | |
| B-factors | ||
| Protein | 32.56 | |
| Water | 45.55 | |
| R.m.s deviations | ||
| Bond lengths (Å) | 0.02 | |
| Bond angles (°) | 1.29 |
Values in parentheses are for highest-resolution shell.
Figure 1.
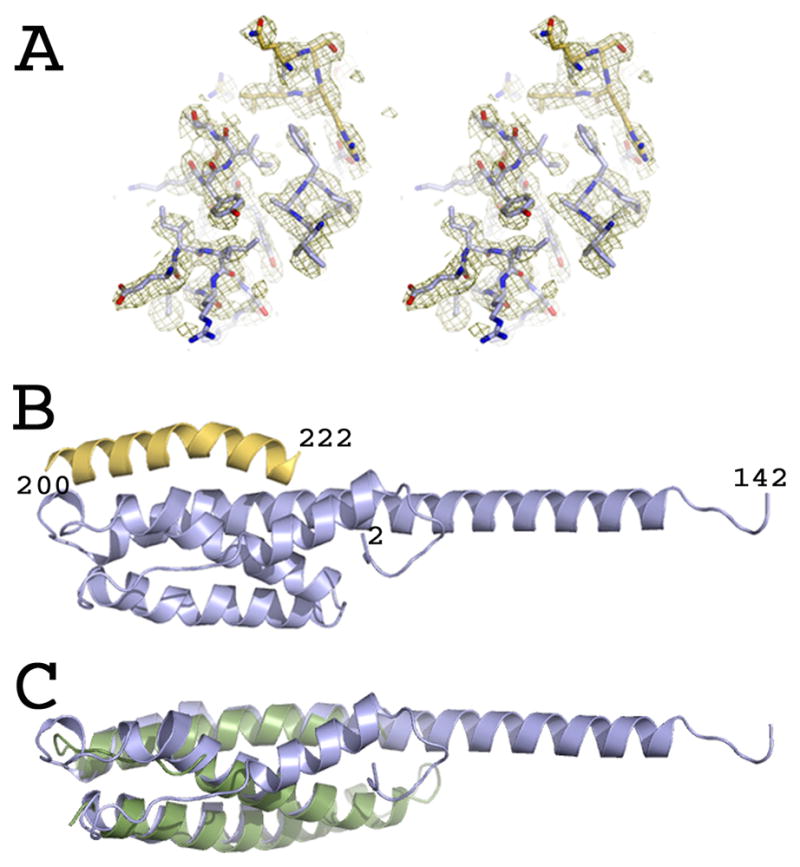
Crystal structure of AMSH in complex with CHMP3.
(A) Stereo image of the experimental electron density map obtained after SAD phasing.
(B) Ribbon representation of AMSHΔC (blue) in complex with CHMP3ΔN residues 200–222 (yellow).
(C) Overlay of the Cα atoms of AMSHΔC (blue) with the VPS4 MIT domain (pdb 2JQ9) (green ribbon). See also Figure S2.
CHMP3ΔN residues 200 to 222 adopt a helical conformation in the complex burying the solvent-accessible surface of 1659 Å2 on AMSHΔC and CHMPΔN, while CHMP3ΔN is unstructured in solution (Figure S2A). It is likely more structured in full-length CHMP3, since the overall helical content is slightly higher for full length CHMP3 (60% α-helical corresponding to 133 residues) compared to CHMP3(9–183) (68% α-helical corresponding to 119 residues) (Figure S2B), although both proteins reveal similar melting temperatures of ~ 60 ºC indicating that the C-terminus does not contribute substantially to the stability of CHMP3 in the auto-inhibited form (Figure S2C). CHMP3ΔN interacts with the MIT domain of AMSH diagonal along a groove made up by helices 3 and 4/5. The orientation of interaction is parallel with respect to the positions of both C-termini (Figure 1B). CHMP3ΔN residues 184–199 do not contribute to the interaction and are disordered in the structure. The complex is stabilized by a hydrogen bond between Glu207 and Tyr80 and salt bridges between Glu203-Lys88, Arg216-Glu104, and Arg221-Glu72. The last C-terminal residue, Ser222 of CHMP3 is capped by AMSHΔC Lys107, which forms a salt bridge with the carboxyl group of Ser222 and hydrogen bonds with the carbonyls of Thr219 and Leu220 (Figure 2A). Hydrophobic contacts play a minor role and include Leu210 interactions with Ile71 and Tyr80 as well as Leu220 contacts with Asn64 and Lys 107 (Figure 2B). Mutagenesis of AMSH interface residues Lys88, Glu104 and Lys107 to Ala confirmed the importance of the salt bridges and hydrogen bonds for interaction. While AMSHΔC-K88A recovered a faint amount of CHMP3 in a pull down, AMSHΔC-E104A and AMSHΔC-K107A failed to pull down CHMP3 (Figure 2C). The effect of the mutations on binding were confirmed by SPR. AMSHΔC-K88A produced a KD of 1.2 μM (ka 2.8±0.094 104 M−1 s−1 and kd of 3.1±0.033 10−3 s−1) and AMSHΔC-E104A a KD of 5.2 μM (ka 0.42±0.016 104 M−1 s−1 and kd of 2.2±0.029 10−3 s−1). Thus both mutations reduced binding by a factor 30 and 130, respectively, compared to the 40 nM KD of wild type AMSHΔC employing the same SPR setup. Notably, the mutations did not change the cellular localization when introduced into GFP-AMSH(D348A) (Figure S3), indicating that they did not alter the overall structure.
Figure 2.

The CHMP3N- AMSHΔC interaction is dominated by polar contacts.
(A) Close-up of the CHMP3-AMSH interactions. Hydrogen bonds and salt bridges along the CHMP3 helical segment mediate high affinity interaction.
(B) Sequence alignment of the C-terminal regions of CHMP3, CHMP1A, CHMP1B and CHMP2A. CHMP3 residues contacting AMSH are indicated by red asterisks (polar contact), blue asterisks (main chain polar contacts) and black asterisks (hydrophobic contacts).
(C) Pull down of CHMP3 by wild type and mutant His-AMSHΔC confirms the essential role of polar interactions. The left panel of the SDS-PAGE shows the input proteins and the right panel the pull down of CHMP3 by wild type and mutant His-AMSHΔC, as indicated. The K88A mutant reveals a strongly reduced interaction with CHMP3 and the E104 and K107A mutants show a complete loss of interaction in this assay. See also Figures S1 and S3.
Previous crystal structures of MIT-CHMP interactions reveal an important role for hydrophobic ESCRT-III MIM residues [53–56], while the AMSHΔC-CHMP3ΔN structure highlights the importance of polar interactions leading to a novel MIM sequence motif (Table 2). Because CHMP1A, CHMP1B and CHMP2A were reported to interact with AMSH [35, 36], the limited sequence conservation with CHMP3 (Figure 2B) predicts that they either bind AMSH similarly, but with lower affinity or they use a different strategy for interaction. Accordingly SPR measurements showed a much lower affinity for CHMP1A and CHMP1B interaction with AMSH (Figure S1D). Both GST-CHMP1AΔN and GST-CHMP1BΔN bound to the analyte MBP-AMSH(1–206) with KDs of 1.49 and 1.16 μM, respectively, based on the association constants (ka) of 3.89±0.06 × 103 M−1 s−1 (CHMP1A) and 2.52±0.03 × 103 M−1 s−1 (CHMP1B) and the dissociation constants (kd) of 5.81±0.09 × 10−3 s−1 (CHMP1A) and 2.92±0.08 × 10−3 s−1 (CHMP1B). Probing AMSH mutants AMSHΔC-K88A and AMSHΔC-E104A for CHMP1B interaction demonstrates that both mutants are still capable to pull down GST-CHMP1BΔN (Figure S1E); this might indicate that AMSH employs different interaction surfaces for CHMP1B and CHMP3. Furthermore the AMSH binding site overlaps with the VPS4 site on CHMP3 (Table 2) [53, 55], consistent with the finding that AMSH CHMP3 and VPS4 binding are exclusive (data not shown).
Table II.
ESCRT-III MIT domain interacting motifs. The motifs have been either reported in the cited literature or deduced from the structures reporting the corresponding contact residues.
Structural homology of the N-terminal domains of AMSH and USP8
A search for homologous structures [57] revealed high similarity to the N-terminal domain of the ubiquitin-specific protease 8 (USP8, also known as UBPY, Ubiquitin-specific protease Y) [58] as evidenced by the r.m.s.d. of 2.9 Å upon overlay of the Cα backbone (Figure 3A). The main difference between both structures is the detachment of the N-terminal segment including α-helix 1 from the core, which mediates dimerization in case of USP8 [58]. The N-terminal region (residues 1–27) of AMSH is only stabilized by a hydrogen bond between His4 and Glu57 and a salt bridge between Arg14 and Glu117 (Figure 3B). Although the N-terminal sequence of AMSH and UBPY diverge substantially (Figure 4), it is possible that the N-terminus of AMSH might be able to detach and adopt a similar conformation as in case of UBPY and vice versa the N-terminus of UBPY might attach to the core as observed for AMSHΔC. Thus the two conformations observed in both structures might be part of a common conformational control mechanism of AMSH and USP8/UBPY.
Figure 3.
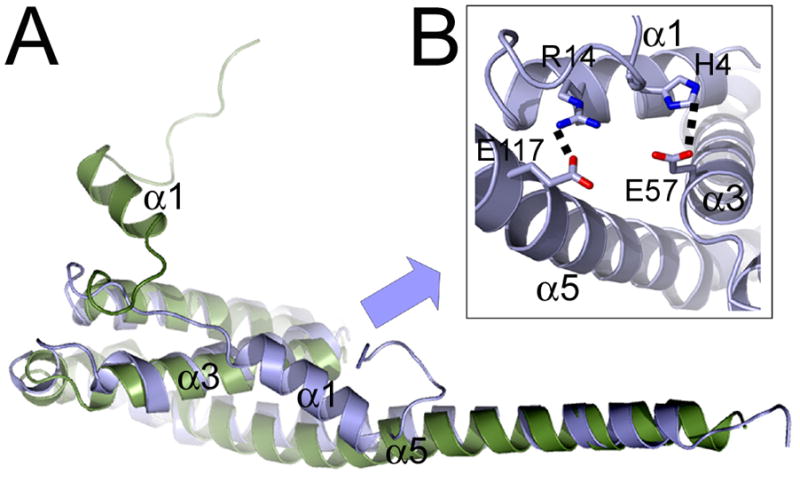
Superpositioning of the Cα atoms of AMSH and UBPY reveal a regulatory role of the N-terminal regions of AMSH and UBPY.
(A) Ribbon representation of the Cα overlay of AMSHΔC (blue) and UBPY (green)(pdb code 2A9U).
(B) Close up of the two main contacts between AMSH helix 1 and helices 3 and 5 of the core.
Figure 4.

Sequence and secondary structure alignment of AMSH, AMSH-LP and UBPY. Human AMSH (accession code AAD05037) residues 1 to 146 were aligned to the corresponding fragments of AMSH-LP (accession code BAC77766) and UBPY (accession code AAI10591). Residues contacting CHMP3 are indicated with blue triangles. Note that the CHMP3 contact residues are conserved between AMSH and AMSH-LP but not UBPY. Secondary structures are shown for AMSH and UBPY (pdb code 2A9U).
The N-terminal helix 1 of AMSH is required for HIV-1 budding
It has been previously shown that expression of catalytically inactive AMSH fused to GFP or YFP alone or in co-expression with full length CHMP3 induces a dominant negative effect on HIV-1 and MLV budding [34, 35]. Expression of catalytic inactive GFP-AMSH(D348A) had a mild effect on the kinetic of HIV-1 release as indicated by the accumulation of the Gag cleavage intermediates CAp2 and p41 (Figure 5A, right panel, lane 3) that define late assembly defects [59, 60]. However co-expression of GFP-AMSH(D348A) with CHMP3-Flag almost completely blocked HIV-1 release as indicated by the absence of CA in the supernatant (Figure 5A, left panel, lane 4) and the enhanced defect in Gag processing (Figure 5A, right panel, lane 4). Notably the intracellular distribution of GFP-AMSH(D348A) resembles that of wild type AMSH-HA when coexpressed with CHMP3-Flag (Figure 6A and B). Mutation of the N-terminal residues His4Ala and Arg14Ala that stabilize the interaction with the core (Figure 3B) within GFP-AMSH(D348A) leads to the loss of the dominant effect when expressed alone or in combination with CHMP3-Flag as indicated by the wild type-like release of CA into the supernatant and normal intracellular Gag processing (Figure 5A, lanes 5 and 6). GFP-AMSH(D348A-H4R14A) expression levels were slightly reduced in comparison to GFP-AMSH(D348A) (Figure 5A, lower panel). GFP-AMSH(D348A-H4R14A) expression was also altered from mostly cytoplasmic staining to accumulation at a perinuclear region without notable concentration of CHMP3-Flag (Figure 6C). Because the previous experiment showed that a strong dominant negative effect of GFP-AMSH(D348A) depends on CHMP3 expression we tested whether GST-CHMP3(151-222) can pull down the N-terminal mutant (GFP-AMSH(D348A-H4R14A)), which is no longer the case. Only a faint band could be detected for the N-terminal mutant in comparison to GFP-AMSH(D348A). Notably GFP-AMSH(D348A-H4R14A) showed a lower expression level than GFP-AMSH(D348A) (Figure 5B).
Figure 5.

Catalytic inactive AMSH and CHMP3 expression inhibit HIV-1 budding.
(A) (Left panel) Expression of catalytically inactive GFP-AMSH(D348A) exerts a strong effect on HIV-1 budding when coexpressed with CHMP3 (lanes 3 and 4). In contrast expression of GFP-AMSH(D348A-H4A-R14A) alone or in combination with CHMP3 has no effect on budding (lanes 5 and 6) comparable to vector expression (lane 1) or CHMP3 expression alone (lane 2). (Right panel) Western blot revealing the intracellular Gag processing. Only expression of GFP-AMSH(D348A) alone and together with CHMP3 show a defect in intracellular Gag processing (lanes 3 and 4), while the N-terminal mutant GFP-AMSH(D348A-H4A-R14A) resembles the negative control (lane 1) and CHMP3 expression (lane 2). (Lower panel) Western blot showing the expression levels of the AMSH constructs.
(B) Pull down of GFP-AMSH(D348A) (lanes 1 and 3) and GFP-AMSH(D348A-H4R14A)- HA (lanes 2 and 4) with GST (lanes 1 and 2) and with GST-CHMP3(151-222) (lanes 3 and 4). The western blot below shows the expression pattern of both GFP-AMSH constructs and the panel on the side shows the protein input used for the pull down.
(C) (Left panel) Expression of catalytically inactive AMSH(HH335,337QQ)-HA exerts a mild effect on HIV-1 budding when coexpressed with CHMP3 (lanes 3 and 4). In contrast expression of AMSH(HH335,337QQ-H4R14A)-HA alone or in combination with CHMP3 has no effect on budding (lanes 5 and 6). (Right panel) Western blot revealing the intracellular Gag processing. Only expression of AMSH(HH335,337QQ)-HA and CHMP3 show a defect in intracellular Gag processing (lane 4), while the N-terminal mutant resembles that of AMSH(HH335,337QQ)-HA expression (lane 3) or the vector control (lane 1). (Lower panel) Western blot revealing similar expression levels for the different AMSH constructs. See also Figure S4.
Figure 6.
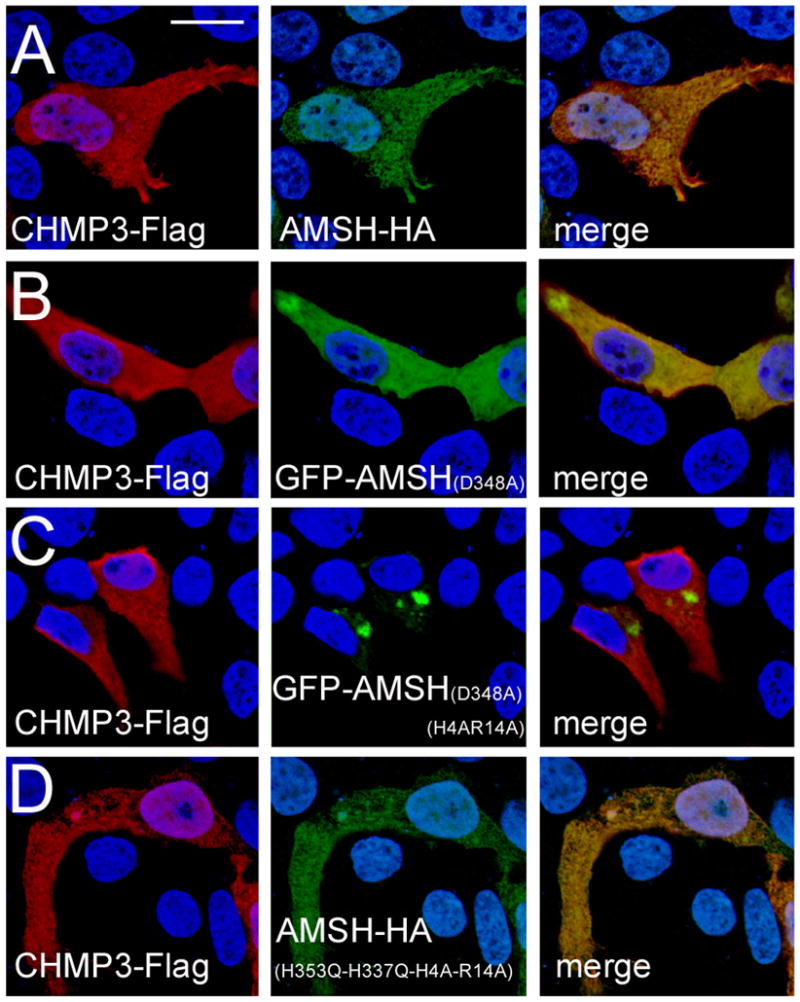
The H4, R14 mutations in combination with GFP alter the cellular localization of AMSH when co-expressed with CHMP3.
(A) CHMP3-Flag and AMSH-HA.
(B) CHMP3-Flag and catalytic inactive GFP-AMSH(D348A).
(C) CHMP3-Flag and catalytic inactive GFP-AMSH(D348A-H4A-R14A).
(D) CHMP3-Flag and inactive AMSH-HA carrying the mutations H335Q and H337Q, which render it catalytic inactive as well as the N-terminal mutations H4A and R14A. The scale bar is 10 μM.
Expression of N-terminally MBP-tagged full length AMSH in E. coli produced polydisperse AMSH, while recombinant AMSH expressed without tag was monodisperse, indicating that N-terminal fusions of AMSH induce the formation of polydisperse AMSH in vitro (Figure S4). This led us to perform the HIV-1 budding assay with catalytic inactive AMSH containing a C-terminal HA-tag instead of a N-terminal GFP-tag in the presence and absence of CHMP3 expression. The results indicate that expression of the catalytic inactive AMSH(HH335,337QQ)-HA or CHMP3 alone has no effect on virus particle production as measured by the detection of CA in the supernatant (Figure 5C, left panel, lanes 1 –3). Only co-expression of both proteins leads to a slight reduction in virion release and defects in intracellular Gag processing (Figure 5C, lanes 3 and 4). This minor assembly defect is lost, when AMSH(HH335,337QQ)-HA containing the N-terminal double mutant (His4Ala, Arg14Ala) is co-expressed with CHMP3 since no reduction in particle production is observed, consistent with the lack of intracellular Gag processing defects (Figure 5C, lanes 5 and 6). The expression levels of AMSH constructs were comparable (Figure 5C, lower panel) and AMSH(HH335,337QQ)-HA shows cytoplasmic staining when co-expressed with CHMP3, similar to wild type AMSH-HA (Figures 6D and E). This indicates that the N-terminal fusion of GFP is responsible for the dominant negative effect of catalytic inactive AMSH when coexpressed with CHMP3 and this function is lost upon destabilization of the N-terminal region, indicating that the His4Ala and Arg14Ala positions are important for supporting the dominant negative effect.
Discussion
Deubiquitinating enzymes (DUBs) AMSH and UBPY targeting ESCRT-III play an important role in ESCRT regulated processes [15, 61, 62] and many ESCRT-III interaction partners recognize sequence motifs located within the C-terminus of ESCRT-III family members [34, 35, 53, 55, 56, 63]. The crystal structure of AMSH in complex with CHMP3 demonstrates that the C-terminal CHMP3 MIM adopts a helical conformation, which is induced by complex formation, since the same peptide is unstructured in solution. However our CD analysis also suggest that part of it might be structured within full-length CHMP3 and thus contribute to the autoinhibited conformation of CHMP3 [34, 40, 64]. The structure of AMSH contains a region that resembles a MIT domain as predicted [36], which interacts with the CHMP3 MIM. However, we propose that the structure does not qualify as a classical MIT domain [63], due to the presence of an N-terminal region including helix 1 and because of the 90 Å-long extension of helix 5 (corresponding to helix 3 of a MIT domain). This is further supported by the poor solubility of the MIT-like domain of AMSH on its own and by AMSHΔC lacking helix 1 in vitro (Solomons et al. unpublished results). However, the general mode of target recognition is similar to that of MIT domains. CHMP3 binds in a helical conformation diagonal along a groove made up by helices 3 (VPS4 MIT helix 2) and 4/5 (VPS4 MIT helix 3). The AMSH-CHMP3 interaction site resembles VPS4 interactions with CHMP1A or yeast Vps2p, which employ helices 2 and 3 for the recognition of a shorter helical motif (MIM 1) [53] [55]. In contrast the spastin MIT domain interacts with a helical segment of CHMP1B via its helices 1 and 3 [56]. The same groove between helices 1 and 3 is employed by VPS4 to bind the CHMP6 MIM2 motif [54] and by Vps4 (Saci1372) from Crenarchaea to interact with the MIM2-like motif present in the ESCRT-III-like protein Saci1373 [65]. The general MIT-MIM contacts range from mostly hydrophobic interactions observed in the VPS4-CHMP1A, Vps4p-Vps2p, VPS4B-CHMP6 and Saci1372 (Vps4-like)-Saci1373 (ESCRT-II-like) complexes [53–55, 65] to a mixture of polar and hydrophobic interactions in case of spastin MIT and CHMP1B [56] to mostly polar contacts dominating the AMSH-CHMP3 complex. Thus the structure of the AMSH-CHMP3 complex extends the diversity of MIT domain interaction surfaces for peptide ligands.
AMSH was reported to interact not only with CHMP3 but also with CHMP1A/B and CHMP2A [35]. SPR measurements show that both CHMP1A and B interact with low micromolar affinities with AMSH. The low the sequence conservation between CHMP3 and CHMP1A/B and the fact that two AMSH mutants that show up to a 130 times lower CHMP3 binding activity still interact with CHMP1B, indicate that CHMP1 and CHMP3 binding sites do not overlap. This is further indirectly supported by the low sequence similarity between AMSH and UBPY that both bind CHMP1A/B [22, 35].
The ~ 60 nM Kd for the CHMP3-AMSH interaction is to date the highest Kd reported for an ESCRT-III MIM-MIT interaction. In contrast spastin-MIT binds to CHMP1A with a Kd of 12 μM Kd [56] ~ 3 times higher than the ~30 μM determined for the VPS4-MIT-MIM1 and MIM2 interactions [53–55, 65]. The high affinity interaction between CHMP3 and AMSH is mainly mediated by 6 salt bridges along the CHMP3 helix compared to 2 salt bridges in case of the spastin-MIT-CHMP1B interaction. Furthermore the total solvent-accessible surface area buried upon complex formation is 1650 Å2 (20 residues) compared to 1946 Å2 (24 residues) of the spastin-CHMP1B complex [56] and 1238 Å2 (15 residues) for the Vps4 MIT-Vps2 complex [55]. The importance of CHMP3 Arg216, which makes a double dent salt bridge with AMSH Glu104 and of an intact CHMP3 C-terminus, has been documented by mutagenesis analysis. Notably, deletion of residues 221 and 222 lead to a loss of interaction [34] consistent with the structural details revealing that CHMP3 Arg221 forms a salt bridge with AMSH Glu72 and the carboxyl group of the C-terminal CHMP3 Ser222 contacts AMSH Lys107 via a salt bridge. Vice versa we show here that mutagenesis of AMSH residues Lys88, Glu104 and Lys107 abrogates CHMP3 binding in vitro.
In yeast, Doa4 DUB activity has been genetically linked to Snf7 (CHMP4) and Bro1, which leads to endosomal recruitment after ESCRT-III assembly [66] [67]. It is thus conceivable that AMSH is also recruited to ESCRT-III polymers assembled on cellular membranes [49, 50]. The helical extension of the MIT domain could then serve as a long arm to position the DUB activity [19] > 20 nm away from the ESCRT-III polymer (Figure 7) thus reaching into the vesicle formed by ESCRT-I and II [48] for deubiquitination of cargo.
Figure 7.
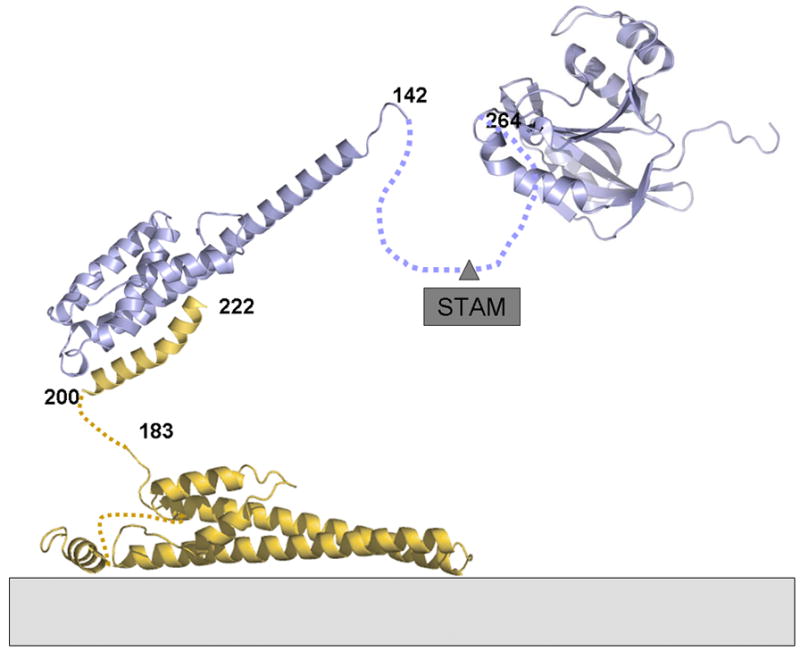
Molecular model of AMSH in complex with membrane associated CHMP3. AMSH can be recruited to membranes early in the ESCRT pathway via ESCRT-0 Stam. This might be independent of ESCRT-III CHMP3 interaction or both processes could be linked. If AMSH interacts with activated CHMP3 present in ESCRT-III polymers on membranes, the DUB activity could have an action radius of > 20 nm due to the long helical arm of AMSH and the flexible linkage of the CHMP3 MIM to the core of CHMP3 mediating ESCRT-III polymerization.
The MIM 1 sequence motif responsible for VPS4 interaction [53] overlaps with the CHMP3-AMSH binding site and superpositioning of the VPS4A-CHMP1B and VPS4B-CHMP2A structures with AMSH-CHMP3 demonstrates the overlap of the binding motifs, which prohibits simultaneous binding of AMSH and VPS4 to CHMP3. Although we do not know the sequence of recruitment, it is most likely that AMSH has to be released from CHMP3 before interacting with VPS4. Since the AMSH interaction is ~ 300 fold tighter than VPS4 binding, a specific mechanism must exist such as potential conformational changes that release AMSH from CHMP3 before it becomes a target for VPS4-driven ESCRT-III disassembly [68].
The structure of AMSHΔC resembles the structure of the N-terminal domain of USP8/UBPY, a DUB acting in the ESCRT pathway [58]. This structural similarity is not evident from the low sequence similarity of AMSH and UBPY. One notable structural difference is however the different conformation of a short N-terminal segment that is attached to the core of the monomer in case of AMSHΔC and is detached in UBPY mediating dimerization in the crystal [58]. N-terminal fusions of catalytic inactive AMSH have been shown previously to exert a dominant negative effect on HIV-1 and MLV budding [34, 35], which was confirmed by our assay. However destabilization of the N-terminus by structure based mutagenesis led to the loss of the dominant negative effect indicating that an intact N-terminus is important for budding inhibition, which correlated with the ability to interact with CHMP3. Since we observed the dramatic effect of the N-terminal MBP fusion on the oligomerization states of AMSH in vitro we reexamined the role of AMSH in HIV-1 budding by using C-terminal-tagged versions of AMSH. This showed indeed that catalytic inactive AMSH-HA has no effect on HIV-1 budding and only a mild effect upon co-expression with CHMP3, which was abrogated by the AMSH mutations that destabilize helix 1 interaction with the core. We conclude from these results that (i) N-terminal fusions of AMSH change the function of AMSH, which renders it dominant negative in combination with its ESCRT-III binding partner CHMP3. This function requires an intact N-terminus and is disturbed by His4 and Arg14 mutations. (ii) Destabilization of the N-terminus is not sufficient to induce a strong dominant negative effect of AMSH-HA, indicating that mutations of His4 and Arg14 interfere with the structural changes required to become dominant negative. In summary we have provided structural principles of a novel MIT-like MIM interaction. The structural and functional analyses demonstrate that conformational plasticity of the N-terminus might play an important role in the regulation of AMSH activity. This might entail activation of the catalytic activity by either STAM binding [21] or other regulatory factors.
Material and Methods
Cloning, expression and purification of AMSH and CHMP3
AMSH containing residues 1 206 (AMSH(1–206) was expressed and purified as described [40]. AMSH(1–206) in complex with full length CHMP3 [40] was subjected to limited proteolysis with trypsin at a 1:500 (w/w) ratio and bands were separated on a 12% SDS-PAGE. A prominent smaller fragment of AMSH was analyzed by mass fingerprinting and mapped to residues 1 to 146 (AMSHΔC). AMSHΔC was cloned into the vector pPROEX HTa (Invitrogen) and into a modified version of the pMalC2G-TEV vector [69] to produce His-tagged AMSHΔC and MBP-AMSHΔC. CHMP3(183–222) was cloned into the vector pBADM-41 (His-MBP fusion protein with TEV protease cleavage site) to produce His-MBP-CHMP3ΔN. Full length AMSH was cloned into pMalC2G-TEV (MBP-AMSH) and tag-less AMSH was cloned into vector pASK-IBA43plus. CHMP1A (residues 123–196) and CHMP1B (residues 126–199) were cloned into vector pGEX-TEV. His-AMSHΔC, MBP-AMSHΔC, His-MBP-CHMP3ΔN, MBP-AMSH, AMSH, GST-CHMP1A and GST-CHMP1B were expressed in E. coli cells BL21 codon+ or E. coli Rosetta. MBP-AMSH and AMSH expression in E. coli was induced at 16 ºC over night. AMSH mutations K88A, E104A and K107A were introduced into His-tagged AMSHΔC and GFP-AMSH(D348A) by using standard mutagenesis procedures.
His-AMSHΔC was solubilized in buffer A (20 mM Tris pH 8.5, 100 mM NaCl) and purified on a Ni2+-sepharose column. His-MBP-CHMP3ΔN was purified in buffer A on an amylase column, which produced mixtures of His-MBP-CHMP3ΔN and truncated forms corresponding to free MBP. For ITC and CD analyses, His-MBP-CHMP3ΔN was further purified by anion exchange chromatography followed gel filtration chromatography on a Superdex-75 (GE Healthcare) column in buffer A. GST-CHMP1A and GST-CHMP1B were purified on a glutathione column in PBS buffer followed by gel filtration chromatography in PBS buffer. His-MBP-CHMP3ΔN and His-AMSHΔC were mixed in a 2:1 molar ratio and the tags were cleaved by TEV protease cleavage at 4°C. His-tags and uncleaved proteins were removed by passing the complex over a Ni2+-sepharose column in buffer A. A final purification step included a gel filtration on a Superdex-75 column in buffer B (10 mM HEPES pH 8.0, 100 mM NaCl). The complex was concentrated to 3.3 mg/ml. Selenomethionine substituted His-AMSHΔC was produced as described [70] and the complex with CHMP3ΔN was purified as described above for the native complex and concentrated to 3.6 mg/ml. MBP-AMSHΔC was purified in buffer A on an amylose affinity column in buffer A coupled to a second purification step on a Superdex 75 gel filtration column in buffer A. MBP-AMSH was purified on an amylose column in buffer C (20 mM Tris pH 8.5, 100 NaCl) followed by gel filtration in buffer C. AMSH was purified on a Q sepharose column in buffer D (25 mM Tris pH 8.5, 25 mM NaCl) by applying a NaCl gradient in buffer D. Further purification included a gel filtration on a superdex 200 column in buffer C. His-tagged AMSHΔC mutants were purified as wild type AMSHΔC in buffer A (20 mM Tris pH 8.5, 100 mM NaCl) on a Ni2+-sepharose column followed by gel filtration on superdex-75 in buffer B. The experimental conditions for the pull downs are described in the supplemenatary material.
Isothermal titration calorimetry and surface plasmon resonance experiments were carried out as described in the supplementary material.
Crystallization, diffraction data collection and structure solution of AMSHΔC in complex with CHMP3ΔN
Initial crystallization conditions for the AMSHΔC-CHMP3ΔN complex at 3.3 mg/ml were determined by using 200 nl sitting drops and 1.9 M sodium malonate as reservoir solution. Crystals were improved by mixing the complex at 7.2 mg/ml with an equal volume of 2.2 M sodium malonate, which resulted in microcrystals. Larger crystals were grown by streak seeding the microcrystals into fresh drops mixed with 1.8 to 2.2 M sodium malonate. Crystals grown in 1.8 M sodium malonate were flash-frozen in liquid nitrogen in the crystallization buffer supplemented with 30 % glycerol as cryo-protectant. A native data set was collected at 1.75 Å resolution at the ESRF beam line (Grenoble) ID-14–4. Selenomethione substituted AMSHΔC in complex with CHMP3ΔN was crystallized as described above for the native complex, except that several rounds of seeding were required to obtain diffraction quality crystals. A single wavelength anomalous dispersion (SAD) data set was collected at 100 K at beam line ID14–4 (ESRF, Grenoble) at the peak wavelength of the selenium K edge (wavelength 0.9795 Å). The data were processed with MOSFLM [71] and Scala [72, 73]. The crystals belong to space group P41 with unit cell dimensions of a, b = 45.97 Å, c= 206.91 Å and two complexes per asymmetric unit. Data were initially analyzed using the Auto-Rickshaw platform at EMBL Hamburg [74]. Selenium sites were localized at a resolution of 2.8 Å with the program SHELXD [75]. The correct hand for the substructure was determined using the programs ABS [76] and SHELXE [77]. Initial phases were calculated after density modification using the program SHELXE [77] and model building was performed with the program ARP/wARP [78]. This initial model was used as a search model for molecular replacement with MOLREP [73] employing the native data to 1.75 Å resolution. Automatic model building was completed with ARP/wARP [78], manual model building using COOT [79] and refinement with the program Refmac [80]. The structure contains AMSH residues 2 to 142 and CHMP3 residues 200 to 222 and was refined to an R factor of 19% and an Rfree of 23.0 %. 99.67% of the residues are within the most favored and allowed regions of a Ramachandran plot [73]. All molecular graphics figures were generated with PYMOL (W Delano; http://www.pymol.org/). The coordinates have been deposited in the protein data bank (PDB code: 2xze).
Analysis of virus particle production
293T cells (1.2 × 106) were seeded into T25 flasks and transfected 24 hr later using a calcium phosphate precipitation technique. The cultures were transfected with 1.5 μg HXBH10, which encodes WT HIV-1, together with GFP-AMSH(D348A) [34] or GFP-AMSH(D348A0H4R14A) and/or pcDNA3.1 vectors expressing CHMP3, AMSH(HH335,337QQ)-HA or AMSH(HH335,337QQ)-HA harboring the H4,R14A mutations or the appropriate empty vector (0.5 μg each). The total amount of transfected DNA was brought to 8 μg with carrier DNA (pTZ18U). Twenty-four hr post transfection, the cells were lysed in RIPA buffer (140 mM NaCl, 8 mM Na2HPO4, 2 mM 1%Nonidet P-40, 0.5% sodium deoxycholate, 0.05% SDS), and the culture NaH2PO4, supernatants were clarified by low speed centrifugation and passaged through 0.45-μm filters. Virions released into the medium were pelleted through 20% sucrose cushions and analyzed by SDS-PAGE and Western blotting with anti-HIV CA antibody 183-H12-5C [81]. Proteins in the cell lysates were detected by Western blotting with anti-HIV CA antibody or rabbit anti-GFP serum (Molecular Probes). The pull down of GFP-AMSH(D348A) and GFP-AMSH(D348A0H4R14A) employing GST-CHMP3(151–222) was performed as described [34].
Immunofluorescence analysis
PcDNA3.1 expression constructs of wild type AMSH-HA, AMSH(HH335,337QQ)-HA carrying the His4 and Arg14 to Ala mutations, CHMP3-Flag and GFP-AMSH(D348A), GFP-AMSH(D348A-H4R14A), GFP-AMSH(D348A-K88A), GFP-AMSH(D348A-E104A) and GFP-AMSH(D348A-K107A) were transfected into 293T cells using standard methods. For indirect immunofluorescence (IIF) 293T cells were cultured on coverslips and fixed with 4% paraformaldehyde for 20 min at 4°C. The GFP constructs were directly visualized and for the other constructs cover slips were incubated with anti-HA-tag or anti-Flag-tag antibodies in PBS for 1h, at RT. Slides were washed three times with PBS, followed by the secondary antibody incubation at RT for 1h (Alexa488 or 594 coupled anti-mouse or anti-rabbit goat antibodies in PBS). After three washes with PBS, slides were mounted in Mowiol and analyzed by confocal microscopy.
Supplementary Material
Acknowledgments
This work was supported by the Agence Nationale de la Recherche (ANR-08-BLAN-0271-01; W.W.) and the DFG SPP1175 (W.W.), the National Institute of Allergy and Infectious Diseases (R37AI029873 to HG), postdoctoral fellowships from the European Molecular Biology Organization (B.H.) and the Fondation pour la Recherche Médicale (PM). We thank N. Miguet for excellent technical assistance and Dr. J. Hurley for providing the CHMP1A cDNA. The HIV-1 p24 monoclonal antibody (183-H12-5C) (provided by Drs. B. Chesebro and K. Wehrly) was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Babst M. A protein’s final ESCRT. Traffic. 2005;6:2–9. doi: 10.1111/j.1600-0854.2004.00246.x. [DOI] [PubMed] [Google Scholar]
- 2.Saksena S, Sun J, Chu T, Emr SD. ESCRTing proteins in the endocytic pathway. Trends in Biochemical Sciences. 2007;32:561–573. doi: 10.1016/j.tibs.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 3.van der Goot FG, Gruenberg J. Intra-endosomal membrane traffic. Trends Cell Biol. 2006;16:514–521. doi: 10.1016/j.tcb.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 4.Hurley J. ESCRT complexes and the biogenesis of multivesicular bodies. Curr Opin Cell Biol. 2008;20(1):4–11. doi: 10.1016/j.ceb.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peel S, Macheboeuf P, Martinelli N, Weissenhorn W. Divergent pathways lead to ESCRT-III catalyzed membrane fission. Trends in Biochemical Sciences. 2011;36(4):199–210. doi: 10.1016/j.tibs.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 6.Morita E, Sundquist WI. Retrovirus budding. Annual Review of Cell and Developmental Biology. 2004;20:395–425. doi: 10.1146/annurev.cellbio.20.010403.102350. [DOI] [PubMed] [Google Scholar]
- 7.Hartlieb B, Weissenhorn W. Filovirus assembly and budding. Virology. 2006;344:64–70. doi: 10.1016/j.virol.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 8.Usami Y, Popov S, Popova E, Inoue M, Weissenhorn W, HGG The ESCRT pathway and HIV-1 budding. Biochem Soc Trans. 2009;37:181–184. doi: 10.1042/BST0370181. [DOI] [PubMed] [Google Scholar]
- 9.Bieniasz PD. The cell biology of HIV-1 virion genesis. Cell Host Microbe. 2009;5:550–558. doi: 10.1016/j.chom.2009.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carlton JG, Martin-Serrano J. Parallels between cytokinesis and retroviral budding: a role for the ESCRT machinery. Science. 2007;316:1908–1912. doi: 10.1126/science.1143422. [DOI] [PubMed] [Google Scholar]
- 11.Morita E, Sandrin V, Chung HY, Morham SG, Gygi SP, Rodesch CK, Sundquist WI. Human ESCRT and ALIX proteins interact with proteins of the midbody and function in cytokinesis. EMBO J. 2007;26:4215–4227. doi: 10.1038/sj.emboj.7601850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carlton JG, Martin-Serrano J. The ESCRT machinery: new functions in viral and cellular biology. Biochem Soc Trans. 2009;37:195–199. doi: 10.1042/BST0370195. [DOI] [PubMed] [Google Scholar]
- 13.Lindas AC, Karlsson EA, Lindgren MT, Ettema TJ, Bernander R. A unique cell division machinery in the Archaea. Proc Natl Acad Sci U S A. 2008;105(48):18942–6. doi: 10.1073/pnas.0809467105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghazi-Tabatabai S, Obita T, Pobbati AV, Perisic O, Samson RY, Bell SD, Williams RL. Evolution and assembly of ESCRTs. Biochem Soc Trans. 2009;37:151–155. doi: 10.1042/BST0370151. [DOI] [PubMed] [Google Scholar]
- 15.Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. 2009;10:550–563. doi: 10.1038/nrm2731. [DOI] [PubMed] [Google Scholar]
- 16.Amerik AY, Nowak J, Swaminathan S, Hochstrasser M. The Doa4 deubiquitinating enzyme is functionally linked to the vacuolar protein-sorting and endocytic pathways. Mol Biol Cell. 2000;11:3365–3380. doi: 10.1091/mbc.11.10.3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nikko E, Andre B. Evidence for a direct role of the Doa4 deubiquitinating enzyme in protein sorting into the MVB pathway. Traffic. 2007;8:566–581. doi: 10.1111/j.1600-0854.2007.00553.x. [DOI] [PubMed] [Google Scholar]
- 18.McCullough J, Clague MJ, Urbe S. AMSH is an endosome-associated ubiquitin isopeptidase. J Cell Biol. 2004;166:487–492. doi: 10.1083/jcb.200401141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sato Y, Yoshikawa A, Yamagata A, Mimura H, Yamashita M, Ookata K, Nureki O, Iwai K, Komada M, Fukai S. Structural basis for specific cleavage of Lys 63-linked polyubiquitin chains. Nature. 2008;455:358–362. doi: 10.1038/nature07254. [DOI] [PubMed] [Google Scholar]
- 20.Alwan HA, van Leeuwen JE. UBPY-mediated epidermal growth factor receptor (EGFR) de-ubiquitination promotes EGFR degradation. J Biol Chem. 2007;282:1658–1669. doi: 10.1074/jbc.M604711200. [DOI] [PubMed] [Google Scholar]
- 21.McCullough J, Row PE, Lorenzo O, Doherty M, Beynon R, Clague MJ, Urbe S. Activation of the endosome-associated ubiquitin isopeptidase AMSH by STAM, a component of the multivesicular body-sorting machinery. Current Biology. 2006;16:160–165. doi: 10.1016/j.cub.2005.11.073. [DOI] [PubMed] [Google Scholar]
- 22.Row PE, Prior IA, McCullough J, Clague MJ, Urbe S. The ubiquitin isopeptidase UBPY regulates endosomal ubiquitin dynamics and is essential for receptor down-regulation. J Biol Chem. 2006;281:12618–12624. doi: 10.1074/jbc.M512615200. [DOI] [PubMed] [Google Scholar]
- 23.Mizuno E, Kobayashi K, Yamamoto A, Kitamura N, Komada M. A deubiquitinating enzyme UBPY regulates the level of protein ubiquitination on endosomes. Traffic. 2006;7:1017–1031. doi: 10.1111/j.1600-0854.2006.00452.x. [DOI] [PubMed] [Google Scholar]
- 24.Nakamura M, Tanaka N, Kitamura N, Komada M. Clathrin anchors deubiquitinating enzymes, AMSH and AMSH-like protein, on early endosomes. Genes Cells. 2006;11:593–606. doi: 10.1111/j.1365-2443.2006.00963.x. [DOI] [PubMed] [Google Scholar]
- 25.Alwan HA, van Zoelen EJ, van Leeuwen JE. Ligand-induced lysosomal epidermal growth factor receptor (EGFR) degradation is preceded by proteasome-dependent EGFR de-ubiquitination. J Biol Chem. 2003;278:35781–35790. doi: 10.1074/jbc.M301326200. [DOI] [PubMed] [Google Scholar]
- 26.Dupre S, Haguenauer-Tsapis R. Deubiquitination step in the endocytic pathway of yeast plasma membrane proteins: crucial role of Doa4p ubiquitin isopeptidase. Mol Cell Biol. 2001;21:4482–4494. doi: 10.1128/MCB.21.14.4482-4494.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma YM, Boucrot E, Villen J, Affar EB, Gygi SP, Gottlinger HG, Kirchhausen T. Targeting of AMSH to endosomes is required for EGF receptor degradation. J Biol Chem. 2007;282(13):9805–9812. doi: 10.1074/jbc.M611635200. [DOI] [PubMed] [Google Scholar]
- 28.Mizuno E, Iura T, Mukai A, Yoshimori T, Kitamura N, Komada M. Regulation of epidermal growth factor receptor down-regulation by UBPY-mediated deubiquitination at endosomes. Mol Biol Cell. 2005;16:5163–5174. doi: 10.1091/mbc.E05-06-0560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reyes-Ibarra AP, Garcia-Regalado A, Ramirez-Rangel I, Esparza-Silva AL, Valadez-Sanchez M, Vazquez-Prado J, Reyes-Cruz G. Calcium-sensing receptor endocytosis links extracellular calcium signaling to parathyroid hormone-related peptide secretion via a Rab11a-dependent and AMSH-sensitive mechanism. Mol Endocrinol. 2007;21:1394–1407. doi: 10.1210/me.2006-0523. [DOI] [PubMed] [Google Scholar]
- 30.Hislop JN, Henry AG, Marchese A, von Zastrow M. Ubiquitination regulates proteolytic processing of G protein-coupled receptors after their sorting to lysosomes. J Biol Chem. 2009;284:19361–19370. doi: 10.1074/jbc.M109.001644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hasdemir B, Murphy JE, Cottrell GS, Bunnett NW. Endosomal deubiquitinating enzymes control ubiquitination and down-regulation of protease-activated receptor 2. J Biol Chem. 2009;284:28453–28466. doi: 10.1074/jbc.M109.025692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sierra MI, Wright MH, Nash P. AMSH interacts with ESCRT-0 to regulate the stability and trafficking of CXCR4. J Biol Chem. 2010;285(18):13990–4004. doi: 10.1074/jbc.M109.061309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mukai A, Mizuno E, Kobayashi K, Matsumoto M, Nakayama KI, Kitamura N, Komada M. Dynamic regulation of ubiquitylation and deubiquitylation at the central spindle during cytokinesis. J Cell Sci. 2008;121:1325–1333. doi: 10.1242/jcs.027417. [DOI] [PubMed] [Google Scholar]
- 34.Zamborlini A, Usami Y, Radoshitzky SR, Popova E, Palu G, Gottlinger H. Release of autoinhibition converts ESCRT-III components into potent inhibitors of HIV-1 budding. Proc Natl Acad Sci U S A. 2006;103:19140–19145. doi: 10.1073/pnas.0603788103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Agromayor M, Martin-Serrano J. Interaction of AMSH with ESCRT-III and deubiquitination of endosomal cargo. J Biol Chem. 2006;281(32):23083–23091. doi: 10.1074/jbc.M513803200. [DOI] [PubMed] [Google Scholar]
- 36.Tsang HT, Connell JW, Brown SE, Thompson A, Reid E, Sanderson CM. A systematic analysis of human CHMP protein interactions: Additional MIT domain-containing proteins bind to multiple components of the human ESCRT III complex. Genomics. 2006;88(3):333–346. doi: 10.1016/j.ygeno.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 37.Tanaka N, Kaneko K, Asao H, Kasai H, Endo Y, Fujita T, Takeshita T, Sugamura K. Possible involvement of a novel STAM-associated molecule “AMSH” in intracellular signal transduction mediated by cytokines. J Biol Chem. 1999;274:19129–19135. doi: 10.1074/jbc.274.27.19129. [DOI] [PubMed] [Google Scholar]
- 38.Kyuuma M, Kikuchi K, Kojima K, Sugawara Y, Sato M, Mano N, Goto J, Takeshita T, Yamamoto A, Sugamura K, Tanaka N. AMSH, an ESCRT-III associated enzyme, deubiquitinates cargo on MVB/late endosomes. Cell Struct Funct. 2007;31:159–172. doi: 10.1247/csf.06023. [DOI] [PubMed] [Google Scholar]
- 39.Agromayor M, Carlton JG, Phelan JP, Matthews DR, Carlin LM, Ameer-Beg S, Bowers K, Martin-Serrano J. Essential role of hIST1 in cytokinesis. Mol Biol Cell. 2009;20:1374–1387. doi: 10.1091/mbc.E08-05-0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lata S, Roessle M, Solomons J, Jamin M, Gottlinger HG, Svergun DI, Weissenhorn W. Structural basis for autoinhibition of ESCRT-III CHMP3. J Mol Biol. 2008;378:818–827. doi: 10.1016/j.jmb.2008.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bajorek M, Schubert HL, McCullough J, Langelier C, Eckert DM, Stubblefield W-MB, Uter NT, Myszka DG, Hill CP, Sundquist WI. Structural basis for ESCRT-III protein autoinhibition. 2009;16:754–762. doi: 10.1038/nsmb.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muziol T, Pineda-Molina E, Ravelli RB, Zamborlini A, Usami Y, Gottlinger H, Weissenhorn W. Structural basis for budding by the ESCRT-III factor CHMP3. Dev Cell. 2006;10:821–830. doi: 10.1016/j.devcel.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 43.Shim S, Kimpler LA, Hanson PI. Structure/Function Analysis of Four Core ESCRT-III Proteins Reveals Common Regulatory Role for Extreme C-Terminal Domain. Traffic. 2007;8:1068–1079. doi: 10.1111/j.1600-0854.2007.00584.x. [DOI] [PubMed] [Google Scholar]
- 44.Ghazi-Tabatabai S, Saksena S, Short JM, Pobbati AV, Veprintsev DB, Crowther RA, Emr SD, Egelman EH, Williams RL. Structure and disassembly of filaments formed by the ESCRT-III subunit Vps24. Structure. 2008;16:1345–1356. doi: 10.1016/j.str.2008.06.010. [DOI] [PubMed] [Google Scholar]
- 45.Lata S, Schoehn G, Jain A, Pires R, Piehler J, Gottlinger HG, Weissenhorn W. Helical structures of ESCRT-III are disassembled by VPS4. Science. 2008;321:1354–1357. doi: 10.1126/science.1161070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pires R, Hartlieb B, Signor L, Schoehn G, Lata S, Roessle M, Moriscot C, Popov S, Hinz A, Jamin M, Boyer V, Sadoul R, Forest E, Svergun DI, Gottlinger HG, Weissenhorn W. A crescent-shaped ALIX dimer targets ESCRT-III CHMP4 filaments. Structure. 2009;17:843–856. doi: 10.1016/j.str.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hanson PI, Roth R, Lin Y, Heuser JE. Plasma membrane deformation by circular arrays of ESCRT-III protein filaments. J Cell Biol. 2008;180(2):389–402. doi: 10.1083/jcb.200707031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wollert T, Hurley JH. Molecular mechanism of multivesicular body biogenesis by ESCRT complexes. Nature. 2010;464(7290):864–9. doi: 10.1038/nature08849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Teis D, Saksena S, Emr SD. Ordered Assembly of the ESCRT-III Complex on Endosomes Is Required to Sequester Cargo during MVB Formation. Dev Cell. 2008;15(4):578–589. doi: 10.1016/j.devcel.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 50.Saksena S, Wahlman J, Teis D, Johnson AE, Emr SD. Functional reconstitution of ESCRT-III assembly and disassembly. Cell. 2009;136:97–109. doi: 10.1016/j.cell.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wollert T, Wunder C, Lippincott-Schwartz J, Hurley JH. Membrane scission by the ESCRT-III complex. Nature. 2009;458:172–177. doi: 10.1038/nature07836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fabrikant G, Lata S, Riches JD, Briggs JA, Weissenhorn W, Kozlov MM. Computational model of membrane fission catalyzed by ESCRT-III. PLoS Comput Biol. 2009;5(11):e1000575. doi: 10.1371/journal.pcbi.1000575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stuchell-Brereton MD, Skalicky JJ, Kieffer C, Karren MA, Ghaffarian S, Sundquist WI. ESCRT-III recognition by VPS4 ATPases. Nature. 2007;449:740–744. doi: 10.1038/nature06172. [DOI] [PubMed] [Google Scholar]
- 54.Kieffer C, Skalicky JJ, Morita E, De Domenico I, Ward DM, Kaplan J, Sundquist WI. Two distinct modes of ESCRT-III recognition are required for VPS4 functions in lysosomal protein targeting and HIV-1 budding. Dev Cell. 2008;15:62–73. doi: 10.1016/j.devcel.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 55.Obita T, Saksena S, Ghazi-Tabatabai S, Gill DJ, Perisic O, Emr SD, Williams RL. Structural basis for selective recognition of ESCRT-III by the AAA ATPase Vps4. Nature. 2007;449:735–739. doi: 10.1038/nature06171. [DOI] [PubMed] [Google Scholar]
- 56.Yang D, Rismanchi N, Renvoise B, Lippincott-Schwartz J, Blackstone C, Hurley JH. Structural basis for midbody targeting of spastin by the ESCRT-III protein CHMP1B. Nat Struct Mol Biol. 2008;15(12):1278–86. doi: 10.1038/nsmb.1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dietmann S, Fernandez-Fuentes N, Holm L. Automated detection of remote homology. Current Opinion in Structural Biology. 2002;12:362–367. doi: 10.1016/s0959-440x(02)00332-9. [DOI] [PubMed] [Google Scholar]
- 58.Avvakumov GV, Walker JR, Xue S, Finerty PJ, Jr, Mackenzie F, Newman EM, Dhe-Paganon S. Amino-terminal dimerization, NRDP1-rhodanese interaction, and inhibited catalytic domain conformation of the ubiquitin-specific protease 8 (USP8) J Biol Chem. 2006;281:38061–38070. doi: 10.1074/jbc.M606704200. [DOI] [PubMed] [Google Scholar]
- 59.Gottlinger H, Dorfman T, Sodroski J, Haseltine W. Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc Natl Acad Sci USA. 1991;88:3195–3199. doi: 10.1073/pnas.88.8.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Garrus JE, von Schwedler UK, Pornillos OW, Morham SG, Zavitz KH, Wang HE, Wettstein DA, Stray KM, Cote M, Rich RL, Myszka DG, Sundquist WI. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell. 2001;107:55–65. doi: 10.1016/s0092-8674(01)00506-2. [DOI] [PubMed] [Google Scholar]
- 61.McDonald B, Martin-Serrano J. No strings attached: the ESCRT machinery in viral budding and cytokinesis. J Cell Sci. 2009;122:2167–2177. doi: 10.1242/jcs.028308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Raiborg C, Stenmark H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature. 2009;458:445–452. doi: 10.1038/nature07961. [DOI] [PubMed] [Google Scholar]
- 63.Scott A, Gaspar J, Stuchell-Brereton MD, Alam SL, Skalicky JJ, Sundquist WI. Structure and ESCRT-III protein interactions of the MIT domain of human VPS4A. Proc Natl Acad Sci U S A. 2005;102:13813–13818. doi: 10.1073/pnas.0502165102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bajorek M, Schubert HL, McCullough J, Langelier C, Eckert DM, Stubblefield WM, Uter NT, Myszka DG, Hill CP, Sundquist WI. Structural basis for ESCRT-III protein autoinhibition. Nat Struct Mol Biol. 2009;16:754–762. doi: 10.1038/nsmb.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Samson RY, Obita T, Freund SM, Williams RL, Bell SD. A role for the ESCRT system in cell division in archaea. Science. 2008;322:1710–1713. doi: 10.1126/science.1165322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Luhtala N, Odorizzi G. Bro1 coordinates deubiquitination in the multivesicular body pathway by recruiting Doa4 to endosomes. J Cell Biol. 2004;166:717–729. doi: 10.1083/jcb.200403139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Amerik A, Sindhi N, Hochstrasser M. A conserved late endosome-targeting signal required for Doa4 deubiquitylating enzyme function. J Cell Biol. 2006;175:825–835. doi: 10.1083/jcb.200605134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Babst M, Wendland B, Estepa EJ, Emr SD. The Vps4p AAA ATPase regulates membrane association of a Vps protein complex required for normal endosome function. EMBO J. 1998;17:2982–2993. doi: 10.1093/emboj/17.11.2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bracher A, Weissenhorn W. Structural basis for the Golgi membrane recruitment of Sly1p by Sed5p. Embo Journal. 2002;21:6114–6124. doi: 10.1093/emboj/cdf608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Doublie S. Preparation of selenomethionyl proteins for phase determination. Methods Enzymol. 1997;276:523–530. [PubMed] [Google Scholar]
- 71.Leslie AGW. Recent changes to the MOSFLM package for processing film and image plate data. Jnt CCP4/ESF-EACMB Newslett Protein Crystallogr. 1992:26. [Google Scholar]
- 72.Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 73.CCP4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:157–163. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 74.Panjikar S, Parthasarathy V, Lamzin VS, Weiss MS, Tucker PA. Auto-Rickshaw: an automated crystal structure determination platform as an efficient tool for the validation of an X-ray diffraction experiment. Acta Crystallogr D Biol Crystallogr. 2005;61:449–457. doi: 10.1107/S0907444905001307. [DOI] [PubMed] [Google Scholar]
- 75.Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta Crystallogr D Biol Crystallogr. 2002;58:1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- 76.Hao Q. ABS: a program to determine absolute configuration and evaluate anomalous scatterer substructure. J Appl Cryst. 2004;37:498–499. [Google Scholar]
- 77.Sheldrick GM. Macromolecular phasing with SHELXE. Zeitschrift für Kristallographie. 2002;217:644–650. [Google Scholar]
- 78.Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 79.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallographica Section D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 80.Murshudov GN, Vagin AA, Dodson EJ. Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Crystallographica Section D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 81.Chesebro B, Wehrly K, Nishio J, SP Macrophage-tropic human immunodeficiency virus isolates from different patients exhibit unusual V3 envelope sequence homogeneity in comparison with T-cell-tropic isolates: definition of critical amino acids involved in cell tropism. J Virol. 1992;66:6547–6554. doi: 10.1128/jvi.66.11.6547-6554.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.