

Abstract
The history of human population size is important to understanding human evolution. Various studies1-5 have found evidence for a founder event (bottleneck) in East Asian and European populations associated with the human dispersal out-of-Africa event around 60 thousand years ago (kya) before present. However, these studies have to assume simplified demographic models with few parameters and do not precisely date the start and stop times of the bottleneck. Here, with fewer assumptions on population size changes, we present a more detailed history of human population sizes between approximately ten thousand to a million years ago, using the pairwise sequentially Markovian coalescent (PSMC) model applied to the complete diploid genome sequences of a Chinese male (YH)6, a Korean male (SJK)7, three European individuals (Venter8, NA12891 and NA128789) and two Yoruba males (NA1850710 and NA19239). We infer that European and Chinese populations had very similar population size histories before 10–20kya. Both populations experienced a severe bottleneck between 10–60kya while African populations experienced a milder bottleneck from which they recovered earlier. All three populations have an elevated effective population size between 60–250kya, possibly due to a population structure11. We also infer that the differentiation of genetically modern humans may have started as early as 100–120kya12, but considerable genetic exchanges may still have occurred until 20–40kya.
The distribution of the time since the most recent common ancestor (TMRCA) between two alleles in an individual provides information about the history of population size change over time. Existing methods for reconstructing the detailed TMRCA distribution have analyzed large samples of individuals at non-recombining loci like mitochondrial DNA13. However, the statistical resolution of inferences from any one locus is poor, and power fades rapidly moving back in time as there are few independent lineages probing deep time depths (in humans, no information at all is available from mitochondrial DNA beyond about 200kya when all humans share a common maternal ancestor11). In contrast, a diploid genome sequence harbors hundreds of thousands of independent loci, each with its own TMRCA between the two alleles an individual carries. In principle it should be possible to reconstruct the TMRCA distribution across the autosomes and chromosome X by studying how the local density of heterozygous sites changes across the genome, reflecting segments of constant TMRCA separated by historical recombination events. To explore whether we could take advantage of this idea to learn about the detailed TMRCA distribution from a diploid whole genome sequence, we proposed the PSMC model, which is a specialization of the sequentially Markovian coalescent model14 to the case of two chromosomes (Figure 1a). The free parameters of this model include the scaled mutation and recombination rates, and piecewise constant ancestral population sizes (Methods). We scaled results to real time assuming 25 years per generation and a neutral mutation rate of 2.5×10−8 per generation15. The consequences of uncertainty in the two scaling parameters will be discussed later in the text.
Figure 1. Illustration of the PSMC model and its application to simulated data.
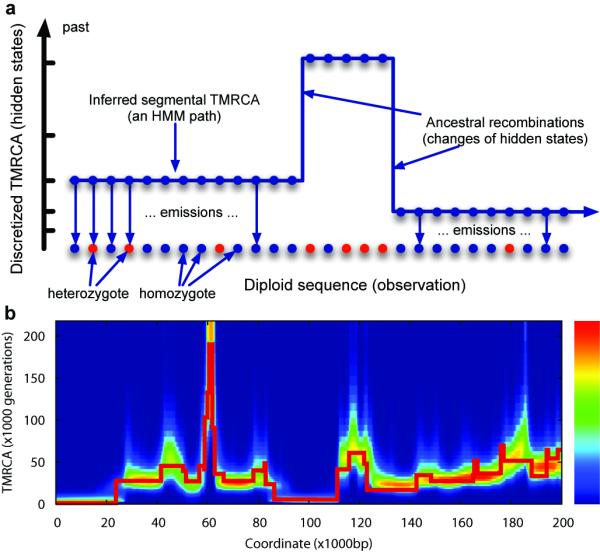
(a) The PSMC infers the local time to the most recent common ancestor (TMRCA) based on the local density of heterozygotes, using a Hidden Markov Model, where the observation is a diploid sequence, the hidden states are discretized TMRCA and the transitions represent ancestral recombination events. (b) We used the ms software to simulate the TMRCA relating the two alleles of an individual across a 200kb region (the thick red line), and inferred the local TMRCA at each locus using the PSMC (the heat map). The inference usually includes the correct time, with the greatest errors at transition points.
To validate our model, we simulated a hundred 30Mbp sequences with a sharp out-of-Africa bottleneck followed by a population expansion, and inferred population size history with PSMC (Figure 2a). PSMC is able to recover the parameters used in the simulation and the variance of the estimate is small between 20kya–3Mya. More recently than 20kya or more anciently than 3Mya few recombination events are left in the present sequence, which reduces the power of PSMC, and therefore the estimated effective population size (Ne) in these time intervals is not as accurate and has large variance. To test the robustness of the model, we introduced variable mutation rates and recombination hotspots in the simulation (Supplementary Text). The inference is still close to the true history (Figure 2b). A uniform rate of SNP ascertainment errors does not change our qualitative results, either (Figure S2). On the other hand, the simulations also reveal a limitation of PSMC in recovering sudden changes in effective population size. For example the instantaneous reduction from 12,000 to 1,200 at 100kya in the simulation was spread over the preceding 50,000 years in the PSMC reconstruction.
Figure 2. PSMC estimate on simulated data.

(a) PSMC estimate on data simulated by msHOT. The blue curve is the population size history used in simulation; the red curve is the PSMC estimate on the originally simulated sequence; the 100 thin green curves are the PSMC estimates on 100 sequences randomly resampled from the original sequence. (b) PSMC estimate on data with variable mutation rate or with hotspots.
We applied the PSMC model to real data from recently published genome sequences (Table 1). Figure 3a reveals that all populations are very similar in estimated Ne history between 150 and 1500kya. YRI differentiates from non-African populations around 100–120kya (at 110kya, NeYRI=15313±559 and NeCHN=12829±485). This evidence of early population differentiation is potentially consistent with the archaeological evidence of anatomically modern humans found in the Near East around 100kya12. European and East Asian populations are nearly identical in estimated Ne beyond 11kya. From a peak of 13,500 at 150kya, the Ne dropped by a factor of 10 to 1,200 between 40–20kya, before a sharp increase whose precise magnitude we do not have power to measure. We also observe a less marked bottleneck in YRI from a peak of 16,100 around 100–150kya to 5,700 at 50kya, recovering earlier16 than the out-of-Africa populations with increases back to 8,700 by 20kya, coinciding with the Last Glacial Maximum. All populations show increased Ne between 60–200kya, about the time of origin of anatomically modern humans17. An alternative to an increase in actual population size during this time would be that there was population structure involving separation and admixture11,16 (Figure S5).
Table 1.
Properties of the input sequences.
| Label | Description | Coverage | # Called bases (bp) |
# Heterozygotes (bp) |
Heterozygosity (×0.001) |
|---|---|---|---|---|---|
| YRI1.A10 | NA18507 autosomes | 40X | 2.14 G | 2.17 M | 1.013 |
| YRI2.A9 | NA19239 autosomes | 29X | 2.11 G | 2.21 M | 1.051 |
| EUR1.A8 | Venter autosomes | 9X | 2.13 G | 1.23 M | 0.578 |
| EUR2.A9 | NA12891 autosomes | 38X | 2.11 G | 1.67 M | 0.791 |
| KOR.A7 | SJK autosomes | 20X | 2.13 G | 1.47 M | 0.690 |
| CHN.A6 | YH autosomes | 30X | 2.19 G | 1.52 M | 0.694 |
| YRI3.X9 | NA19240 X chromosome | 38X | 106 M | 71.6 k | 0.673 |
| EUR3.X9 | NA12878 X chromosome | 35X | 110 M | 48.0 k | 0.436 |
| KOR-CHN.X | SJK-YH combined X chr. | - | 102 M | 39.7 k | 0.390 |
| YRI1-EUR1.X | NA18507-Venter combined X chr. | - | 83 M | 55.6 k | 0.670 |
| YRI1-KOR.X | NA18507-KOR combined X chr. | - | 100 M | 66.9 k | 0.669 |
| YRI1-CHN.X | NA18507-YH combined X chr. | - | 106 M | 69.5 k | 0.657 |
Coverage equals the average number of reads covering HapMap3 loci. A base is said to be called if it passes all filters described (Methods). The relatively lower coverage for EUR1.A leads to higher sampling bias at heterozygotes, which leads to underestimated heterozygosity but can be corrected by adjusting the neutral mutation rate in scaling (Supplementary Section S1.2).
Figure 3. PSMC estimate on real data.

(a) The population sizes inferred from autosomes of six individuals. 5%, 10% and 29% of heterozygotes are assumed to be missing in CHN.A, KOR.A and EUR1.A, respectively. (b) The population sizes inferred from male-combined X chromosomes and the simulated African-Asian combined sequences from the best-fit model by Schaffner et al. Sizes inferred from X chromosome data are scaled by 4/3; the neutral mutation rate on X, which is used in time scaling, is estimated with the ratio of male-to-female mutation rate α equal to 2 (Methods).
We also see in all populations an increase in estimated Ne beyond 1Mya, with a sharp increase beyond 3Mya. Although it is tempting to read into this the transition from the previously estimated larger Ne at the time of the split from chimpanzee18, our method may also be subject to artifacts in this region due to regions of balancing selection or clustered false heterozygotes related to segmental duplications (Figure S3).
Analyzing a European female X chromosome (EUR3.X) yields a history similar to that from autosomes scaled by 0.75, as expected for the X chromosome (Figure 3b). We do not observe a more severe bottleneck on the X chromosome19. To investigate the relationship between African and non-African populations, we combined X chromosomes from YRI and a non-African to construct a pseudo-diploid genome. From Figure 3b, we can see that although African and non-African populations might have started to differentiate as early as 100–120kya, they largely remain as one population until approximately 60–80kya, the time point when the YRI1-EUR1.X curve clearly leaves EUR3.X. This supports the recent analysis of the relationship between the Neandertal genome and that of modern humans20, which concluded that West Africans and non-Africans descended from a homogeneous ancestral population in the last 100,000 years with subsequent minor admixture out of Africa from Neandertals, rather than an alternative explanation of ancient (>300,000 year old) sub-structure separating West African and non-African populations.
From Figure 3b, we also notice surprisingly that there is a low Ne between African and non-African populations until approximately 20kya, suggesting substantial genetic exchanges between these populations long after the initial separation. Complete separation would correspond to very large or effectively infinite Ne, as seen below 20kya. To explore whether the inferred recent gene flow is a modeling artifact, we simulated complete divergence at 60kya according to the Schaffner et al. model21, and saw increased rather than reduced Ne in the period 20–60kya (brown line in Figure 3b). To explore further, we extracted from YRI1-KOR.X segments that PSMC indicates coalesce more recently than 50kya. These comprise 220 segments covering 31.2Mbp (>20% X chromosome). We observe 1,363 base-pair differences in 20.7Mbp of callable sequence in these segments, corresponding to an average divergence time of 37.4kya. In contrast, if we apply the same process to the simulated data from the Schaffner et al. model, the apparently recently diverged segments cover only 0.4% of the simulated chromosome. The human-macaque divergence in the 220 segments was only 4% lower than the chromosome average, so regional variability in mutation rates cannot explain these results. In summary the existence of long segments of low divergence between YRI1 and KOR suggests substantial genetic exchange between West African and non-African populations up until 20–40kya, and is not consistent with a simple separation approximately 60kya.
The evidence for continued gene flow between Africans and non-Africans prior to the separation of Europeans and East Asians (Supplementary Section S4.2) is more recent than the archaeologically documented time of the out-of-Africa dispersal since there are fossils in both Europe and Australasia that date to >40 kya22. An important caveat to this result is uncertainty of the per-year mutation rate 1.0×10−9 (=2.5×10−8/25). While this mutation rate agrees well with the rates estimated between primates averaged over millions of years (Supplementary Section S3.1), generation intervals as high as 29 years per generation over the last few thousands of years23 and present mutation rates lower than 2.5×10−8 per generation9 are possible in principle, and these could make our recent date estimates somewhat older, although it is difficult to imagine a date of final gene flow of as old as 60kya as being consistent with the data due to these inaccuracies. Our analyses can also not exclude the possibility that the divergence time inferred from X chromosomes may not be representative due to sex-biased demographic processes19, highlighting the importance of repeating this analysis on autosomal data once haploid whole genome sequences become available24. Intriguingly, a recent study using an orthogonal type of data (analysis of allele frequencies) also inferred that gene flow between Africans and non-Africans continued until strikingly recently, in the case of that study, until 17–26kya25. An important goal for future work is to determine whether these recent dates reflect real history, and if so to obtain more detail about the timing and scale of the events involved.
In this paper we have introduced a novel method to infer the history of effective population size from genome wide diploid sequence data. It is relatively straightforward to apply, with less potential ascertainment bias in comparison to existing methods that use selective genotyping data or the resequencing data from a few loci. Furthermore, our method is computationally tractable and typically uses much more primary sequence data than the existing methods, which allows us to estimate population size at each time going back in history, rather than assume a parametric structure of times, divergences and size changes. The results described above concerning the timing and depth of the out-of-Africa bottleneck are broadly consistent with previous studies though our results are more detailed (Supplementary Section S4.2). The hypothesis that there was significant ongoing genetic exchange throughout the bottleneck is surprising in light of current views about human migrations; however, it is not inconsistent with the archaeological literature, and should motivate further research. There is the potential to extend this type of SMC-HMM approach to data from multiple individuals, which would access more recent times, but this will require inference over a substantially more complex hidden state space of trees on the haplotypes, with each Markov path representing an ancestral recombination graph14. In addition, beyond humans, there is the potential to apply the method to investigate the population size history of other species for which a single diploid genome sequence has been obtained (Supplementary Section S2.2).
Methods Summary
Illumina short reads were obtained from Short Read Archive and capillary reads from TraceDB. Reads were aligned to the human reference genome with BWA26. The consensus sequences were called by SAMtools27 and then divided into non-overlapping 100bp bins with a bin scored heterozygous if there is a heterozygote in the bin or being homozygous otherwise. The resultant bin sequences were taken as the input of the PSMC estimate. Coalescent simulation was done by ms28 and cosi21. The simulated sequences were binned in the same way.
The free parameters in the discrete PSMC-HMM model are the scaled mutation rate, recombination rate and piecewise constant population sizes. The time interval each size parameter spans was manually chosen. The estimation-maximization iteration started from a constant-sized population history. The estimation step was done analytically; Powell’s direction set method is used for the maximization step. Parameter values stablized by the 20th iteration, and these were taken as the final estimate. All parameters are scaled to a constant that is further determined under the assumption of a neutral mutation rate 2.5×10−8.
Supplementary Material
Acknowledgement
We are grateful to David Bentley from Illumina and Jun Wang from Beijing Genomics Institute for early access to the sequencing data. We thank Avril Coghlan for the idea of bootstrapping, and Nick Patterson, Molly Przeworski, David Reich, and members of the Durbin research group for helpful discussions and critiques. This work was funded by the Wellcome Trust.
APPENDIX
Online Methods
Read alignment
Alignment for individuals from the 1000 Genomes Project (NA12878, NA12891, NA19239 and NA19240) was obtained from the project FTP site. Illumina sequence reads for NA18507, YH, and SJK were obtained from SRA (AC:ERA000005, SRA000271 and SRA008175, respectively) and Sanger sequencing reads for Craig Venter from NCBI TraceDB. These sequence reads were mapped by BWA (0.5.5) against the human reference genome build 36 including unassembled contigs and the genome of Epstein-Barr Virus (AC:NC_007605), with pseudoautosomal regions on Y masked. For Illumina short reads, BWA option ‘−q 15’ was applied to enable trimming of low-quality bases at the 3′-end. Base qualities of SJK reads were overestimated and thus were recalibrated using GATK29 after alignment with known SNPs in dbSNP-129 discarded. For capillary reads, the BWA-SW algorithm with the default options was used.
Calling the consensus sequence
The diploid consensus sequence for an autosome was obtained by the ‘pileup’ command of the SAMtools software package and then processed with the following loci marked as missing data: a) read depth over twice above or below half of the average read depth estimated on HapMap3 genotyping loci; b) the root-mean-square mapping quality of reads covering the locus below 25; c) 10bp around predicted short INDELs, d) inferred consensus quality below a threshold (20 for Illumina data and 10 for capillary data), or e) where fewer than 18 out of the 35 overlapping 35-mers from the reference sequence can be mapped elsewhere with zero or one mismatch.
The X chromosome consensus was derived in a similar way but with pseudoautosomal regions filtered as missing data. The X chromosomes of males are haploid and thus the few heterozygotes that were called were discarded as errors. The pseudo-diploid X chromosomes of males were combined by marking a difference as a heterozygote.
The consensus sequences were further divided into 100bp non-overlapping bins where each bin is represented as a ‘.’ (missing) if 90 or more bases are filtered or uncalled; as ‘1’ (heterozygous) if over 10bp are called and there is at least one heterozygote; or as ‘0’ (homozygous) otherwise. The sequence of bin values was taken as the input of the PSMC inference.
Coalescent simulation
100 sequences of 30Mbp were simulated by ms with piecewise constant history as shown in Figure 2a. To simulate variable in mutation rate, the local mutation rate averaged in a 20kbp window between human and macaque was calculated from the EPO cross-species alignment obtained from Ensembl v50. In the simulation, the local coalescent trees were simulated with ms but mutations were generated based on the relative local mutation rate on a 30Mbp segment randomly drawn from the human-macaque alignment. The program msHOT was used to simulate sequences with recombination hotspots. The location and size of hotspots were randomly drawn from the hotspot map obtained from HapMap (release 21); the scaled recombination rate in hotspots was 10 times higher than that in non-hotspot regions.
The cosi software package was used to simulate sequences under the best-fit model by Schaffner et al.21. This model considers variable recombination rates, recombination hotspots and migration between African and non-African populations.
Overview of the PSMC model
In the PSMC-HMM, the observation is a binary sequence of 0s, 1s and dots as described above. The emission probability from state t is e(1|t)=e−θt, e(0|t)=1-e−θt and e(.|t)=1; the transition probability from s to t is:
where θ is the scaled mutation rate, ρ the scaled recombination rate, δ(·) is the Dirac delta function and
is the transition probability conditional on there being a recombination event, where λ(t)=Ne(t)/N0 is the relative population size at state t. The discrete-state HMM is constructed by dividing coalescent time into intervals and integrating emission and transition probabilities in the intervals, which can be done analytically given a piecewise-constant function λ(t). The stationary distribution of TMRCA can also be analytically derived. Details are available in the Supplementary Text.
Scaling to real time
The estimated TMRCA is in units of 2N0 time and λ(t) is scaled to N0 as well. The value of N0 cannot be determined from the model itself. To estimate N0, a neutral mutation rate μA=2.5×10−8 on autosomes15 was used and thus N0A=θ/4μA. Given the ratio of male-to-female mutation rate30 α=2, the neutral mutation rate of X chromosomes was derived as μX=μA·[2(2+α)]/[3(1+α)]=2.2×10−8. Missing heterozygotes uniformly at a probability p is equivalent to reducing the neutral mutation rate from μ to μ’=μ·(1-p). False negatives due to the lack of coverage can thus be corrected. Generations were converted to years under the assumption of 25 years per generation.
Parameter estimate with PSMC
Given a maximum TMRCA in the 2N0 scale Tmax and the number of atomic time intervals n, let the boundaries of these intervals be ti=0.1exp[i/n·log(1+10Tmax)]-0.1, i=0,…,n. To reduce the complexity of the search space, blocks of adjacent atomic intervals were combined to have the same population size parameter via a user-specified pattern. On autosome and simulated data, Tmax=15, n=64 and the pattern is ‘1*4+25*2+1*4+1*6’ which means the first population size parameter spans the first 4 atomic time intervals, each of the next 25 parameters spans 2 intervals, the 27th spans 4 intervals and the last parameter spans the last 6 time intervals. On X chromosome data, Tmax=15, n=60, and the pattern is ‘1*6+2*4+1*3+13*2+1*3+2*4+1*6’.
In the EM parameter estimate, the initial population size parameters were all set as 1, representing a constant-sized history; the scaled mutation rate was calculated to match the observed heterozygosity; the initial value of the scaled recombination rate was arbitrarily set as a quarter of the mutation rate. At the maximization step, Powell’s direction set method was used to numerically minimize the Q function in the EM algorithm. Parameters at the 20th EM iteration were taken as the final results.
Bootstrapping was applied by breaking the consensus sequences into 5Mbp segments and randomly sampling with replacement a set of segments such that the total length of the sampled segments are close to the size of the human reference genome.
Further discussion of methods and parameters is given in the Supplementary Text.
References for online methods
- 29.McKenna A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miyata T, Hayashida H, Kuma K, Mitsuyasu K, Yasunaga T. Male-driven molecular evolution: a model and nucleotide sequence analysis. Cold Spring Harb Symp Quant Biol. 1987;52:863–867. doi: 10.1101/sqb.1987.052.01.094. [DOI] [PubMed] [Google Scholar]
Footnotes
Author information The method is implemented in the PSMC software package that is freely available at http://github.com/lh3/psmc.
The authors declare no competing financial interests.
References
- 1.Reich DE, et al. Linkage disequilibrium in the human genome. Nature. 2001;411:199–204. doi: 10.1038/35075590. [DOI] [PubMed] [Google Scholar]
- 2.Marth GT, Czabarka E, Murvai J, Sherry ST. The allele frequency spectrum in genome-wide human variation data reveals signals of differential demographic history in three large world populations. Genetics. 2004;166:351–372. doi: 10.1534/genetics.166.1.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Plagnol V, Wall JD. Possible ancestral structure in human populations. PLoS Genet. 2006;2:e105. doi: 10.1371/journal.pgen.0020105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keinan A, Mullikin JC, Patterson N, Reich D. Measurement of the human allele frequency spectrum demonstrates greater genetic drift in East Asians than in Europeans. Nat Genet. 2007;39:1251–1255. doi: 10.1038/ng2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fagundes NJR, et al. Statistical evaluation of alternative models of human evolution. Proc Natl Acad Sci. 2007;104:17614–17619. doi: 10.1073/pnas.0708280104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang J, et al. The diploid genome sequence of an Asian individual. Nature. 2008;456:60–65. doi: 10.1038/nature07484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahn S-M, et al. The first Korean genome sequence and analysis: full genome sequencing for a socio-ethnic group. Genome Res. 2009;19:1622–1629. doi: 10.1101/gr.092197.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levy S, et al. The Diploid Genome Sequence of an Individual Human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–73. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bentley DR, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Behar DM, et al. The dawn of human matrilineal diversity. Am J Hum Genet. 2008;82:1130–1140. doi: 10.1016/j.ajhg.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mellars P. Going east: new genetic and archaeological perspectives on the modern human colonization of Eurasia. Science. 2006a;313:796–800. doi: 10.1126/science.1128402. [DOI] [PubMed] [Google Scholar]
- 13.Atkinson QD, Gray RD, Drummond AJ. mtDNA variation predicts population size in humans and reveals a major Southern Asian chapter in human prehistory. Mol Biol Evol. 2008;25:468–474. doi: 10.1093/molbev/msm277. [DOI] [PubMed] [Google Scholar]
- 14.McVean GAT, Cardin NJ. Approximating the coalescent with recombination. Philos Trans R Soc Lond B Biol Sci. 2005;360:1387–1393. doi: 10.1098/rstb.2005.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nachman MW, Crowell SL. Estimate of the mutation rate per nucleotide in humans. Genetics. 2000;156:297–304. doi: 10.1093/genetics/156.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mellars P. Why did modern human populations disperse from Africa ca. 60,000 years ago? A new model. Proc Natl Acad Sci. 2006b;103:9381–9386. doi: 10.1073/pnas.0510792103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wall JD, Hammer MF. Archaic admixture in the human genome. Curr Opin Genet Dev. 2006;16:606–610. doi: 10.1016/j.gde.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 18.Hobolth A, Christensen OF, Mailund T, Schierup MH. Genomic relationships and speciation times of human, chimpanzee, and gorilla inferred from a coalescent hidden Markov model. PLoS Genet. 2007;3:e7. doi: 10.1371/journal.pgen.0030007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keinan A, Mullikin JC, Patterson N, Reich D. Accelerated genetic drift on chromosome X during the human dispersal out of Africa. Nat Genet. 2009;41:66–70. doi: 10.1038/ng.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Green RE, et al. A draft sequence of the Neandertal genome. Science. 2010;328:710–22. doi: 10.1126/science.1188021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schaffner SF, et al. Calibrating a coalescent simulation of human genome sequence variation. Genome Res. 2005;15:1576–1583. doi: 10.1101/gr.3709305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mellars P. A new radiocarbon revolution and the dispersal of modern humans in Eurasia. Nature. 2006c;439:931–5. doi: 10.1038/nature04521. [DOI] [PubMed] [Google Scholar]
- 23.Fenner JN. Cross-cultural estimation of the human generation interval for use in genetics-based population divergence studies. Am J Phys Anthropol. 2005;128:415–23. doi: 10.1002/ajpa.20188. [DOI] [PubMed] [Google Scholar]
- 24.Kitzman JO, et al. Haplotype-resolved genome sequencing of a Gujarati Indian individual. Nat Biotechnol. 2010 doi: 10.1038/nbt.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gutenkunst RN, Hernandez RD, Williamson SH, Bustamante CD. Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genet. 2009;5:e1000695. doi: 10.1371/journal.pgen.1000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hudson RR. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics. 2002;18:337–338. doi: 10.1093/bioinformatics/18.2.337. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.