

Abstract
The CRISPRs (Clustered Regularly Interspaced Short Palindromic Repeats) found in prokaryotic genomes confer small RNA-mediated protection against viruses and other invaders. CRISPR loci contain iterations of a short repeat sequence alternating with small segments of varying invader-derived sequences. Distinct families of CRISPR-associated Cas proteins function to cleave within the repeat sequence of CRISPR transcripts and produce the individual invader-targeting crRNAs. Here we report the crystal structure of Pyrococcus furiosus Cas6 bound with a repeat RNA at 3.2 Å resolution. In contrast to other Cas families of endonucleases, Cas6 clasps nucleotides 2–9 of the repeat RNA using its two ferredoxin-like domains, and the enzyme-anchored 5’ end tethers the distal cleavage site of the RNA between nucleotides 22 and 23 to the predicted enzyme active site on the opposite side of the ferrodoxin-like domains. Our findings suggest a wrap-around mechanism for CRISPR RNA recognition and cleavage by Cas6 and related processing endonucleases.
Introduction
The CRISPR-Cas system confers adaptive and heritable immunity against invading genetic elements in bacteria and archaea (Bolotin et al., 2005; Horvath and Barrangou, 2010; Karginov and Hannon, 2010; Marraffini and Sontheimer, 2010a; Mojica et al., 2005; Sorek et al., 2008; van der Oost et al., 2009; Waters and Storz, 2009). This recently identified microbial defense system has been exploited in pathogen detection (Driscoll, 2009; Mokrousov et al., 2007), microbial ecology and evolution studies (Andersson and Banfield, 2008; Heidelberg et al., 2009; Snyder et al., ; Tyson and Banfield, 2008) and maintenance of domesticated bacteria (Barrangou et al., 2007; Mills et al., 2009), and may have the potential to aid in combating the spread of bacterial pathogens and antimicrobial resistance. CRISPR loci are found in many archaeal and bacterial genomes, including clinically important pathogens (Haft et al., 2005; Jansen et al., 2002; Makarova et al., 2006). A CRISPR locus typically consists of short (~30–40-nucleotide) variable sequences interspersed between short (~30–40-nucleotide) conserved repeat sequences, and is accompanied by one or more of the evolutionarily-linked sets of CRISPR-associated (cas) genes that encode Cas proteins. The variable (alternatively “spacer”) sequences are derived from past invaders and provide the specificity for recognition of and resistance against future infection (Barrangou et al., 2007; Bolotin et al., 2005; Brouns et al., 2008; Marraffini and Sontheimer, 2008; Mojica et al., 2005; Pourcel et al., 2005). CRISPR locus transcripts are processed to small CRISPR (cr)RNAs (also called prokaryotic silencing (psi)RNAs) that contain individual invader-targeting sequences, and incorporated into effector complexes that silence invaders presumably via interaction with DNA or RNA (Brouns et al., 2008; Carte et al., 2008; Hale et al., 2009; Marraffini and Sontheimer, 2008). Each of the sets or modules of Cas proteins is thought to function with the crRNAs and the core Cas proteins to affect acquisition of invader-derived sequences and invader silencing. Indeed, an effector complex comprised of the Cmr (Cas RAMP module) proteins was recently shown to cleave RNA targets recognized by the crRNAs (Hale et al., 2009). In some organisms it has been found that silencing occurs at the DNA level, and though the mechanism of silencing is not known in these cases, evidence indicates that the CRISPR-Cas pathway discriminates between self and non-self DNA based on the absence or presence of extended complementarity outside of the invader-targeting sequence of the crRNAs (Marraffini and Sontheimer, 2010b).
CRISPR RNA processing is essential to the function of the defense pathway (Brouns et al., 2008). This essential function is executed by distinct Cas proteins in organisms with different modules of cas genes and CRISPR repeat sequences. In Pyrococcus furiosus, CRISPR RNA is processed by the Cas6 protein, while in Escherichia coli and Pseudomonas aeruginosa, processing is carried out by Cse3 (also called CasE) and Csy4, respectively (Brouns et al., 2008; Carte et al., 2008; Haurwitz et al., 2010). Cas6 is a “core” Cas protein that is commonly found in organisms with four of the eight subtypes of cas genes defined by Haft et al. (Tneap, Hmari, Apern and Mtube), including diverse archaea and bacteria (Haft et al., 2005). Cse3 and Csy4 are found in bacteria with the Ecoli and Ypest subtype CRISPR systems, respectively (Haft et al., 2005). The three known processing endonucleases share little sequence homology. However, their three dimensional structures all exhibit the ferredoxin fold that is also known to be RNA Recognition Motif (RRM) (Maris et al., 2005), suggesting a related mechanism of RNA processing. The RNA-boundCsy4 contains a single while both Cas6 and Cse3 contain a tandem ferredoxin fold (Carte et al., 2008; Ebihara et al., 2006; Haurwitz et al., 2010).
In the organisms where it has been investigated, mature crRNAs have been found to retain 8 nucleotides of the repeat sequence upstream of the invader targeting sequence (Brouns et al., 2008; Hale et al., 2009; Haurwitz et al., 2010; Marraffini and Sontheimer, 2008). Accordingly, the identified CRISPR RNA endonucleases, Cas6, Cse3 and Csy4, each cleave 8 nucleotides upstream of the repeat/invader-targeting sequence junction (Brouns et al., 2008; Carte et al., 2008; Haurwitz et al., 2010). There is no significant similarity between the primary sequences of the CRISPR repeat RNAs recognized and cleaved by the three enzymes. The CRISPR repeat RNA that is cleaved by P. aeruginosa Csy4 contains palindromic sequences that are predicted to form a stable hairpin structure immediately upstream of the cleavage site (Kunin et al., 2007), and the recent co-crystal revealed that Csy4 interacts specifically with the P. aeruginosa repeat RNA hairpin to place the cleavage site at the base of the hairpin within the enzyme active site (Haurwitz et al., 2010). The E. coli Cse3 substrate RNA is also predicted to form an internal hairpin that terminates at the cleavage site (Brouns et al., 2008). However, the P. furiosus repeat RNA recognized and cleaved by Cas6 belongs to a different CRISPR repeat class that is predicted to be non-structured (Kunin et al., 2007). Moreover, biochemical studies indicate that while sequences near the cleavage site (e.g. A22 and A23, see Figure 1A) are important for Cas6 cleavage, distal sequences near the 5’ terminus of the CRISPR repeat (G1-A12, see Figure 1A) are necessary and sufficient for Cas6 binding (Carte et al., 2008), suggesting the necessity for a distinct RNA recognition and cleavage site positioning mechanism.
Fig. 1. Overview of CRISPR RNA processing steps and structural properties of Cas6.

A, crRNA biogenesis. The primary CRISPR transcript contains repeats (R, red blocks) interspaced by guide sequences (G, grey blocks). The sequence of the repeat (RNA used in this study) is shown. Cas6 cleaves within each repeat (white dotted lines), releasing individual crRNA units (1X intermediates) that are further trimmed at the 3’ end by an unknown exonuclease(s). B, Overview of the Cas6-RNA complex structure shown in two orientations. The protein is shown in teal and RNA is in red. The α-helix α2 that contains the putative catalytic residue, His46, is indicated. Nucleotides of the repeat RNA are numbered from the 5’ end.
See also Figures S1 and S2.
In order to better understand the mechanisms of CRISPR RNA recognition and cleavage, we undertook further structural and biochemical studies of Cas6. We determined the crystal structures of P. furiosus (Pf) Cas6 bound to a Pf CRISPR repeat RNA and two of its 5’ fragments. In addition, we performed extensive mutagenesis studies that support structural observations. Both structural and biochemical data suggest that Cas6, unlike previously described other CRISPR processing endonucleases, binds unstructured precursor crRNA in a wrap around model. Specific recognition of the 5’ end of the RNA by Cas6 tethers the cleavage end of the RNA to the active site of Cas6.
Results and Discussion
Overview
We obtained crystals of the previously characterized Pf Cas6 protein (Carte et al., 2008) bound with the full-length Pf CRISPR repeat RNA (30mer), the first 26 nucleotides of the repeat RNA (26mer), and the first 15 nucleotides of the repeat RNA (15mer) (Figure 1A), respectively. The 30mer and 26mer RNAs contained 2’-deoxy modifications at positions 22 and 23 designed to prevent their cleavage by Cas6. Even though the three complexes are crystallized in two different space groups, their structures are indistinguishable within experimental error and the structure of the highest resolution (3.2 Å) (the 30mer-bound complex) is described here. We determined the structures by a seleno-methionine single wavelength anomalous diffraction method. Structural determination procedures are described in detail in the Methods and Materials section. The Cas6-RNA complex structure contains Pf Cas6 residues 1–8, 15–141, and 147–240 (wild-type length 264 amino acids) and nucleotides 2–10 of the 30-nucleotide Pf CRISPR repeat RNA. The rest of the residues/nucleotides could not be modeled owing to structural disorder.
The bound RNA molecule was first located by electron densities computed using the experimental amplitudes and phases of the protein coordinates prior to introduction of the RNA (Figure S1). In addition, the positions of two nucleotides, U2 and U8, were unambiguously determined by anomalous difference Fourier methods using data collected from Cas6-RNA complex crystals containing 5-bromouridine substitutions at these two positions (Figure S2). Although two Cas6-RNA complexes are in the asymmetric unit, the two protein-RNA complexes form a small interaction interface (595 Å2). Based on statistical observations on crystal contacts, the small interface is suggestive of a monomeric or a weak dimeric Cas6-RNA complex in solution (Janin et al., 1988).
The overall structure of the Cas6-RNA complex reveals that Cas6 interacts with single-stranded RNA. This structural discovery is consistent with the lack of a stable structure of the RNA both in the present and absence of Cas6 as observed by melting curve analysis (Figure S3). Unlike the well described interactions between RRM and single stranded RNA, the RNA does not interact with the characteristic β-sheet of the RRM domains of Cas6. Rather, the single-stranded RNA is bound at the groove formed between the two RRM domains. Detailed interactions between nucleotides of the RNA and Cas6 are described below.
Molecular basis of interaction of Cas6 with CRISPR RNA
In previous work we determined that block substitution of the first 8 nucleotides of the 30-nucleotide Pf CRISPR repeat sequence disrupted binding of Cas6 and that an RNA comprised of the first 12 nucleotides of the repeat was sufficient for Cas6 binding (Carte et al., 2008). In addition, we determined the structure of the Cas6 protein (Carte et al., 2008), however the molecular basis for the interaction of Cas6 with CRISPR RNA was not known.
In the co-crystal structure determined here, we find that nucleotides 2–10 of the single-stranded Pf CRISPR repeat RNA lie along the positively charged central cleft formed by the two ferredoxin folds of Cas6 (Figure 2B). The overall binding surface is wider at the two ends and narrows through the middle of the cleft (Figure 2B), suggesting a more important role played by nucleotides bound in the middle than at the two ends of the surface in RNA recognition. Pf CRISPR repeat nucleotides C5 and A6 are bound in the narrowest part of the Cas6 cleft and are held tightly in place by surrounding residues. These two nucleotides form an impressive network of interactions with the protein via both base and backbone functional groups (Figure 3A). The interactions involve several well-conserved, positively charged amino acids (Figure 3A and Figure 4). Arg64, Lys188, and Lys190 interact with the sugar phosphate backbone groups of C5 and A6. Arg64 also stacks on the nucleobase of C5 (Figure 3A and Figure 4). Strikingly, the nucleobases of C5 and A6 contact only protein backbone atoms (Figure 3A and Figure 4), suggesting the importance of shape recognition in this region.
Fig. 2. Structural comparison of free and RNA-bound Cas6, the surface properties of the complex, and the effect of G-rich loop mutations.
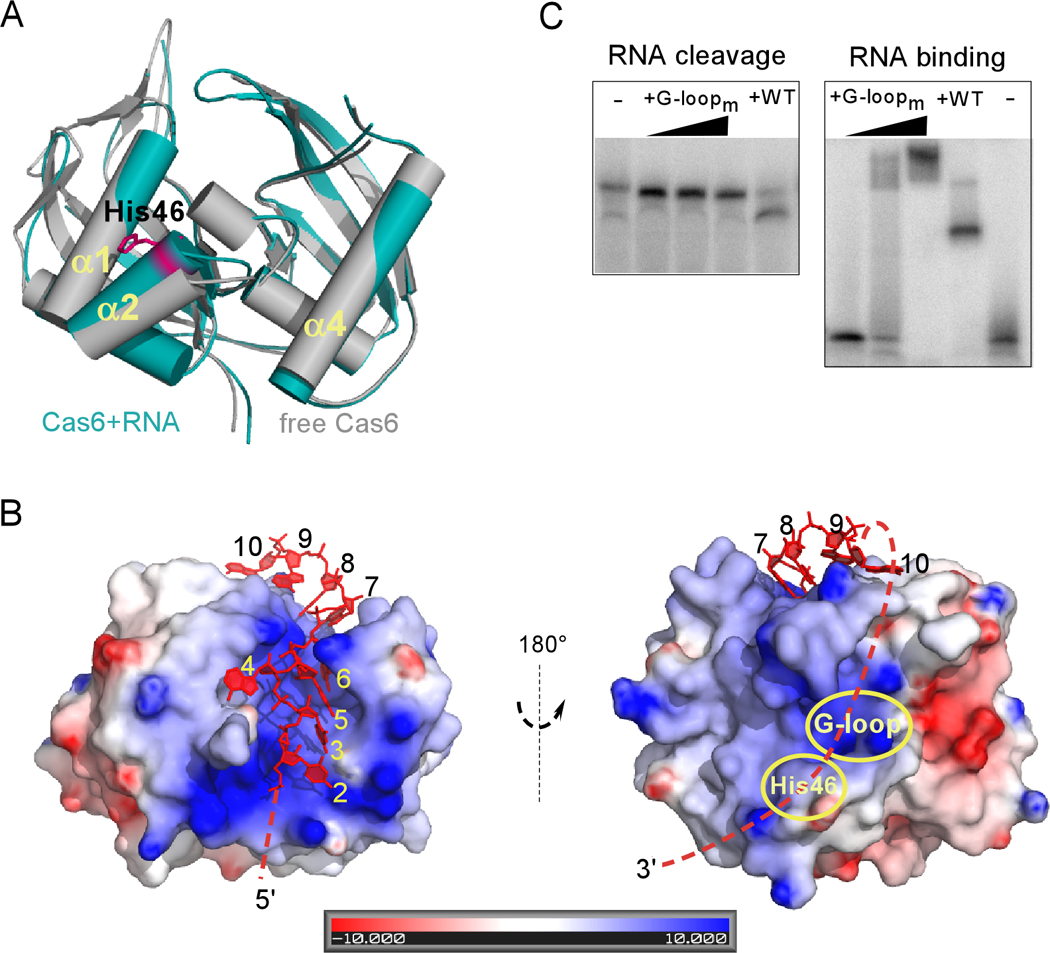
A, Superimposed structures of Cas6 bound to RNA (teal) and free Cas6 (grey). The putative catalytic residue (His46) is indicated in magenta. B, Electrostatic potentials of Cas6 in the RNA-bound complex are shown. The left view is the same as that in A. Cas6 binds nucleotides 2–10 of the repeat RNA (red) and is predicted to tether the 3’ end of the repeat RNA (dashed line) to its cleavage site (indicated by His46). Note that the potential path overlaps with the conserved Cas6-specific G-rich loop (G-loop). C, Impact on RNA cleavage (left) and binding (right) of Cas6 by alanine substation of Gly223, Gly225, Gly231, and Gly233 (G-loopm). The three concentrations of the G-loopm protein used in the cleavage and binding experiments were 0.5 µM, 1 µM and 10 µM, respectively.
Fig. 3. Key interaction features of the Cas6-RNA complex.

The protein is represented by surface (upper panels) or stick models (lower panels) and the RNA is represented by red stick models. Contacts shorter than 3.4 Å between RNA and protein atoms are indicated by dashed lines. A, Interactions between Cas6 residues and nucleotides 2–6. B. Interactions between Cas6 residues and nucleotides 7–10.
Fig. 4. Detailed interactions between repeat RNA nucleotides and Cas6 residues.

RNA nucleotides are represented by red sticks and protein residues are by grey-blue-red sticks. Nucleotides 4 and 9 involve simple interactions and are thus not included.
The two ends of the RNA binding cleft make more moderate contacts with the bound RNA, but nonetheless enhance RNA recognition and interaction (Figure 2B). Nucleotides U2 and U3 each make direct contacts with the protein (Figure 3A). The O2 atom of U3 is within 3.5 Å of Phe80 and Glu62. A4 is flipped out and makes a stacking interaction with the backbone of resides 174–175 of a symmetry-related Cas6 and Glu238 (Figure 4). Nucleotides A7, A9 and A10 also make several direct contacts with the protein (Figure 4). Cas6 specifically recognizes U8 within a deep protein pocket (Figure 3B). The Watson-Crick edge of the nucleobase of U8 lies against the backbone of residues Lys190 and Arg191. In addition, Tyr20, A7, U8 and to some extent, A9 form a continuous base stacking interaction that stabilizes the bound RNA in this region (Figure 3B).
The contacts observed in the CRISPR RNA-Cas6 protein complex indicate that Cas6 specifically interacts with nucleotides U3, C5, A6, and U8 of the Pf CRISPR repeat. These four nucleotides are conserved in the seven CRISPR repeats found in P. furiosus (Grissa et al., 2007). Consistent with key roles in recognition and binding CRISPR RNAs deduced from interactions observed in the co-structure, we find that substitution of C5, A6, and A7 (by G5, U6 and U7) abolishes RNA binding by Cas6 in gel shift assays (Figure 5A and 5B, mutant H). Deletion of G1-U3 or substitution of U2 and U3 (by A2 and A3) also abolishes CRISPR RNA binding by Cas6 (Figure 5A and 5B, mutants K and G). Disruption of recognition and binding by each of these mutations also prevents Cas6 cleavage (Figure 5B). At the same time, deletion of G1 or G1-U2 does not significantly impact binding or cleavage activity (Figure 5A and 5B, mutants I and J). The findings presented here delineate the mechanism of CRISPR RNA recognition and binding by Cas6 and underscore the importance of the interaction of Cas6 with the primary binding site at the 5’-end of the CRISPR repeat sequence.
Fig. 5. Analysis of Cas6 binding and cleavage of a series of CRISPR repeat RNA mutants and a proposed model of Cas6 function.

A, Sequences of the wild-type (A) and mutant repeat RNAs (B–R) used in Cas6 cleavage and binding studies. Mutant B (2’-deoxy modification on positions 22 and 23) is used as a non-cleavable substrate control as previously established by Carte et al. (Carte et al., 2008). Substituted, inserted, or modified nucleotides are shown in red. Deletions are indicated by spaces. “d” denotes 2’-deoxy modification. Results are summarized on the right. For mutations near the cleavage site, the experimentally observed cleavage position is denoted. B, Cleavage (upper panels) and binding (lower panels) of wild-type (A) and mutant RNAs (B–K) by Cas6. C, Cleavage and mass spectrometry analysis of the wild-type and mutant RNAs (L–O). D, Cleavage of repeat RNAs containing mutation A23U. Sizes of cleavage products are indicated. Analysis of the 21 and 22 nt cleavage products by MALDI-TOF mass spectrometric methods (Bruker, autoflex III) is shown in the right panel. The red trace is the product of cleavage of the wild-type RNA and the black trace is that from cleavage of the A23U mutant RNA. The predicted molecular weights of the 21 and 22 nt products, both with 2’,3’ cyclic phosphate termini, are indicated. E, Depiction of a beads-on-a-string model of CRISPR RNA processing by Cas6. CRISPR repeats are shown in red and invader-derived guide sequences are shown in grey. Individual Cas6 proteins bind the 5’ regions of the CRISPR repeats and the 3’ regions of the repeats wrap around Cas6 to the cleavage site (scissors).
See also Figure S3.
CRISPR RNA binding tethers the RNA cleavage site to the putative active site of Cas6
Our biochemical and structural data indicate that recognition and cleavage of CRISPR RNAs by Cas6 occur at physically distant sites. The co-crystal structure described here reveals that Cas6 specifically binds CRISPR RNA repeat nucleotides 2–10, twelve nucleotides upstream of the cleavage site. Likewise, the CRISPR RNA binding site of Cas6 identified here is on the opposite face of the protein from the putative active site (Figure 1B, bottom panel and Figure 2B). Sequence analysis and comparison with other riboendonucleases suggests that Tyr31, His46, and Lys52 comprise the catalytic triad of Pf Cas6 (Carte et al., 2008). Although their direct involvement in catalysis has not been established, mutation of His46 and Tyr31 abolish detectable RNA cleavage activity without affecting RNA binding, and mutation of Lys52 also significantly reduces RNA cleavage activity but not binding (Carte et al., in press). Likewise, in Cse3 and Csy4, mutation of a conserved histidine in a similar location was deleterious to catalysis (Brouns et al., 2008; Haurwitz et al., 2010). Furthermore, in Csy4, the relevant histidine forms close contacts with the scissile phosphate of the RNA substrate, supporting its role in catalysis (Haurwitz et al., 2010). Interestingly, superimposition of the structures of the RNA-bound and free Cas6 proteins shows that while the arrangement of most regions of the protein is similar (0.94 Å RMSD for 221 Cαatoms), α-helix α2, which holds the putative catalytic residue His46, is rotated ~6° towards α-helix α1 in the RNA-bound structure (Figure 2A) (Carte et al., 2008) (PDBid: 3I4H). The observed shift indicates that RNA binding induces a change in the predicted Cas6 catalytic region located on the opposite face of the protein.
The insights provided by the Cas6-CRISPR RNA structure suggest a path for the unobserved region of the repeat RNA, connecting nucleotide 10, bound on the CRISPR RNA recognition face of Cas6, with nucleotides 22 and 23 in the putative active site containing His46 (Figure 2B). The hypothetical path is lined with positive electrostatic potential and passes over the signature Cas6-specific G-rich loop (Figure 2B). Consistently, mutation of Gly223, Gly225, Gly231, and Gly233 to alanine completely abolished the RNA cleavage activity while maintaining a weak RNA binding activity of Cas6 (Figure 2C). The length of the intervening RNA is theoretically more than sufficient to bridge the distance between the Cas6 RNA binding and active sites. The fact that this region of the RNA is not observed in the Cas6-RNA structures suggests that interactions of these nucleotides with the protein are weak or transient.
In order to further investigate the role of the unobserved nucleotides that link the regions of the CRISPR repeat that interact with the Cas6 binding and putative cleavage sites, we examined the consequences of mutations in this region on binding and cleavage by Cas6. In previous work we had concluded that substitutions, insertions and deletions of 4 nucleotides in the region between the binding and cleavage sites disrupted cleavage (Carte et al., 2008), however the relevant mutations analyzed in that work each disrupted nucleotides A9 and A10, which we now know are constituents of the Cas6 binding site (Figure 2B). The previous results confirm the importance of the Cas6-A9 and A10 interactions identified here, but do not clearly address the role of the intervening sequence. Therefore we generated RNAs with mutations in the region between nucleotides 14–19 of the repeat. Deletion of 2–4 nucleotides from the linker region did not prevent cleavage or binding (Figure 5A, 5B and 5C, mutants E, L, and O, and data not shown). However, deletion of either 5 or 6 nucleotides of the CRISPR repeat between the Cas6 binding and cleavage sites prevented cleavage (Figure 5A and 5C, mutants M and N) but not binding (data not shown). At the same time, insertion of 2 nucleotides in the linker region did not significantly affect Cas6 binding or cleavage (Figure 5A and 5B, mutant F). These results are consistent with the two site model for Cas6 recognition and cleavage of CRISPR RNAs that has arisen from this work and suggest that a linker of at least 8 nucleotides is required to bridge the Cas6 binding and cleavage sites.
We further probed the regions of the CRISPR repeat RNA at and downstream of the cleavage site, which are not observed in the co-structure. Consistent with previous results (Carte et al., 2008), we found that removal of nucleotides 27–30 or 25–30, downstream of the cleavage site (between nucleotides A22 and A23), did not disrupt binding by Cas6 (Figure 5A & 5B, mutant C and D). However, analysis of cleavage revealed that the repeat RNA lacking nucleotides 25–30 was not cleaved by Cas6 (Figure 5A & 5B, mutant D), indicating that nucleotides U25 and/or G26 are important for cleavage. At the cleavage site, mutation of A23 to uridine shifted Cas6 cleavage upstream one position to after nucleotide 21 as demonstrated by gel analysis and mass spectrometry (Figure 5A & 5D, mutant Q), and further 2’-deoxy modification of A22 did not alter the shifted cleavage site (Figure 5A & 5D, mutant P). Interestingly however, 2’-deoxy modification of G21 in the context of the A23U mutation shifted the cleavage site back to after position 22 (Figure 5A & 5D, mutant R), suggesting a plasticity of the enzyme active site.
The recently characterized Csy4 CRISPR RNA endonuclease is found in a set of bacteria with similar CRISPR repeat sequences that are predicted to form an internal hairpin (Kunin et al., 2007). The co-crystal structure of Csy4 and an RNA substrate analog showed that a region of Csy4 adjacent to the active site specifically interacts with the RNA hairpin, positioning the RNA cleavage site near the catalytic His (Haurwitz et al., 2010). The results presented here support a very different model for CRISPR RNA processing by Cas6 (Figure 5E). Cas6 is tightly linked with 6 of the 12 CRISPR repeat sequence clusters identified by Kunin et al. (Kunin et al., 2007). The repeat sequences found in bacteria and archaea with Cas6 are predicted to be non-structured or to have intermediate folding potential (Kunin et al., 2007). Our findings suggest that this type of CRISPR repeat RNA wraps around the surface of the processing endonuclease for recognition and cleavage. Cas6 binds eight nucleotides near the 5’ end of the repeat RNA tightly and specifically on the “front” surface, which tethers the downstream cleavage site to the Cas6 active site on the “back” surface. Weak interactions may occur along the surface of Cas6 between the RNA binding and cleavage sites. RNA binding induces a change in the predicted Cas6 active site that could play a role in promoting cleavage. Processing of CRISPR transcripts by Cas6 could occur simultaneously at multiple repeats as depicted, stochastically, or sequentially. The distinct tethered, wrap-around mechanism of coupling CRISPR RNA recognition and cleavage at distant sites in Cas6 highlights the remarkable diversity in CRISPR machineries.
Materials and methods
Protein preparation
Pf Cas6 was cloned and expressed as previously described (Carte et al., 2008). Selenomethionine (Se-Met) labeled PfCas6 was produced according to a procedure by Ramakrishnan and Biou (Ramakrishnan and Biou, 1997) and purified as described (Carte et al., 2008). The G-loop mutant contains alanine substitutions at positions Gly223, Gly225, Gly231, and Gly233 and was purified by Ni-affinity chromatography and dialyzed to the same storage buffer as Cas6.
Crystallization
Pf repeat RNAs consisting of nucleotides 1–15, 1–26, and 1–30 (see Figure 1A) were ordered from Integrated DNA Technology (Coralville, IA). The 26mer and 30mer RNAs contained 2’-deoxy modification at positions 22 and 23 to prevent cleavage during crystallization. Crystals of Pf Cas6-RNA complexes were obtained by the vapor diffusion method in hanging drops. Pf Cas6 or the selenomethionine-labeled protein was combined with the repeat RNA or a fragment in 1:1.2 molar ratio at a protein concentration of 30 mg/ml and the protein-RNA complex was mixed in a 1:1 volume ratio with a reservoir solution containing 20 mM MgCl2, 50 mM NaHepes (pH7.0), 1.4 M ammonium sulfate, 4% polypropylene glycol 400 (PEG400), 20 mM NiCl2, and 100 mM NaCl. Crystals usually appeared in two days and grew to full size in a week, regardless of the length of the RNA used. For diffraction studies, crystals were cryo-protected with the same crystallization well solution supplemented with 10% ethylene glycol prior to being plunged into liquid N2 and mounted to the goniometer head. X-ray diffraction data were collected at the Southeast Regional Collaborative Access Team (SER-CAT) beamline 22ID or 22BM via remote access. Despite crystal sizes of typically 400µm × 400µm × 300µm, all Pf Cas6-RNA crystals diffracted anisotropically with the worst direction to ~4 Å and the best to 3.2 Å. The data was processed with HKL2000 (Otwinowski and Minor, 1997). The crystals containing Pf Cas6 and any one of the three RNAs appear sporadically in space group of either P3221 with unit cell dimensions a = b = 96.6 Å, c = 165.5 Å or P3121 with unit cell dimensions a = b = 96.0 Å, c = 82.8 Å. The unit cell of the P3221 space group crystals is twice as large as that of the P3121 crystals and thus contains two protein-RNA complexes in each asymmetric unit. The two crystals containing 15mer to 30mer RNAs are otherwise identical and have a Matthews coefficient of 2.75–3.13 A3/Dalton that indicates a solvent content of 55.9%–60.6%. The crystal structure was determined from a crystal in P3221 space group containing the full-length (30mer) RNA.
Structure determination
The structure of the 30mer-bound Pf Cas6 complex in P3221 space group was determined by a SAD (single wavelength anomalous diffraction) method. The Se-SAD data produced sufficient quality for the placement and modeling the Cas6 protein. However, anisotropy of the data introduced noise in density that prevented unambiguous identification of RNA nucleotides and ultimately high B-factors of the refined structure (Table S1). To ensure the placement of the nucleotides, we obtained crystals and diffraction data for the endonuclease bound with the repeat RNA with site-specific substitutions of 5-bromouridine at positions 2 and 8. The two bromide positions were unambiguously identified using difference anomalous peaks computed from the diffraction data. Iterative model building and structure refinement were done with the PHENIX program suite (Adams et al., 2002), COOT (Emsley and Cowtan, 2004), and O (Jones et al., 1991) before RNA nucleotides were introduced. To correctly place RNA nucleotides, anomalous diffraction data were collected from crystals containing 5-bromouridine substituted-RNA (purchased from Thermo Scientific Dharmacon RNAi Technology) at positions 2 and 8 respectively, and anomalous difference map peaks were obtained (Figure S1). The final model was validated by PROCHECK (Laskowski et al., 1993). The refinement statistics are shown in Table 1. The structures in the P3121 space group could be readily obtained by molecular replacement using a Cas6 structure as the search probe and no differences from that determined from the P3221 were found other than the switch of non-crystallographic to crystallographic symmetry.
Table 1.
Data collection and refinement statistics
| Data collection statistics | |
| Space group | P3221 |
| a | 96.6 |
| b | 96.6 |
| c | 165.5 |
| Resolution range (Å) | 100.0–3.2 (3.3–3.2) |
| No. of observed unique reflections | 14419 (557) |
| Redundancy | 25.68 (12.3) |
| Completeness (%) | 92.1 (68.8) |
| I/σ(I) | 37 (2.1) |
| Rsym(%) | 9.9 (40.1) |
| Refinement statistics | |
| Resolution range (Å) | 100.0–3.2 (3.3–3.2) |
| Rwork (%) | 26.6 |
| Rfree (%) | 31.7 |
| Model information | |
| No. of protein-RNA complexes | 2 |
| No. of protein/RNA atoms | 3822/401 |
| No. of amino-acid/nucleotide | 488/19 |
| Root-mean-square-deviation (RMSD) | |
| Bond length(Å) | 0.011 |
| Bond angle(°) | 1.680 |
| Average B-factors | |
| Protein/RNA | 159.1/200.6 |
| Ramachandran plot of protein residues in | |
| most favored region (%) | 74.0 |
| additionally allowed region (%) | 21.6 |
| generously allowed region (%) | 4.2 |
| disallowed region (%) | 0.2 |
values in parenthesis are those for the last resolution shell.
RNA-binding and cleavage reactions
RNA cleavage reactions were initiated by addition of 1 µM enzyme to a 20 µL solution containing 0.05 pmol of 5’ end 32P-radiolabeled RNA, 0.02 M HEPES-KOH (pH 7.0), 0.25 M KCl, 0.075 M DTT, 0.0015 M MgCl2, 5µg E. coli tRNA, and 10% glycerol at 70°C. Half of the reaction was loaded directly onto a non-denaturing 10% polyacrylamide gel to assess RNA binding and the other half was loaded onto a denaturing 15% polyacrylamide gel in 7M urea to assess RNA cleavage. Radiolabeled RNAs were visualized by phosphorimaging.
Mass spectrometry
MALDI-TOF mass spectrometry was used to confirm the cleavage products for the wild-type (wt) and mutant (A23U) RNAs. The RNA cleavage reaction was carried out as described above and was followed by phenol extraction and ethanol precipitation to isolate the RNA components. The sample was resuspended in 10 µL water and purified using a C18 ZipTip (ZipTip® Pipette Tips, Millipore) before being spotted on a matrix. The matrix solution contains 70 mg/ml 3-hydroxypicolinic acid and picolinic acid (9:1) and 50% ACN. The negative ions/reflector mode of the Autoflex III mass spectrometer (Bruker Daltonics) was utilized.
Supplementary Material
Acknowledgments
This work was supported by NIH grants R01 GM66958 to H.L. and R01 GM54682 to M.T. and R.T. X-ray diffraction data were collected from the Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory. Supporting institutions for APS beamlines may be found at http://necat.chem.cornell.edu/ and www.ser-cat.org/members.html. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of a Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
ACCESSION NUMBERS:
Coordinates and structure factors have been deposited in the Protein Data Bank with accession number 3PKM
References
- Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- Andersson AF, Banfield JF. Virus population dynamics and acquired virus resistance in natural microbial communities. Science. 2008;320:1047–1050. doi: 10.1126/science.1157358. [DOI] [PubMed] [Google Scholar]
- Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- Bolotin A, Quinquis B, Sorokin A, Ehrlich SD. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology. 2005;151:2551–2561. doi: 10.1099/mic.0.28048-0. [DOI] [PubMed] [Google Scholar]
- Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, Dickman MJ, Makarova KS, Koonin EV, van der Oost J. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321:960–964. doi: 10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carte J, Pfister NT, Compton MM, Terns RM, Terns MP. Binding and cleavage of CRISPR RNA by Cas6. RNA. doi: 10.1261/rna.2230110. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carte J, Wang R, Li H, Terns RM, Terns MP. Cas6 is an endoribonuclease that generates guide RNAs for invader defense in prokaryotes. Genes Dev. 2008;22:3489–3496. doi: 10.1101/gad.1742908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll JR. Spoligotyping for molecular epidemiology of the Mycobacterium tuberculosis complex. Methods Mol Biol. 2009;551:117–128. doi: 10.1007/978-1-60327-999-4_10. [DOI] [PubMed] [Google Scholar]
- Ebihara A, Yao M, Masui R, Tanaka I, Yokoyama S, Kuramitsu S. Crystal structure of hypothetical protein TTHB192 from Thermus thermophilus HB8 reveals a new protein family with an RNA recognition motif-like domain. Protein Sci. 2006;15:1494–1499. doi: 10.1110/ps.062131106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Grissa I, Vergnaud G, Pourcel C. The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics. 2007;8:172. doi: 10.1186/1471-2105-8-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haft DH, Selengut J, Mongodin EF, Nelson KE. A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput Biol. 2005;1 doi: 10.1371/journal.pcbi.0010060. e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale CR, Zhao P, Olson S, Duff MO, Graveley BR, Wells L, Terns RM, Terns MP. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell. 2009;139:945–956. doi: 10.1016/j.cell.2009.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haurwitz RE, Jinek M, Wiedenheft B, Zhou K, Doudna JA. Sequence- and structure-specific RNA processing by a CRISPR endonuclease. Science. 2010;329:1355–1358. doi: 10.1126/science.1192272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidelberg JF, Nelson WC, Schoenfeld T, Bhaya D. Germ warfare in a microbial mat community: CRISPRs provide insights into the co-evolution of host and viral genomes. PLoS One. 2009;4 doi: 10.1371/journal.pone.0004169. e4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath P, Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010;327:167–170. doi: 10.1126/science.1179555. [DOI] [PubMed] [Google Scholar]
- Janin J, Miller S, Chothia C. Surface, subunit interfaces and interior of oligomeric proteins. J Mol Biol. 1988;204:155–164. doi: 10.1016/0022-2836(88)90606-7. [DOI] [PubMed] [Google Scholar]
- Jansen R, Embden JD, Gaastra W, Schouls LM. Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol. 2002;43:1565–1575. doi: 10.1046/j.1365-2958.2002.02839.x. [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for binding protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Karginov FV, Hannon GJ. The CRISPR system: small RNA-guided defense in bacteria and archaea. Mol Cell. 2010;37:7–19. doi: 10.1016/j.molcel.2009.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunin V, Sorek R, Hugenholtz P. Evolutionary conservation of sequence and secondary structures in CRISPR repeats. Genome Biol. 2007;8:R61. doi: 10.1186/gb-2007-8-4-r61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- Makarova KS, Grishin NV, Shabalina SA, Wolf YI, Koonin EV. A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol Direct. 2006;1:7. doi: 10.1186/1745-6150-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maris C, Dominguez C, Allain FH. The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. The FEBS journal. 2005;272:2118–2131. doi: 10.1111/j.1742-4658.2005.04653.x. [DOI] [PubMed] [Google Scholar]
- Marraffini LA, Sontheimer EJ. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science. 2008;322:1843–1845. doi: 10.1126/science.1165771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marraffini LA, Sontheimer EJ. CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat Rev Genet. 2010a;11:181–190. doi: 10.1038/nrg2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marraffini LA, Sontheimer EJ. Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature. 2010b;463:568–571. doi: 10.1038/nature08703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills S, Griffin C, Coffey A, Meijer WC, Hafkamp B, Ross RP. CRISPR analysis of bacteriophage-insensitive mutants (BIMs) of industrial Streptococcus thermophilus- implications for starter design. J Appl Microbiol. 2009 doi: 10.1111/j.1365-2672.2009.04486.x. [DOI] [PubMed] [Google Scholar]
- Mojica FJ, Diez-Villasenor C, Garcia-Martinez J, Soria E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol. 2005;60:174–182. doi: 10.1007/s00239-004-0046-3. [DOI] [PubMed] [Google Scholar]
- Mokrousov I, Limeschenko E, Vyazovaya A, Narvskaya O. Corynebacterium diphtheriae spoligotyping based on combined use of two CRISPR loci. Biotechnol J. 2007;2:901–906. doi: 10.1002/biot.200700035. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Vol 276. San Diego: Academic Press; 1997. [DOI] [PubMed] [Google Scholar]
- Pourcel C, Salvignol G, Vergnaud G. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology. 2005;151:653–663. doi: 10.1099/mic.0.27437-0. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan V, Biou V. Treatment of multiwavelength anomalous diffraction data as a special case of multiple isomorphous replacement. Methods Enzymol. 1997;276:538–557. [PubMed] [Google Scholar]
- Snyder JC, Bateson MM, Lavin M, Young MJ. Use of cellular CRISPR (clusters of regularly interspaced short palindromic repeats) spacer-based microarrays for detection of viruses in environmental samples. Appl Environ Microbiol. 76:7251–7258. doi: 10.1128/AEM.01109-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorek R, Kunin V, Hugenholtz P. CRISPR--a widespread system that provides acquired resistance against phages in bacteria and archaea. Nat Rev Microbiol. 2008;6:181–186. doi: 10.1038/nrmicro1793. [DOI] [PubMed] [Google Scholar]
- Tyson GW, Banfield JF. Rapidly evolving CRISPRs implicated in acquired resistance of microorganisms to viruses. Environ Microbiol. 2008;10:200–207. doi: 10.1111/j.1462-2920.2007.01444.x. [DOI] [PubMed] [Google Scholar]
- van der Oost J, Jore MM, Westra ER, Lundgren M, Brouns SJ. CRISPR-based adaptive and heritable immunity in prokaryotes. Trends Biochem Sci. 2009;34:401–407. doi: 10.1016/j.tibs.2009.05.002. [DOI] [PubMed] [Google Scholar]
- Waters LS, Storz G. Regulatory RNAs in bacteria. Cell. 2009;136:615–628. doi: 10.1016/j.cell.2009.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.