

Abstract
This protocol describes how to perform basic statistical analysis in a population-based genetic association case-control study. The steps described involve the (i) appropriate selection of measures of association and relevance of disease models; (ii) appropriate selection of tests of association; (iii) visualization and interpretation of results; (iv) consideration of appropriate methods to control for multiple testing; and (v) replication strategies. Assuming no previous experience with software such as PLINK, R or Haploview, we describe how to use these popular tools for handling single-nucleotide polymorphism data in order to carry out tests of association and visualize and interpret results. This protocol assumes that data quality assessment and control has been performed, as described in a previous protocol, so that samples and markers deemed to have the potential to introduce bias to the study have been identified and removed. Study design, marker selection and quality control of case-control studies have also been discussed in earlier protocols. The protocol should take ~1 h to complete.
INTRODUCTION
A genetic association case-control study compares the frequency of alleles or genotypes at genetic marker loci, usually single-nucleotide polymorphisms (SNPs) (see Box 1 for a glossary of terms), in individuals from a given population—with and without a given disease trait—in order to determine whether a statistical association exists between the disease trait and the genetic marker. Although individuals can be sampled from families (‘family-based’ association study), the most common design involves the analysis of unrelated individuals sampled from a particular outbred population (‘population-based association study’). Although disease-related traits are usually the main trait of interest, the methods described here are generally applicable to any binary trait.
BOX 1. GLOSSARY.
Admixture
The result of interbreeding between individuals from different populations.
Cochran-Armitage trend test
Statistical test for analysis of categorical data when categories are ordered. It is used to test for association in a 2 × k contingency table (k > 2). In genetic association studies, because the underlying genetic model is unknown, the additive version of this test is most commonly used.
Confounding
A type of bias in statistical analysis that occurs when a factor exists that is causally associated with the outcome under study (e.g., case-control status) independently of the exposure of primary interest (e.g., the genotype at a given locus) and is associated with the exposure variable but is not a consequence of the exposure variable.
Covariate
Any variable other than the main exposure of interest that is possibly predictive of the outcome under study; covariates include confounding variables that, in addition to predicting the outcome variable, are associated with exposure.
False discovery rate
The proportion of non-causal or false positive significant SNPs in a genetic association study.
False positive
Occurs when the null hypothesis of no effect of exposure on disease is rejected for a given variant when in fact the null hypothesis is true.
Family-wise error rate
The probability of one or more false positives in a set of tests. For genetic association studies, family-wise error rates reflect false positive findings of associations between allele/genotype and disease.
Hardy-Weinberg equilibrium (HWE)
Given a minor allele frequency of p, the probabilities of the three possible unordered genotypes (a/a, A/a, A/A) at a biallelic locus with minor allele A and major allele a, are (1 – p)2, 2p (1 – p), p2. In a large, randomly mating, homogenous population, these probabilities should be stable from generation to generation.
Linkage disequilibrium (LD)
The population correlation between two (usually nearby) allelic variants on the same chromosome; they are in LD if they are inherited together more often than expected by chance.
r2
A measure of LD between two markers calculated according to the correlation between marker alleles.
Odds ratio
A measure of association derived from case-control studies; it is the ratio of the odds of disease in the exposed group compared with the non-exposed.
Penetrance
The risk of disease in a given individual. Genotype-specific penetrances reflect the risk of disease with respect to genotype.
Population allele frequency
The frequency of a particular allelic variant in a general population of specified origin.
Population stratification
The presence of two or more groups with distinct genetic ancestry.
Relative risk
The risk of disease or of an event occurring in one group relative to another.
Single-nucleotide polymorphism (SNP)
A genetic variant that consists of a single DNA base-pair change, usually resulting in two possible allelic identities at that position.
Following previous protocols on study design, marker selection and data quality control1–3, this protocol considers basic statistical analysis methods and techniques for the analysis of genetic SNP data from population-based genome-wide and candidate-gene (CG) case-control studies. We describe disease models, measures of association and testing at genotypic (individual) versus allelic (gamete) level, single-locus versus multilocus methods of association testing, methods for controlling for multiple testing and strategies for replication. Statistical methods discussed relate to the analysis of common variants, i.e., alleles with a minor allele frequency (MAF) > 1%; different analytical techniques are required for the analysis of rare variants4. All methods described are proven and used routinely in our research group5,6.
Conceptual basis for statistical analysis
The success of a genetic association study depends on directly or indirectly genotyping a causal polymorphism. Direct genotyping occurs when an actual causal polymorphism is typed. Indirect genotyping occurs when nearby genetic markers that are highly correlated with the causal polymorphism are typed. Correlation, or non-random association, between alleles at two or more genetic loci is referred to as linkage disequilibrium (LD). LD is generated as a consequence of a number of factors and results in the shared ancestry of a population of chromosomes at nearby loci. The shared ancestry means that alleles at flanking loci tend to be inherited together on the same chromosome, with specific combinations of alleles known as haplotypes. In genome-wide association (GWA) studies, common SNPs are typically typed at such high density across the genome that, although any single SNP is unlikely to have direct causal relevance, some are likely to be in LD with any underlying common causative variants. Indeed, most recent GWA arrays containing up to 1 million SNPs use known patterns of genomic LD from sources such as HapMap7 to provide the highest possible coverage of common genomic variation8. CG studies usually focus on genotyping a smaller but denser set of SNPs, including functional polymorphisms with a potentially higher previous probability of direct causal relevance2.
A fundamental assumption of the case-control study is that the individuals selected in case and control groups provide unbiased allele frequency estimates of the true underlying distribution in affected and unaffected members of the population of interest. If not, association findings will merely reflect biases resulting from the study design1.
Models and measures of association
Consider a genetic marker consisting of a single biallelic locus with alleles a and A (i.e., a SNP). Unordered possible genotypes are then a/a, a/A and A/A. The risk factor for case versus control status (disease outcome) is the genotype or allele at a specific marker. The disease penetrance associated with a given genotype is the risk of disease in individuals carrying that genotype. Standard models for disease penetrance that imply a specific relationship between genotype and phenotype include multiplicative, additive, common recessive and common dominant models. Assuming a genetic penetrance parameter γ (γ > 1), a multiplicative model indicates that the risk of disease is increased γ-fold with each additional A allele; an additive model indicates that risk of disease is increased γ-fold for genotype a/A and by 2γ-fold for genotype A/A; a common recessive model indicates that two copies of allele A are required for a γ-fold increase in disease risk, and a common dominant model indicates that either one or two copies of allele A are required for a γ-fold increase in disease risk. A commonly used and intuitive measure of the strength of an association is the relative risk (RR), which compares the disease penetrances between individuals exposed to different genotypes. Special relationships exist between the RRs for these common models9 (see Table 1).
TABLE 1.
Disease penetrance functions and associated relative risks.
| Penetrance |
Relative risk |
||||
|---|---|---|---|---|---|
| Disease model | a/a | A/a | A/A | A/a | A/A |
| Multiplicative | f0 | f0γ | f0γ2 | γ | γ2 |
| Additive | f0 | f0γ | 2f0γ | γ | 2γ |
| Common recessive | f0 | f0 | f0γ | 1 | γ |
| Common dominant | f0 | f0γ | f0γ | γ | γ |
Shown are disease penetrance functions for genotypes a/a, A/a and A/A and associated relative risks for genotypes A/a and A/a compared with baseline genotype a/a for standard disease models when baseline disease penetrance associated with genotype a/a is f0 0 and genetic penetrance parameter is γ> 19.
RR estimates based on penetrances can only be derived directly from prospective cohort studies, in which a group of exposed and unexposed individuals from the same population are followed up to assess who develops disease. In a case-control study, in which the ratio of cases to controls is controlled by the investigator, it is not possible to make direct estimates of disease penetrance, and hence of RRs. In this type of study, the strength of an association is measured by the odds ratio (OR). In a case-control study, the OR of interest is the odds of disease (the probability that the disease is present compared with the probability that it is absent) in exposed versus non-exposed individuals. Because of selected sampling, odds of disease are not directly measurable. However, conveniently, the disease OR is mathematically equivalent to the exposure OR (the odds of exposure in cases versus controls), which we can calculate directly from exposure frequencies10. The allelic OR describes the association between disease and allele by comparing the odds of disease in an individual carrying allele A to the odds of disease in an individual carrying allele a. The genotypic ORs describe the association between disease and genotype by comparing the odds of disease in an individual carrying one genotype to the odds of disease in an individual carrying another genotype. Hence, there are usually two genotypic ORs, one comparing the odds of disease between individuals carrying genotype A/A and those carrying a/a and the other comparing the odds of disease between individuals carrying genotype a/A and those carrying genotype a/a. Beneficially, when disease penetrance is small, there is little difference between RRs and ORs (i.e., RR ≈ OR). Moreover, the OR is amenable to analysis by multivariate statistical techniques that allow extension to incorporate further SNPs, risk factors and clinical variables. Such techniques include logistic regression and other types of log-linear models11.
To work with observations made at the allelic (gamete) rather than the genotypic (individual) level, it is necessary to assume (i) that there is Hardy-Weinberg equilibrium (HWE) in the population, (ii) that the disease has a low prevalence ( < 10%) and (iii) that the disease risks are multiplicative. Under the null hypothesis of no association with disease, the first condition ensures that there is HWE in both controls and cases. Under the alternative hypothesis, the second condition further ensures that controls will be in HWE and the third condition further ensures that cases will also be in HWE. Under these assumptions, allelic frequencies in affected and unaffected individuals can be estimated from case-control studies. The OR comparing the odds of allele A between cases and controls is called the allelic RR (γ*). It can be shown that the genetic penetrance parameter in a multiplicative model of penetrance is closely approximated by the allelic RR, i.e., γ ≈ γ* (ref. 10).
Tests for association
Tests of genetic association are usually performed separately for each individual SNP. The data for each SNP with minor allele a and major allele A can be represented as a contingency table of counts of disease status by either genotype count (e.g., a/a, A/a and A/A) or allele count (e.g., a and A) (see Box 2). Under the null hypothesis of no association with the disease, we expect the relative allele or genotype frequencies to be the same in case and control groups. A test of association is thus given by a simple χ2 test for independence of the rows and columns of the contingency table.
Box 2. CONTINGENCY TABLES AND ASSOCIATED TESTS.
The risk factor for case versus control status (disease outcome) is the genotype or allele at a specific marker. The data for each SNP with minor allele a and major allele A in case and control groups comprising n individuals can be written as a 2 × k contingency table of disease status by either allele (k = 2) or genotype (k = 3) count.
Allele count
| Allele | a | A | Total |
|---|---|---|---|
| Cases | m11 | m12 | m1. |
| Controls | m21 | m22 | m2. |
| Total | m.1 | m.2 | 2n |
The allelic odds ratio is estimated by .
If the disease prevalence in a control individual carrying an a allele can be estimated and is denoted as P0, then the relative risk of disease in individuals with an A allele compared with an a allele is estimated by .
An allelic association test is based on a simple χ2 test for independence of rows and columns where has a χ2 distribution with 1 d.f. under the null hypothesis of no association.
Genotype count
| Genotype | a/a | A/a | A/A | Total |
|---|---|---|---|---|
| Case | n11 | n12 | n13 | n1. |
| Controls | n21 | n22 | n23 | n2. |
| Total | n.1 | n.2 | n.3 | n |
The genotypic odds ratio for genotype A/A relative to genotype a/a is estimated by . The genotypic odds ratio for genotype A/a relative to genotype a/a is estimated by .
- If the disease prevalence in a control individual carrying an a/a genotype can be estimated and is denoted as P0, then the relative risk of disease in individuals with an A/A [A/a] genotype compared with an a/a genotype is estimated by
A genotypic association test is based on a simple χ2 test for independence of rows and columns where has a χ2 distribution with 2 d.f. under the null hypothesis of no association. To test for a dominant (recessive) effect of allele A, counts for genotypes a/A and A/A (a/a and A/a) can be combined and the usual 1 d.f. χ2-test for independence of rows and columns can be applied to the summarized 2 × 2 table.
- A Cochran-Armitage trend test of association between disease and marker is given by
where w = (w1, w2, w3) are weights chosen to detect particular types of association. For example, to test whether allele A is dominant over allele a w = (0,1,1) is optimal; to test whether allele A is recessive to allele a, the optimal choice is w = (0,0,1). In genetic association studies, w = (0,1,2) is most often used to test for an additive effect of allele A. T2 has a χ2 distribut ion with 1 d.f. under the null hypothesis of no association.
In a conventional χ2 test for association based on a 2 × 3 contingency table of case-control genotype counts, there is no sense of genotype ordering or trend: each of the genotypes is assumed to have an independent association with disease and the resulting genotypic association test has 2 degrees of freedom (d.f.). Contingency table analysis methods allow alternative models of penetrance by summarizing the counts in different ways. For example, to test for a dominant model of penetrance, in which any number of copies of allele A increase the risk of disease, the contingency table can be summarized as a 2 × 2 table of genotype counts of A/A versus both a/A and a/a combined. To test for a recessive model of penetrance, in which two copies of allele A are required for any increased risk, the contingency table is summarized into genotype counts of a/a versus a combined count of both a/A and A/A genotypes. To test for a multiplicative model of penetrance using contingency table methods, it is necessary to analyze by gamete rather than individual: a χ2 test applied to the 2 × 2 table of case-control allele counts is the widely used allelic association test. The allelic association test with 1 d.f. will be more powerful than the genotypic test with 2 d.f., as long as the penetrance of the heterozygote genotype is between the penetrances of the two homozygote genotypes. Conversely, if there is extreme deviation from the multiplicative model, the genotypic test will be more powerful. In the absence of HWE in controls, the allelic association test is not suitable and alternative methods must be used to test for multiplicative models. See the earlier protocol on data quality assessment and control for a discussion of criteria for retaining SNPs showing deviation from HWE3. Alternatively, any penetrance model specifying some kind of trend in risk with increasing numbers of A alleles, of which additive, dominant and recessive models are all examples, can be examined using the Cochran-Armitage trend test12,13. The Cochran-Armitage trend test is a method of directing χ2 tests toward these narrower alternatives. Power is very often improved as long as the disease risks associated with the a/A genotype are intermediate to those associated with the a/a and A/A genotypes. In genetic association studies in which the underlying genetic model is unknown, the additive version of this test is most commonly used. Table 2 summarizes the various tests of association that use contingency table methods. Box 2 outlines contingency tables and associated tests in statistical detail.
TABLE 2.
Tests of association using contingency table methods.
| Test | Degrees of freedom (d.f.) |
contingency table description | PLINK keyword |
|---|---|---|---|
| Genotypic association |
2 | 2 × 3 table of N case-control by genotype (a/a, a/A, A/A) counts |
GENO |
| Dominant model | 1 | 2 × 2 table of N case-control by dominant genotype pattern of inheritance (a/a, not a/a) counts |
DOM |
| Recessive model | 1 | 2 × 2 table of N case-control by recessive genotype pattern of inheritance (not A/A, A/A) counts |
REC |
| Cochran-Armitage trend test |
1 | 2 × 3 table of N case-control by genotype (a/a, a/A, A/A) counts |
TREND |
| Allelic association | 1 | 2 × 2 table of 2N case-control by allele (a, A) counts |
ALLELIC |
d.f. for tests of association based on contingency tables along with associated PLINK keyword are shown for allele and genotype counts in case and control groups, comprising N individuals at a bi-allelic locus with alleles a and A.
Tests of association can also be conducted with likelihood ratio (LR) methods in which inference is based on the likelihood of the genotyped data given disease status. The likelihood of the observed data under the proposed model of disease association is compared with the likelihood of the observed data under the null model of no association; a high LR value tends to discredit the null hypothesis. All disease models can be tested using LR methods. In large samples, the χ2 and LR methods can be shown to be equivalent under the null hypothesis14.
More complicated logistic regression models of association are used when there is a need to include additional covariates to handle complex traits. Examples of this are situations in which we expect disease risk to be modified by environmental effects such as epidemiological risk factors (e.g., smoking and gender), clinical variables (e.g., disease severity and age at onset) and population stratification (e.g., principal components capturing variation due to differential ancestry3), or by the interactive and joint effects of other marker loci. In logistic regression models, the logarithm of the odds of disease is the response variable, with linear (additive) combinations of the explanatory variables (genotype variables and any covariates) entering into the model as its predictors. For suitable linear predictors, the regression coefficients fitted in the logistic regression represent the log of the ORs for disease gene association described above. Linear predictors for genotype variables in a selection of standard disease models are shown in Table 3.
TABLE 3.
Linear predictors for genotype variables in a selection of standard disease models.
| Genotype | Model |
|||
|---|---|---|---|---|
| Multiplicative | Genotypic | Recessive | Dominant | |
| a/a | b 0 | b 0 | b 0 | b 0 |
| a/A | b0 + b1 | b0 + b1 + b2 | b 0 | b0 + b1 |
| A/A | b0 + 2b1 | b0 + 2b1 | b0 + b1 | b0 + b1 |
| Interpretation | b1 provides an estimate of the log odds ratio for disease risk associated with each additional A allele (also called the haplotype relative risk). If b1 is significant, then there is a multiplicative contribution to disease risk in that the odds ratio for disease risk increases multiplicatively for every additional A allele | b1 and b2 provide estimates of the log odds ratio for disease risk in individuals with genotypes a/A and A/A, respectively, relative to an individual with genotype a/a. A likelihood ratio test of whether both b1 and b2 are significant is equivalent to the conventional 2 d.f. test for association in a 2 × 3 contingency table | b1 provides an estimate of the log odds ratio for disease risk in an individual with at least 1 A allele (genotype A/A or a/A) compared with an individual with no A alleles (genotype a/a). A test of whether b1 is significant corresponds to a 1 d.f. test for association in a 2 × 2 contingency table of disease outcome by genotype classified as A/A or not | b1 provides an estimate of the log odds ratio for disease risk in an individual with two A alleles (genotype A/A) compared with an individual with one or two A alleles (genotype a/A or a/a). A test of whether b1 is significant corresponds to a 1 d.f. test for association in a 2 × 2 contingency table of disease outcome by genotype classified as a/a or not |
Multiple testing
Controlling for multiple testing to accurately estimate significance thresholds is a very important aspect of studies involving many genetic markers, particularly GWA studies. The type I error, also called the significance level or false-positive rate, is the probability of rejecting the null hypothesis when it is true. The significance level indicates the proportion of false positives that an investigator is willing to tolerate in his or her study. The family-wise error rate (FWER) is the probability of making one or more type I errors in a set of tests. Lower FWERs restrict the proportion of false positives at the expense of reducing the power to detect association when it truly exists. A suitable FWER should be specified at the design stage of the analysis1. It is then important to keep track of the number of statistical comparisons performed and correct the individual SNP-based significance thresholds for multiple testing to maintain the overall FWER. For association tests applied at each of n SNPs, per-test significance levels of α* for a given FWER of α can be simply approximated using Bonferroni (α* = α/n) or Sidak15,16 (α* = 1 − (1 – α)1/n) adjustments. When tests are independent, the Sidak correction is exact; however, in GWA studies comprising dense sets of markers, this is unlikely to be true and both corrections are then very conservative. A similar but slightly less-stringent alternative to the Bonferroni correction is given by Holm17. Alternatives to the FWER approach include false discovery rate (FDR) procedures18,19, which control for the expected proportion of false positives among those SNPs declared significant. However, dependence between markers and the small number of expected true positives make FDR procedures problematic for GWA studies. Alternatively, permutation approaches aim to render the null hypothesis correct by randomization: essentially, the original P value is compared with the empirical distribution of P values obtained by repeating the original tests while randomly permuting the case-control labels20. Although Bonferroni and Sidak corrections provide a simple way to adjust for multiple testing by assuming independence between markers, permutation testing is considered to be the ‘gold standard’ for accurate correction20. Permutation procedures are computationally intensive in the setting of GWA studies and, moreover, apply only to the current genotyped data set; therefore, unless the entire genome is sequenced, they cannot generate truly genome-wide significance thresholds. Bayes factors have also been proposed for the measurement of significance6. For GWA studies of dense SNPs and resequence data, a standard genome-wide significance threshold of 7.2 × 10− 8 for the UK Caucasian population has been proposed by Dudbridge and Gusnanto21. Other thresholds for contemporary populations, based on sample size and proposed FWER, have been proposed by Hoggart et al22. Informally, some journals have accepted a genome-wide significance threshold of 5 × 10− 7 as strong evidence for association6; however, most recently, the accepted standard is 5 × 10− 8 (ref. 23). Further, graphical techniques for assessing whether observed P values are consistent with expected values include log quantile-quantile P value plots that highlight loci that deviate from the null hypothesis24.
Interpretation of results
A significant result in an association test rarely implies that a SNP is directly influencing disease risk; population association can be direct, indirect or spurious. A direct, or causal, association occurs when different alleles at the marker locus are directly involved in the etiology of the disease through a biological pathway. Such associations are typically only found during follow-up genotyping phases of initial GWA studies, or in focused CG studies in which particular functional polymorphisms are targeted. An indirect, or non-causal, association occurs when the alleles at the marker locus are correlated (in LD) with alleles at a nearby causal locus but do not directly influence disease risk. When a significant finding in a genetic association study is true, it is most likely to be indirect. Spurious associations can occur as a consequence of data quality issues or statistical sampling, or because of confounding by population stratification or admixture. Population stratification occurs when cases and controls are sampled disproportionately from different populations with distinct genetic ancestry. Admixture occurs when there has been genetic mixing of two or more groups in the recent past. For example, genetic admixture is seen in Native American populations in which there has been recent genetic mixing of individuals with both American Indian and Caucasian ancestry25. Confounding occurs when a factor exists that is associated with both the exposure (genotype) and the disease but is not a consequence of the exposure. As allele frequencies and disease frequencies are known to vary among populations of different genetic ancestry, population stratification or admixture can confound the association between the disease trait and the genetic marker; it can bias the observed association, or indeed can cause a spurious association. Principal component analyses or multidimensional scaling methods are commonly used to identify and remove individuals exhibiting divergent ancestry before association testing. These techniques are described in detail in an earlier protocol3. To adjust for any residual population structure during association testing, the principal components from principal component analyses or multidimensional scaling methods can be included as covariates in a logistic regression. In addition, the technique of genomic control26 can be used to detect and compensate for the presence of fine-scale or within-population stratification during association testing. Under genomic control, population stratification is treated as a random effect that causes the distribution of the χ2 association test statistics to have an inflated variance and a higher median than would otherwise be observed. The test statistics are assumed to be uniformly affected by an inflation factor λ, the magnitude of which is estimated from a set of selected markers by comparing the median of their observed test statistics with the median of their expected test statistics under an assumption of no population stratification. Under genomic control, if λ > 1, then population stratification is assumed to exist and a correction is applied by dividing the actual association test χ2 statistic values by λ. As λ scales with sample size, λ1,000, the inflation factor for an equivalent study of 1,000 cases and 1,000 controls calculated by rescaling λ, is often reported27. In a CG study, λ can only be determined if an additional set of markers specifically designed to indicate population stratification are genotyped. In a GWA study, an unbiased estimation of λ can be determined using all of the genotyped markers; the effect on the inflation factor of potential causal SNPs in such a large set of genomic control markers is assumed to be negligible.
Replication
Replication occurs when a positive association from an initial study is confirmed in a subsequent study involving an independent sample drawn from the same population as the initial study. It is the process by which genetic association results are validated. In theory, a repeated significant association between the same trait and allele in an independent sample is the benchmark for replication. However, in practice, so-called replication studies often comprise findings of association between the same trait and nearby variants in the same gene as the original SNP, or between the same SNP and different high-risk traits. A precise definition of what constitutes replication for any given study is therefore important and should be clearly stated28.
In practice, replication studies often involve different investigators with different samples and study designs aiming to independently verify reports of positive association and obtain accurate effect-size estimates, regardless of the designs used to detect effects in the primary study. Two commonly used strategies in such cases are an exact strategy, in which only marker loci indicating a positive association are subsequently genotyped in the replicate sample, and a local strategy, in which additional variants are also included, thus combining replication with fine-mapping objectives. In general, the exact strategy is more balanced in power and efficiency; however, depending on local patterns of LD and the strength of primary association signals, a local strategy can be beneficial28.
In the past, multistage designs have been proposed as cost-efficient approaches to allow the possibility of replication within a single overall study. The first stage of a standard two-stage design involves genotyping a large number of markers on a proportion of available samples to identify potential signals of association using a nominal P value threshold. In stage two, the top signals are then followed up by genotyping them on the remaining samples while a joint analysis of data from both stages is conducted29,30. Significant signals are subsequently tested for replication in a second data set. With the ever-decreasing costs of GWA genotyping, two-stage studies have become less common.
Software
Standard statistical software (such as R (ref. 31) or SPSS) can be used to conduct and visualize all the analyses outlined above. However, many researchers choose to use custom-built GWA software. In this protocol we use PLINK32, Haploview33 and the customized R package car34. PLINK is a popular and computationally efficient software program that offers a comprehensive and well-documented set of automated GWA quality control and analysis tools. It is a freely available open source software written in C++, which can be installed on Windows, Mac and Unix machines (http://pngu.mgh.harvard.edu/~purcell/plink/index.shtml). Haploview (http://www.broadinstitute.org/haploview/haploview) is a convenient tool for visualizing LD; it interfaces directly with PLINK to produce a standard visualization of PLINK association results. Haploview is most easily run through a graphical user interface, which offers many advantages in terms of display functions and ease of use. car (http://socserv.socsci.mcmaster.ca/jfox/) is an R package that contains a variety of functions for graphical diagnostic methods.
The next section describes protocols for the analysis of SNP data and is illustrated by the use of simulated data sets from CG and GWA studies (available as gzipped files from http://www.well.ox.ac.uk/ggeu/NPanalysis/ or .zip files as Supplementary Data 1 and Supplementary Data 2). We assume that SNP data for a CG study, typically comprising on the order of thousands of markers, will be available in a standard PED and MAP file format (for an explanation of these file formats, see http://pngu.mgh.harvard.edu/~purcell/plink/data.shtml#ped) and that SNP data for a GWA study, typically comprising on the order of hundreds of thousands of markers, will be available in a standard binary file format (for an explanation of the binary file format, see http://pngu.mgh.harvard.edu/~purcell/plink/data.shtml#bed). In general, SNP data for either type of study may be available in either format. The statistical analysis described here is for the analysis of one SNP at a time; therefore, apart from the requirement to take potentially differing input file formats into account, it does not differ between CG and GWA studies.
MATERIALS
EQUIPMENT
Computer workstation with Unix/Linux operating system and web browser
PLINK32 software for association analysis (http://pngu.mgh.harvard.edu/~purcell/plink/download.shtml).
Unzipping tool such as WinZip (http://www.winzip.com) or gunzip (http://www.gzip.org)
Statistical software for data analysis and graphing such as R (http://cran.r-project.org/) and Haploview33 (http://www.broadinstitute.org/haploview/haploview).
SNPSpD35 (Program to calculate the effective number of independent SNPs among a collection of SNPs in LD with each other; http://genepi.qimr.edu.au/general/daleN/SNPSpD/)
Files: genome-wide and candidate-gene SNP data (available as gzipped files from http://www.well.ox.ac.uk/ggeu/NPanalysis/ or .zip files as Supplementary Data 1 and Supplementary Data 2)
PROCEDURE
Identify file formats ● TIMING ~5 min
1| For SNP data available in standard PED and MAP file formats, as in our CG study, follow option A. For SNP data available in standard binary file format, as in our GWA study, follow option B. The instructions provided here are for unpacking the sample data provided as gzipped files at http://www.well.ox.ac.uk/ggeu/NPanalysis/. If using the .zip files provided as supplementary Data 1 or supplementary Data 2, please proceed directly to step 2.
▲ CRITICAL STEP The format in which genotype data are returned to investigators varies according to genome-wide SNP platforms and genotyping centers. We assume that genotypes have been called by the genotyping center, undergone appropriate quality control filters as described in a previous protocol3 and returned as clean data in a standard file format.
- Standard PED and MAP file format
- Download the file ‘cg-data.tgz’.
-
Type ‘tar -xvzf cg-data.tgz’ at the shell prompt to unpack the gzipped .tar file and create files ‘cg.ped’ and ‘cg.map’.▲ CRITICAL STEP The simulated data used here have passed standard quality control filters: all individuals have a missing data rate of < 20%, and SNPs with a missing rate of > 5%, a MAF < 1% or an HWE P value < 1 × 10− 4 have already been excluded. These filters were selected in accordance with procedures described elsewhere3 to minimize the influence of genotype-calling artifacts in a CG study.
- Standard binary file format
- Download the file ‘gwa-data.tgz’.
-
Type ‘tar -xvzf gwa-data.tgz’ at the shell prompt to unpack the gzipped .tar file and obtain the standard binary files ‘gwa.bed’, ‘gwa.bim’ and ‘gwa.fam’ and the covariate file ‘gwa-covar.’▲ CRITICAL STEP We assume that covariate files are available in a standard file format. For an explanation of the standard format for covariate files, see http://pngu.mgh.harvard.edu/~purcell/plink/data.shtml#covar.▲ CRITICAL STEP Optimized binary BED files contain the genotype information and the corresponding BIM/FAM files contain the map and pedigree information. The binary BED file is a compressed file that allows faster processing in PLINK and takes less storage space, thus facilitating the analysis of large-scale data sets32.▲ CRITICAL STEP The simulated data used here have passed standard quality control: all individuals have a missing data rate of < 10%. SNPs with a missing rate > 10%, a MAF < 1% or an HWE P value < 1 × 10− 5 have already been excluded. These filters were selected in accordance with procedures described elsewhere3 to minimize the influence of genotype-calling artifacts in a GWA study.? TROUBLESHOOTING
Basic descriptive summary ● TIMING ~5 min
2| To obtain a summary of MAFs in case and control populations and an estimate of the OR for association between the minor allele (based on the whole sample) and disease in the CG study, type ‘plink --file cg --assoc --out data’. In any of the PLINK commands in this protocol, replace the ‘--file cg’ option with the ‘--bfile gwa’ option to use the binary file format of the GWA data rather than the PED and MAP file format of the CG data.
▲ CRITICAL STEP PLINK always creates a log file called ‘data.log’, which includes details of the implemented commands, the number of cases and controls in the input files, any excluded data and the genotyping rate in the remaining data. This file is very useful for checking the software is successfully completing commands.
▲ CRITICAL STEP The options in a PLINK command can be specified in any order.
? TROUBLESHOOTING
3| Open the output file ‘data.assoc’. It has one row per SNP containing the chromosome [CHR], the SNP identifier [SNP], the base-pair location [BP], the minor allele [A1], the frequency of the minor allele in the cases [F_A] and controls [F_U], the major allele [A2] and statistical data for an allelic association test including the χ2-test statistic [CHISQ], the asymptotic P value [P] and the estimated OR for association between the minor allele and disease [OR].
? TROUBLESHOOTING
Single SNP tests of association ● TIMING ~5 min
4| When there are no covariates to consider, carry out simple χ2 tests of association by following option A. For inclusion of multiple covariates and covariate interactions, follow option B.
- Simple χ2 tests of association
-
Create a file containing output from single SNP χ2 tests of association in the CG data by typing ‘plink --file cg --model --out data’. The command for the GWA data is ‘plink --bfile gwa --model --out data’.▲ CRITICAL STEP Genotypic, dominant and recessive tests will not be conducted if any one of the cells in the table of case control by genotype counts contains less than five observations. This is because the χ2 approximation may not be reliable when cell counts are small. For SNPs with MAFs < 5%, a sample of more than 2,000 cases and controls would be required to meet this threshold and more than 50,000 would be required for SNPs with MAF < 1%. To change the threshold, use the ‘--cell’ option. For example, we could lower the threshold to 3 and repeat the χ2 tests of association by typing ‘plink --file cg --model --cell 3 --out data’.
- Open the output file ‘data.model’. It contains five rows per SNP, one for each of the association tests described in Table 2. Each row contains the chromosome [CHR], the SNP identifier [SNP], the minor allele [A1], the major allele [A2], the test performed [TEST: GENO (genotypic association); TREND (Cochran-Armitage trend); ALLELIC (allelic association); DOM (dominant model); and REC (recessive model)], the cell frequency counts for cases [AFF] and controls [UNAFF], the χ2 test statistic [CHISQ], the degrees of freedom for the test [DF] and the asymptotic P value [P].
-
- Test of association using logistic regression
-
Create a file containing output of association tests based on logistic regression assuming a multiplicative model and including covariates in the GWA data by typing ‘plink --bfile gwa --logistic --covar gwa.covar --out data’.▲ CRITICAL STEP To specify a genotypic, dominant or recessive model in place of a multiplicative model, include the model option --genotypic, --dominant or --recessive, respectively. To include sex as a covariate, include the option --sex. To specify interactions between covariates, and between SNPs and covariates, include the option --interaction. Open the output file ‘data.assoc.logistic’. If no model option is specified, the first row for each SNP corresponds to results for a multiplicative test of association. If the ‘--genotypic’ option has been selected, the first row will correspond to a test for additivity and the subsequent row to a separate test for deviation from additivity. If the ‘--dominant’ or ‘--recessive’ model options have been selected, then the first row will correspond to tests for a dominant or recessive model of association, respectively. If covariates have been included, each of these P values is adjusted for the effect of the covariates. The C ≥ 0 subsequent rows for each SNP correspond to separate tests of significance for each of the C covariates included in the regression model. Finally, if the ‘--genotypic’ model option has been selected, there is a final row per SNP corresponding to a 2 d.f. LR test of whether both the additive and the deviation from additivity components of the regression model are significant. Each row contains the chromosome [CHR], the SNP identifier [SNP], the base-pair location [BP], the minor allele [A1], the test performed [TEST: ADD (multiplicative model or genotypic model testing additivity), GENO_2DF (genotypic model), DOMDEV (genotypic model testing deviation from additivity), DOM (dominant model) or REC (recessive model)], the number of missing individuals included [NMISS], the OR, the coefficient z-statistic [STAT] and the asymptotic P value [P].▲ CRITICAL STEP ORs for main effects cannot be interpreted directly when interactions are included in the model; their interpretation depends on the exact combination of variables included in the model. Refer to a standard text on logistic regression for more details36.? TROUBLESHOOTING
-
Data visualization ● TIMING ~5 min
5| To create quantile-quantile plots to compare the observed association test statistics with their expected values under the null hypothesis of no association and so assess the number, magnitude and quality of true associations, follow option A. Note that quantile-quantile plots are only suitable for GWA studies comprising hundreds of thousands of markers. To create a Manhattan plot to display the association test P values as a function of chromosomal location and thus provide a visual summary of association test results that draw immediate attention to any regions of significance, follow option B. To visualize the LD between sets of markers in an LD plot, follow option C. Manhattan and LD plots are suitable for both GWA and CG studies comprising any number of markers. Otherwise, create customized graphics for the visualization of association test output using customized simple R31 commands37 (not detailed here)).
- Quantile-quantile plot
- Start R software.
- Create a quantile-quantile plot ‘chisq.qq.plot.pdf’ with a 95% confidence interval based on output from the simple χ2 tests of association described in Step 4A for trend, allelic, dominant or recessive models, wherein statistics have a χ2 distribution with 1 d.f. under the null hypothesis of no association. Create the plot by typing
data < -read.table(“[path_to]/data.model”, header = TRUE); pdf(“[path_to]/chisq.qq.plot.pdf”); library(car); obs < - data[data$TEST = = “[model]”,]$CHISQ; qqPlot(obs, distribution = ”chisq”, df = 1, xlab = ”Expected chi-squared values”, ylab = “Observed test statistic”, grid = FALSE); dev.off()’,
where [path_to] is the appropriate directory path and [model] identifies the association test output to be displayed, and where [model] can be TREND (Cochran-Armitage trend); ALLELIC (allelic association); DOM (dominant model); or REC (recessive model). For simple χ2 tests of association based on a genotypic model, in which test statistics have a χ2 distribution with 2 d.f. under the null hypothesis of no association, use the option [df] = 2 and [model] = GENO. - Create a quantile-quantile plot ‘pvalue.qq.plot.pdf’ based on – log10 P values from tests of association using logistic regression described in Step 4B by typing
‘data < - read.table(“[path_to]/data.assoc.logistic”, header = TRUE); pdf(“[path_to]/pvalue.qq.plot.pdf”); obs < - −log10(sort(data[data$TEST = = ”[model]”,]$P)); exp < - −log10( c(1:length(obs)) /(length(obs) + 1)); plot(exp, obs, ylab = “Observed (−logP)”, xlab = ”Expected(−logP) “, ylim = c(0,20), xlim = c(0,7)) lines(c(0,7), c(0,7), col = 1, lwd = 2) ; dev.off()’,
where [path_to] is the appropriate directory path and [model] identifies the association test output to be displayed and where [model] is ADD (multiplicative model); GENO_2DF (genotypic model); DOMDEV (genotypic model testing deviation from additivity); DOM (dominant model); or REC (recessive model).
- Manhattan plot
- Start Haploview. In the ‘Welcome to Haploview’ window, select the ‘PLINK Format’ tab. Click the ‘browse’ button and select the SNP association output file created in Step 4. We select our GWA study χ2 tests of association output file ‘data.model’. Select the corresponding MAP file, which will be the ‘.map’ file for the pedigree file format or the ‘.bim’ file for the binary file format. We select our GWA study file ‘gwa.bim’. Leave other options as they are (ignore pairwise comparison of markers > 500 kb apart and exclude individuals with > 50% missing genotypes). Click ‘OK’.
- Select the association results relevant to the test of interest by selecting ‘TEST’ in the dropdown tab to the right of ‘Filter:’, ‘ = ’ in the dropdown menu to the right of that and the PLINK keyword corresponding to the test of interest in the window to the right of that. We select PLINK keyword ‘ALLELIC’ to visualize results for allelic tests of association in our GWA study. Click the gray ‘Filter’ button. Click the gray ‘Plot’ button. Leave all options as they are so that ‘Chromosomes’ is selected as the ‘X-Axis’. Choose ‘P’ from the drop-down menu for the ‘Y-Axis’ and ‘−log10′ from the corresponding dropdown menu for ‘Scale:’. Click ‘OK’ to display the Manhattan plot.
- To save the plot as a scalable vector graphics file, click the button ‘Export to scalable vector graphics:’ and then click the ‘Browse’ button (immediately to the right) to select the appropriate title and directory.
- LD plot
- Start R.
- Using the standard MAP file, create the locus information file required by Haploview for the CG data by typing
‘cg.map < - read.table(“[path_to]/cg.map”); write.table(cg.map[,c(2,4)],“[path_to]/cg.hmap”, col.names = FALSE, row.names = FALSE, quote = FALSE)
where [path_to] is the appropriate directory path. - Start Haploview. In the ‘Welcome to Haploview’ window, select the ‘LINKAGE Format’ tab. Click the ‘browse’ button to enter the ‘Data File’ and select the PED file ‘cg.ped’. Click the ‘browse’ button to enter the ‘Locus Information File’ and select the file ‘cg.hmap’. Leave other options as they are (ignore pairwise comparison of markers > 500 kb apart and exclude individuals with > 50% missing genotypes). Click ‘OK’. Select the ‘LD Plot’ tab.
-
To save the plot as a portable network graphics (PNG) file, click on the ‘File’ button; from the drop-down menu, select ‘Export current tab to PNG’. The appropriate title and directory can then be selected.? TROUBLESHOOTING
Adjustment for multiple testing ● TIMING ~5 min
6| For CG studies, typically comprising hundreds of thousands of markers, control for multiple testing using Bonferroni’s adjustment (follow option A); Holm, Sidak or FDR (follow option B) methods; or permutation (follow option C). Although Bonferroni, Holm, Sidak and FDR are simple to implement, permutation testing is widely recommended for accurately correcting for multiple testing and should be used when computationally possible. For GWA studies, select an appropriate genome-wide significance threshold (follow option D).
- Bonferroni’s Adjustment
-
Consider the total number of markers tested in the CG. For a FWER α = 0.05, derive the per-test significance rate α* by dividing α by the number of markers tested. In our CG study, we have 40 markers; therefore, α* = 0.05/40 = 0.00125. Markers with P values less than α* are then declared significant.▲ CRITICAL STEP If some of the SNPs are in LD so that there are fewer than 40 independent tests, the Bonferroni correction will be too conservative. Use LD information from HapMap and SNPSpD (http://genepi.qimr.edu.au/general/daleN/SNPSpD/)35 to estimate the effective number of independent SNPs1. Derive the per-test significance rate α* by dividing α by the effective number of independent SNPs.
-
- Holm, Sidak and FDR
- To obtain significance values adjusted for multiple testing for trend, dominant and recessive tests of association, include the --adjust option along with the model specification option --model-[x] (where [x] is ‘trend’, ‘rec’ or ‘dom’ to indicate whether trend, dominant or recessive test association P values, respectively, are to be adjusted for) in any of the PLINK commands described in Step 4A. For example, adjusted significance values for a Cochran-Armitage trend test of association in the CG data are obtained by typing ‘plink --file cg --adjust --model-trend --out data’. Obtain significance values adjusted for an allelic test of association by typing ‘plink --file cg --assoc –adjust --out data’.
- Open the output file ‘data.model.[x].adjusted’ for adjusted trend, dominant or recessive test association P values or ‘data.assoc.adjusted’ for adjusted allelic test of association P values. These files have one row per SNP containing the chromosome [CHR], the SNP identifier [SNP], the unadjusted P value [UNADJ] identical to that found in the original association output file, the genomic-control–adjusted P value [GC], the Bonferroni-adjusted P value [BONF], the Holm step-down–adjusted P value [HOLM], the Sidak single-step–adjusted P value [SIDAK_SS], the Sidak step-down–adjusted P value [SIDAK_SD], the Benjamini and Hochberg FDR control [FDR_BH] and the Benjamini and Yekutieli FDR control [FDR_BY]. To maintain a FWER or FDR of α = 0.05, only SNPs with adjusted P values less than α are declared significant.
- Permutation
- To generate permuted P values, include the --mperm option along with the number of permutations to be performed and the model specification option –model-[x] (where [x] is ‘gen’, ‘trend’, ‘rec’ or ‘dom’ to indicate whether genotypic, trend, dominant or recessive test association P values are to be permuted) in any of the PLINK commands described in Step 4A. For example, permuted P values based on 1,000 replicates for a Cochran-Armitage trend test of association are obtained by typing ‘plink --file cg --model --mperm 1000 --model-trend --out data’ and permuted P values based on 1,000 replicates for an allelic test of association are obtained by typing ‘plink --file cg --assoc –mperm 1000 --out data’.
- Open the output file ‘data.model.[x].mperm’ for permuted P values for genotypic, trend, dominant or recessive association tests or ‘data.assoc.mperm’ for permuted P values for allelic tests of association. These files have one row per SNP containing the chromosome [CHR], the SNP identifier [SNP], the point-wise estimate of the SNP’s significance [EMP1] and the family-wise estimate of the SNP’s significance [EMP2]. To maintain a FWER of α = 0.05, only SNPs with family-wise estimated significance of less than α are declared significant.
- Genome-wide significance threshold ● TIMING ~5 min
-
Obtain per-SNP significance thresholds for a given FWER from Hoggart et al22. In our GWA study of 2,000 cases and 2,000 controls from a Caucasian population, the standard per-SNP significance threshold for a FWER of α = 0.05 is estimated at 12 × 10−8 using linear interpolation between the given value of 11 × 10−8 for studies with 1,000 cases and 1,000 controls and 15 × 10−8 for studies with 5,000 cases and 5,000 controls.? TROUBLESHOOTING
-
Population stratification ● TIMING ~5 min
7| For CG studies, typically comprising hundreds of thousands of markers, calculate the inflation factor λ (follow option A). For GWA studies, obtain an unbiased evaluation of the inflation factor λ by using all testing SNPs (follow option B).
- Calculate the inflation factor λ for CG studies
-
Assuming that PED and MAP files for null loci are available, obtain the inflation factor by specifying the null marker loci data files instead of the CG data files and including the --adjust option along with the model specification option --model-[x] (where [x] is ‘trend’, ‘rec’ or ‘dom’ to indicate whether an inflation factor based on a trend, dominant or recessive test of association, respectively, is to be calculated) in any of the PLINK commands described in Step 4A. For example, the inflation factor corresponding to a Cochran-Armitage trend test of association is obtained by typing ‘plink --file null --model --adjust --model-trend --out data’; the inflation factor corresponding to an allelic test of association is obtained by typing ‘plink --file null --assoc --adjust --out data’, where files ‘null.ped’ and ‘null.map’ are PED and MAP files for the case and control individuals at the null marker loci.▲ CRITICAL STEP To assess the inflation factor in CG studies, an additional set of null marker loci, which are common SNPs not associated with the disease and not in LD with CG SNPs, must be available. We do not have any null loci data files available for our CG study.Open the PLINK log file ‘data.log’ that records the inflation factor.
-
- Calculate the inflation factor λ for GWA studies
- To obtain the inflation factor, include the --adjust option in any of the PLINK commands described in Step 4B. For example, the inflation factor based on logistic regression tests of association for all SNPs and assuming multiplicative or genotypic models in the GWA study is obtained by typing ‘plink --bfile gwa --genotypic --logistic --covar gwa.covar --adjust --out data’.
-
Open the PLINK log file ‘data.log’, which records the inflation factor. The inflation factor for our GWA study is 1, indicating that no population stratification is detected in our GWA data.▲ CRITICAL STEP When the sample size is large, the inflation factor λ1000, for an equivalent study of 1,000 cases and 1,000 controls, can be calculated by rescaling λ according to the following formula
? TROUBLESHOOTING
For general help on the programs and websites used in this protocol, refer to the relevant websites:
PLINK: http://pngu.mgh.harvard.edu/~purcell/plink/download.shtml R: http://cran.r-project.org/ Haploview: http://www.broadinstitute.org/haploview/haploview
Step 1: If genotypes are not available in standard PED and MAP or binary file formats, both Goldsurfer2 (Gs2; see refs. 38,39) and PLINK have the functionality to read other file formats (e.g., HapMap, HapMart, Affymetrix, transposed file sets and long-format file sets) and convert these into PED and MAP or binary file formats.
Steps 2–6: The default missing genotype character is ‘0′. PLINK can recognize a different character as the missing genotype by using the ‘--missing-genotype’ option. For example, specify a missing genotype character of ‘N’ instead of ‘0′ in Step 2 by typing ‘plink --file cg --assoc --missing-genotype N --out data’.
● TIMING
None of the programs used take longer than a few minutes to run. Displaying and interpreting the relevant information are the rate-limiting steps.
ANTICIPATED RESULTS
CG study
Summary of results
Table 4 shows the unadjusted P value for an allelic test of association in the CG region, as well as corresponding adjusted P values for SNPs with significant P values. Here we have defined a P value to be significant if at least one of the adjusted values is smaller than the threshold required to maintain a FWER of 0.05. The top four SNPs are significant according to every method of adjustment for multiple testing. The last SNP is only significant according to the FDR method of Benjamini and Hochberg, and statements of significance should be made with some caution.
TABLE 4.
SNPs in the CG study showing the strongest association signals.
| Unadjusted |
Adjusted |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Chr | SNP | Allelic test of association |
Genomic control |
Bonferroni | Holm | Sidak single step |
Sidak step-down |
FDR BH | FDR BY | Family-wise permutation |
| 3 | rs1801282 | 3.92E – 14 | 2.22E – 05 | 1.45E – 12 | 1.61E – 12 | 1.61E – 12 | 1.61E – 12 | 1.61E – 12 | 6.92E – 12 | 9.90E – 03 |
| 3 | rs12636454 | 5.54E – 07 | 4.99E – 03 | 2.05E – 05 | 2.22E – 05 | 2.27E – 05 | 2.22E – 05 | 1.14E – 05 | 4.89E – 05 | 9.90E – 03 |
| 3 | rs4135247 | 1.27E – 05 | 1.44E – 02 | 4.71E – 04 | 4.96E – 04 | 5.21E – 04 | 4.96E – 04 | 1.64E – 04 | 7.05E – 04 | 9.90E – 03 |
| 3 | rs2120825 | 1.60E – 05 | 1.56E – 02 | 5.92E – 04 | 6.08E – 04 | 6.56E – 04 | 6.08E – 04 | 1.64E – 04 | 7.05E – 04 | 9.90E – 03 |
| 3 | rs3856806 | 3.62E – 03 | 1.03E – 01 | 1.34E – 01 | 1.34E – 01 | 1.38E – 01 | 1.26E – 01 | 2.97E – 02 | 1.28E – 01 | 9.90E – 02 |
Shown are adjusted and unadjusted P values for those SNPs with significant P values in an allelic test of association according to at least one method of adjustment for multiple testing. Chr, chromosome; FDR, false discovery rate; BH, Benjamini and Hochberg; BY, Benjamini and Yekutieli.
LD plot
Figure 1 shows an LD plot based on CG data. Numbers within diamonds indicate the r2 values. SNPs with significant P values (P value < 0.05 and listed in Table 4) in the CG study are shown in white boxes. Six haplotype blocks of LD across the region have been identified and are marked in black. The LD plot shows that the five significant SNPs belong to three different haplotype blocks with the region studied: three out of five significantly associated SNPs are located in Block 2, which is a 52-kb block of high LD (r2 > 0.34). The two remaining significant SNPs are each located in separate blocks, Block 3 and Block 5. Results indicate possible allelic heterogeneity (the presence of multiple independent risk-associated variants). Further fine mapping would be required to locate the precise causal variants.
Figure 1.

LD plot. LD plot showing LD patterns among the 37 SNPs genotyped in the CG study. The LD between the SNPs is measured as r2 and shown (× 100) in the diamond at the intersection of the diagonals from each SNP. r2 = 0 is shown as white, 0 < r2 < 1 is shown in gray and r2 = 1 is shown in black. The analysis track at the top shows the SNPs according to chromosomal location. Six haplotype blocks (outlined in bold black line) indicating markers that are in high LD are shown. At the top, the markers with the strongest evidence for association (listed in Table 4) are boxed in white.
GWA study
Quantile-quantile plot
Figure 2 shows the quantile-quantile plots for two different tests of association in the GWA data, one based on χ2 statistics from a test of allelic association and another based on − log10 P values from a logistic regression under a multiplicative model of association. These plots show only minor deviations from the null distribution, except in the upper tail of the distribution, which corresponds to the SNPs with the strongest evidence for association. By illustrating that the majority of the results follow the null distribution and that only a handful deviate from the null we suggest that we do not have population structure that is unaccounted for in the analysis. These plots thus give confidence in the quality of the data and the robustness of the analysis. Both these plots are included here for illustration purposes only; typically only one (corresponding to the particular test of association) is required.
Figure 2.
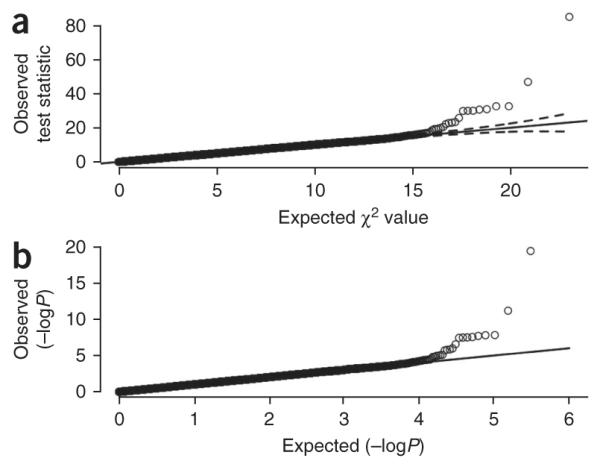
Quantile-quantile plots. Quantile-quantile plots of the results from the GWA study of (a) a simple χ2 allelic test of association and (b) a multiplicative test of association based on logistic regression for all 306,102 SNPs that have passed the standard quality control filters. The solid line indicates the middle of the first and third quartile of the expected distribution of the test statistics. The dashed lines mark the 95% confidence interval of the expected distribution of the test statistics. Both plots show deviation from the null distribution only in the upper tails, which correspond to SNPs with the strongest evidence for association.
Manhattan plot
Figure 3 shows a Manhattan plot for the allelic test of association in the GWA study. SNPs with significant P values are easy to distinguish, corresponding to those values with large log10 P values. Three black ellipses mark regions on chromosomes 3, 8 and 16 that reach genome-wide significance (P < 5 × 10−8). Markers in these regions would then require further scrutiny through replication in an independent sample for confirmation of a true association.
Figure 3.

Manhattan plot. Manhattan plot of simple χ2 allelic test of association P values from the GWA study. The plot shows –log10 P values for each SNP against chromosomal location. Values for each chromosome (Chr) are shown in different colors for visual effect. Three regions are highlighted where markers have reached genome-wide significance (P value < 5 × 10−8).
Supplementary Material
ACKNOWLEDGMENTS
G.M.C. is funded by the Wellcome Trust. F.H.P. is funded by the Welcome Trust. C.A.A. is funded by the Wellcome Trust (WT91745/Z/10/Z). A.P.M. is supported by a Wellcome Trust Senior Research Fellowship. K.T.Z. is supported by a Wellcome Trust Research Career Development Fellowship.
Footnotes
Note: Supplementary information is available in the HTML version of this article.
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/.
References
- 1.Zondervan KT, Cardon LR. Designing candidate gene and genome-wide case-control association studies. Nat. Protoc. 2007;2:2492–2501. doi: 10.1038/nprot.2007.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pettersson FH, et al. Marker selection for genetic case-control association studies. Nat. Protoc. 2009;4:743–752. doi: 10.1038/nprot.2009.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson CA, et al. Data quality control in genetic-case control association studies. Nat. Protoc. 2010;5:1564–1573. doi: 10.1038/nprot.2010.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morris AP, Zeggini E. An evaluation of statistical approaches to rare variant analysis in genetic association studies. Genet. Epidemiol. 2010;34:188–193. doi: 10.1002/gepi.20450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cho EY, et al. Genome-wide association analysis and replication of coronary artery disease in South Korea suggests a causal variant common to diverse populations. Heart Asia. 2010;2:104–108. doi: 10.1136/ha.2009.001370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 8.Anderson CA, et al. Evaluating the effects of imputation on the power, coverage, and cost efficiency of genome-wide SNP platforms. Am. J. Hum. Genet. 2008;83:112–119. doi: 10.1016/j.ajhg.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Camp NJ. Genomewide transmission/disequilibrium testing—consideration of the genotypic relative risks at disease loci. Am. J. Hum. Genet. 1997;61:1424–1430. doi: 10.1086/301648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Balding DJ, Bishop M, Cannings C. Handbook of Statistical Genetics. John Wiley & Sons Ltd.; 2003. [Google Scholar]
- 11.Bishop YMM, Fienberg SE, Holland PW. Discrete Multivariate Analysis: Theory and Practice. MIT Press; 1975. p. 557. [Google Scholar]
- 12.Cochran WG. Some methods for strengthening the common chi-squared test. Biometrics. 1954;10 [Google Scholar]
- 13.Armitage P. Tests for linear trends in proportions and frequencies. Biometrics. 1955;11:375–386. [Google Scholar]
- 14.Rice JA. Mathematical Statistics and Data Analysis. Duxbury Press; 1995. [Google Scholar]
- 15.Sidak Z. On multivariate normal probabilities of rectangles: their dependence on correlations. Ann. Math. Statist. 1968;39:1425–1434. [Google Scholar]
- 16.Sidak Z. On probabilities of rectangles in multivariate Student distributions: their dependence on correlations. Ann. Math. Statist. 1971;42:169–175. [Google Scholar]
- 17.Holm S. A simple sequentially rejective multiple test procedure. Scand. J. Stat. 1979;6:65–70. [Google Scholar]
- 18.Benjamini Y, Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. J. Royal Statist. Soc. Series B-Methodological. 1995;57:289–300. [Google Scholar]
- 19.Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 2001;29:1165–1188. [Google Scholar]
- 20.Westfall PH, Young SS. Resampling-Based Multiple Testing: Examples and Methods for P-value Adjustment. xvii. John Wiley & Sons; 1993. p. 340. [Google Scholar]
- 21.Dudbridge F, Gusnanto A. Estimation of significance thresholds for genomewide association scans. Genet. Epidemiol. 2008;32:227–234. doi: 10.1002/gepi.20297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoggart CJ, Clark TG, De Iorio M, Whittaker JC, Balding DJ. Genome-wide significance for dense SNP and resequencing data. Genet. Epidemiol. 2008;32:179–185. doi: 10.1002/gepi.20292. [DOI] [PubMed] [Google Scholar]
- 23.Pe’er I, Yelensky R, Altshuler D, Daly MJ. Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet. Epidemiol. 2008;32:381–385. doi: 10.1002/gepi.20303. [DOI] [PubMed] [Google Scholar]
- 24.Weir BS, Hill WG, Cardon LR. Allelic association patterns for a dense SNP map. Genet. Epidemiol. 2004;27:442–450. doi: 10.1002/gepi.20038. [DOI] [PubMed] [Google Scholar]
- 25.Knowler WC, Williams RC, Pettitt DJ, Steinberg AG. Gm3;5,13,14 and type 2 diabetes mellitus: an association in American Indians with genetic admixture. Am. J. Hum. Genet. 1988;43:520–526. [PMC free article] [PubMed] [Google Scholar]
- 26.Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 27.de Bakker PI, et al. Practical aspects of imputation-driven meta-analysis of genome-wide association studies. Hum. Mol. Genet. 2008;17:R122–R128. doi: 10.1093/hmg/ddn288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clarke GM, Carter KW, Palmer LJ, Morris AP, Cardon LR. Fine mapping versus replication in whole-genome association studies. Am. J. Hum. Genet. 2007;81:995–1005. doi: 10.1086/521952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Skol AD, Scott LJ, Abecasis GR, Boehnke M. Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat. Genet. 2006;38:209–213. doi: 10.1038/ng1706. [DOI] [PubMed] [Google Scholar]
- 30.Skol AD, Scott LJ, Abecasis GR, Boehnke M. Optimal designs for two-stage genome-wide association studies. Genet. Epidemiol. 2007;31:776–788. doi: 10.1002/gepi.20240. [DOI] [PubMed] [Google Scholar]
- 31.R Development Core Team . A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; 2009. [Google Scholar]
- 32.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 34.Fox J. An R and S-Plus Companion to Applied Regression. xvi. Sage Publications; 2002. p. 312. [Google Scholar]
- 35.Nyholt DR. A simple correction for multiple testing for single-nucleotide polymorphisms in linkage disequilibrium with each other. Am. J. Hum. Genet. 2004;74:765–769. doi: 10.1086/383251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hosmer DW, Lemeshow S. Applied Logistic Regression. xii. Wiley; 2000. p. 373. [Google Scholar]
- 37.Dalgaard P. Introductory Statistics with R. xvi. Springer; 2008. p. 363. [Google Scholar]
- 38.Pettersson F, Jonsson O, Cardon LR. GOLDsurfer: three dimensional display of linkage disequilibrium. Bioinformatics. 2004;20:3241–3243. doi: 10.1093/bioinformatics/bth341. [DOI] [PubMed] [Google Scholar]
- 39.Pettersson F, Morris AP, Barnes MR, Cardon LR. Goldsurfer2 (Gs2): a comprehensive tool for the analysis and visualization of genome wide association studies. BMC Bioinformatics. 2008;9:138. doi: 10.1186/1471-2105-9-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.