

Summary
The evolution of eukaryotes was accompanied by an increased need for intracellular communication and cellular specialization. Thus, a more complex collection of secreted and membrane proteins had to be synthesized, modified, and folded. The endoplasmic reticulum (ER) thereby became equipped with devoted enzymes and associated factors that both catalyze the production of secreted proteins and remove damaged proteins. A means to modify ER function to accommodate and destroy misfolded proteins also evolved. Not surprisingly, a growing number of human diseases are linked to various facets of ER function. Each of these topics will be discussed in this article, with an emphasis on recent reports in the literature that employed diverse models.
Introduction
Most secreted proteins in eukaryotes are initially targeted to the Endoplasmic Reticulum (ER) where they are inserted, or translocated, into the ER lumen or lipid bilayer, depending respectively on whether they are soluble or possess transmembrane domains. Because up to one-third of all proteins interact with the ER, and because some specialized cells produce large amounts of secreted proteins, the flux through this compartment can be formidable. Fortunately, the ER provides a safe-haven for protein folding; the compartment is oxidative, which facilitates disulfide bond formation, and is loaded with proteins that augment folding. These molecular chaperones facilitate protein folding and prevent the formation of off-pathway aggregates.
Most secreted proteins fold and are modified in an error-free manner, but stress, mutations, or stochastic errors during synthesis can decrease the folding yield or rate of folding. Resident chaperones might then be unable to prevent the generation of toxic unfolded species. Indeed, toxic proteins in the ER are observed in some disease states, and can initiate apoptosis [1,2]. However, if misfolded proteins transiently accumulate, or if a stress is sufficiently brief, the problems that accompany protein unfolding may be repaired. Either the ER increases its ability to handle misfolded proteins, or misfolded proteins are destroyed. The first pathway is the unfolded protein response (UPR), and the second is ER associated degradation (ERAD).
In this review, we discuss the protein folding machineries in the ER and then turn our attention to recent highlights on the mechanisms of UPR induction and the ERAD pathway. We conclude with a few notable links between ER function and disease.
The Protein Folding Machinery in the ER
ER protein folding is a complex interplay between a polypeptide's primary structural information and linked cellular networks. Folding begins as synthesis initiates on cytosolic ribosomes, and ends when the native protein is packaged for ER exit (Figure 1). As the signal recognition particle recognizes a signal sequence, the ribosome-nascent chain complex is targeted to the cytosolic face of the “translocon”. This aligns the ribosome exit tunnel with an aqueous channel formed by the Sec61 αβγ complex [3,4] such that the elongating polypeptide passes directly into the ER lumen coincident with synthesis. For membrane proteins, transmembrane segment compaction is stimulated within the ribosome exit tunnel. This shortens nascent polypeptide length [5,6], and triggers a dynamic rearrangement that closes the lumenal end of the translocon, terminates translocation, and redirects protein movement to the cytosol [7,8]. For polytopic membrane proteins, transmembrane helix formation is coordinated with orientation across the membrane, membrane integration, and helical packing in the bilayer [9], and involves changes in transmembrane segment boundaries and topology after targeting [10,11]. Future studies will surely determine how other early folding events are facilitated.
Figure 1. Early events during protein folding and quality control in the mammalian ER.

(1) Nascent polypeptides enter the ER through the Sec61 translocon complex, which contains ∼20 components (some of which are shown). Next, a core oligosaccharide, GlcNAC2Man9Glc3, is cotranslationally transferred from a dolichol lipid precursor (orange oval) to an NXS/T (X≠P) consensus site by OST. Terminal glucoses (green triangles) are trimmed by glucosidase I and II (Glc I and Glc II) to a GlcNAC2Man9Glc1 structure that binds the membrane protein, calnexin (CNX). Non-glycosylated substrates either bind BiP and undergo non-lectin mediated folding, and/or are posttranslationally glycosylated by the OST(B) isoform and thereby enter the CNX binding cycle. If translocation is aborted, substrates are delivered to cytosolic chaperones (e.g., Hsp70) and may be degraded. (2) Upon release from CNX, N-linked glycans are further deglucosylated by GlcII, and the substrate either folds spontaneously, is transferred to BiP-, PDI-, and/or Grp94-containing chaperone complexes, or is recognized as misfolded by UGT1 and reglucosylated. Reglucosylation enables rebinding to CNX and its lumenal homolog calreticulin (CRT), which stimulate isomerization and disulfide bond formation via the associated ERp57 PDI homolog. (3) When folded, the substrate is packaged into COPII-coated vesicles for transport to the Golgi. (4) During this process, certain terminal mannose residues (yellow circles) may be subjected to removal by ER Mannosidase I (ManI) and several EDEM homologs. Mannose trimming reduces the affinity for UGT1 and ultimately generates a GlcNAC2Man5 structure that binds additional lectins, including XTP3-B and OS-9 (not pictured), which bring the substrate to the retrotranslocation machinery (5) for retro-translocation to the cytosol, ubiquitination by E3 ligases, and degradation by the 26S proteasome. (6) In times of ER stress or excess protein load, misfolded substrates containing GlcNAC2Man9 glycans can also be delivered to the Golgi and sorted for lysosomal degradation, or modified by Golgi ManII and returned to the ER for degradation via ERAD.
Secreted proteins entering the ER immediately encounter a network of chaperone systems that minimize aggregation, facilitate native structure formation, and ensure oligomeric assembly [12] (Figure 1). These systems involve: i) non-covalent interactions with Hsp40 (J-proteins), Hsp70 (Grp78/BiP/Kar2) and Hsp90 (Grp94) chaperones, (ii) lectin-based chaperones, such as calnexin (CNX) and calreticulin (CRT), and (iii) protein disulfide isomerases (PDIs). The relative contribution to folding of each component is dictated by the properties and requirements of individual clients. Because nascent polypeptides are delivered into the ER in a relatively unstructured state [13], ER chaperones are organized in both space and time. For example, ERj1 is a type I membrane protein in mammals that binds the exit tunnel of 80S ribosomes and contains a lumenal J-domain that stimulates the ATPase activity of BiP, thereby facilitating client loading [14,15]. In the absence of BiP, ERj1 binds ribosomes with high affinity and allosterically inhibits translation, whereas BiP binding reduces ribosome affinity and allows translation to continue [16]. Thus, ERj1 recruits BiP to the translocon and signals the ribosome that chaperones are available on the opposite side of the membrane. Two translocon components, Sec63 (a J-protein) and its partner Sec62, may perform a similar role [17].
An early modification on nascent proteins involves transfer of a core oligosaccharide (GlcNAC2Man9Glc3) to Asn-X-Ser/Thr. N-linked glycosylation improves protein solubility, decreases aggregation, provides a binding site for CNX and CRT, facilitates PDI interaction, and can be a marker for ERAD [18]. Glycan attachment is catalyzed by the SST3 subunit(s) of oligosaccharyltransferase (OST) [19], a heterogeneous membrane complex that associates with the translocon [20,21]. Translocon residence helps OST scan for consensus sites and attach sugars cotranslationally. However, not all consensus sites are utilized due to premature folding, disulfide bond formation, and/or peptide release from the ribosome. Ruiz-Canada et al. demonstrated that glycosylation efficiency is also improved by the existence of two OST forms, containing either SST3A or SST3B subunits [22]. Both catalyze cotranslational glycosylation, with SST3A having less activity and more selectivity. OST containing the SST3B isoform can also glycosylate substrates after release from the transloocn, thereby recognizing sequons missed during cotranslational scanning. Glycosylation is further improved by a different OST subunit, also with two homologs, OST3/6 [23]. Each contains a thioredoxin-like fold capable of forming mixed disulfides that improve glycosylation for a subset of clients. An important future question is how other OST subunits, whose activities remain unknown, contribute to these events.
After glycan attachment, the terminal glucose is removed by glucosidase I (Glc I), followed by removal of the second glucose by Glc II (Figure 1). This generates GlcNAC2Man9Glc1 that forms a high affinity ligand for CNX and CRT [24-26]. The membrane protein CNX is associated with the translocon and binds substrates cotranslationally. In contrast, CRT is a soluble protein and interacts primarily with secreted proteins after ribosome release. Once bound, CNX/CRT retain substrates in the ER, prevent aggregation and degradation, and facilitate folding by recruiting a PDI, ERp57. Removal of the last glucose by Glc II generates GlcNAC2Man9, which does not bind CNX/CRT. Folded substrates can exit the ER, whereas nearly native substrates with exposed hydrophobic patches, usually near the N-linked site, are transferred to BiP or reglucosylated by UDP-Glc glycoprotein glycosyltransferase (UGT) and enter a second CNX/CRT cycle. The crystal structure of CRT complexed with a Man3Glc1 tetrasaccharide recently revealed the basis for specificity of GlcNAC2Man9Glc1 over GlcNAC2Man9 whereby the terminal glucose fits into CRT via extensive hydrogen bonding [27]. Interestingly, the bound glucose is inaccessible to Glc II, which raises the question of how the substrate is released prior to glucose removal. The structural homology between CRT and CNX indicates that glycan binding is mediated by identical mechanisms, suggesting that their different specificities are due to protein-protein interactions [28] or their distinct locations in the ER.
Deglucosylation and reglucosylation are in kinetic competition with ER/Golgi mannosidases. These enzymes catalyze the slow removal of mannose, which decreases reglucosylation by UGT [18] and generates a ligand for a different set of ER lectins. Among them, EDEM, a mannosidase [29-31], and Yos9 (in yeast)[32] and OS-9 and XTP3-B (in mammals)[33,34] target substrates to an ER quality control (ERQC) compartment. The net result is a molecular clock in which glucose trimming and reattachment provide proteins a limited time, and multiple chances to fold, after which the profolding program is replaced by a prodegradative program.
Because the vectorial nature of translocation allows formation of non-native disulfide bonds, many initial disulfides are subsequently isomerized by the PDIs. The Cys-X-X-Cys motif in PDI's thioredoxin domain accepts oxidizing equivalents, primarily from Ero1 and Erv2 (but other mechanisms have recently been uncovered [35]). Regardless, oxidizing equivalents are transferred to clients via mixed disulfide intermediates. Mammalian cells contain ∼20 PDIs (yeast contain 5) that vary in substrate specificity as well as oxidation, reduction, and isomerization activities [36]. While mammalian PDI is ubiquitously expressed and interacts with a range of substrates, other homologs show remarkable selectivity [37].
Disulfide trapping, has revealed that mammalian PDIs, ERP57, ERP18, and ERp64 interact with Ero1 and exhibit distinct yet overlapping substrate specificity [38]. The PDI P5 also forms a noncovalent interaction with BiP and mixed disulfides with BiP clients. BiP binding is strengthened by P5 oxidation, suggesting that P5 recruitment facilitates the refolding of oxidized BiP substrates. By analogy, the recruitment of ERp57 to CNX/CRT likely explains its preference for facilitating glycoprotein folding, as demonstrated by siRNA knockdown experiments [39]. In yeast, PDI interacts with the EDEM homologue, suggesting that PDI chaperones this enzyme and/or that the two proteins function together during protein folding [30,40]. Structural studies have also provided new clues into PDI specificity. Yeast PDI forms a twisted “U”, formed by four thioredoxin domains, of which a and a' domains contain active CXXC motifs [41]. Substrate binding occurs at a flexible hydrophobic pocket on the b' domain, which can be partially occluded by a linker peptide [42]. The flexible nature of this site in PDI may explain its broad substrate specificity. In contrast, the b' domain of ERp57 forms a high affinity binding site for calnexin [43], while ERp72 contains a related structure that lacks this site [44].
One might predict that ERQC prevents the export of unfolded proteins. However, recent studies suggest that ER retention and export are competing processes. In yeast, an ERAD substrate exits the ER if appended to a strong exit signal [45]. Also, misfolded variants of a mammalian prion, which are neither retained nor degraded in the ER, traffic to the Golgi and are sorted to the endo-lysosomal system [46]. This may be explained by the discovery of bipartite degradation motifs that require a locally unfolded peptide region and an adjacent mannose-trimmed glycan [47]. In the absence of glycan, misfolded substrates may lack an ERAD determinant and become subject to Golgi quality control. But, some substrates undergo mannose trimming in the Golgi and return to the ER for ERAD [48,49]. Overall, ERQC is one of several mechanisms to protect the cell against proteotoxicity. Folding, trafficking and degradation are therefore highly integrated, utilize common machinery, and generate complex signals that determine a protein's fate in multiple compartments.
The Unfolded Protein Response: An End-Run Around Folding Problems in the ER
Through UPR induction, the stresses arising from the accumulation of misfolded ER proteins can be ameliorated. The UPR is initiated by a single (in yeast) or three-pronged (in mammals) signal transduction pathway. Common to both systems is IRE1, which encodes a single-pass membrane protein that in yeast activates synthesis of the Hac1 transcription factor by splicing a translational inhibitory region within the Hac1 mRNA [50]. Once translated, Hac1 binds to promoters that contain a UPR element. A significant number of secondary UPR targets are also activated, but the identities of all contributing transcription factors are unclear [51]. The transcriptional response increases the levels of lumenal chaperones and of components required for ERAD, protein transport to other compartments, and lipid biosynthesis [52]. ER expansion might provide the most critical means to ameliorate stress [53]. The UPR appears to be triggered from select regions of the ER because Ire1 clusters upon activation [54], which may reflect self-assembly [55] and a more efficient response. Overall, the UPR reduces the concentration and toxicity associated with misfolded proteins in the ER.
The accumulation of misfolded proteins in the ER—especially those with disulfide bonds—generates oxidative stress, and some of the enzymes that mitigate oxidative stress are UPR targets. The use of a real-time reporter has confirmed, in yeast, that misfolded proteins trigger oxidative stress [56]. Moreover, disulfide-containing, misfolded proteins in the ER arrest cell growth when the UPR is disabled, an effect that arises from oxidative stress [57].
Can ER stressed yeast recover? ER stress prevents the inheritance of cortical ER by daughter cells and delays cytokinesis. However, all is not lost: once cell division is reinitiated, the selfless mother cell retains the stressed ER but passes undamaged ER on to the daughter [58].
In mammals, IRE1 is joined by two additional UPR transducers, PERK and ATF6 (Figure 2). The downstream target of Ire1 is the XBP1 transcription factor, which like Hac1 induces the synthesis of products that repair the ER. However, the more specialized demands in mammals are evident by PERK and ATF6, which transiently inhibit new protein synthesis and further increase the levels of components that ameliorate stress. At later times, these transducers can initiate an apoptotic program [1]. Indeed, there is a growing appreciation that the UPR plays a role in several disease states, as exemplified by observations in mouse models. For example, chemical chaperones and anti-oxidants that attenuate ER stress respectively reduce complications from type 2 diabetes and improve the folding and secretion of a protein linked to hemophilia [59,60]. Drosophila and mammalian IRE1 also possesses a nonspecific RNAase activity, termed regulated Ire1-dependent decay (RIDD) [61]. ER-targeted mRNAs, which mostly encode secretory pathway cargo, are destroyed when the ER cannot handle more proteins. Recent data suggest that RIDD is regulated by conditions that perhaps favor XBP1 splicing over message destruction [62,63].
Figure 2. The mammalian UPR is triggered by a multi-pronged signal transduction pathway.

The accumulation of misfolded proteins in the ER and/or liberation of BiP from IRE1, PERK, and ATF6, activate a series of down-stream events. IRE1 induces the splicing of the XBP-1 message, which is then translated into an active transcription factor. There are actually two isoforms of IRE1 (α and β) but for simplicity only a single, generic protein is depicted. PERK phosphorylates eIF2α, which inhibits general protein synthesis but the ATF4 transcription factor can still be translated because of the presence of a short, upstream ORF that is skipped when eIF2α is phosphorylated. In contrast, ATF6, when activated, is released from the ER and migrates to the Golgi where it is clipped, thus releasing an active transcription factor, ATF6-p50. The immediate effects of UPR induction help the ER cope with an increase in the concentration of misfolded proteins. However, prolonged UPR induction triggers apoptosis through the ATF4-dependent transcription of the CHOP/Gadd153 transcription factor. CHOP both increases the expression of pro-apoptotic genes and inhibits the expression of Bcl2, an antiapoptotic protein.
Initially, it was thought that Ire1 sensed the levels of misfolded proteins indirectly because Ire1 and BiP coprecipitate in cell lysates [64]. Thus, as BiP is recruited from Ire1 to accommodate increased levels of misfolded proteins, Ire1 dimerizes and is activated via transphosphorylation. Consistent with this model, BiP diffusion within the ER decreases as misfolded proteins accumuluate, and this event serves as one of the first signs that the UPR will be activated [65]. However, the yeast Ire1 crystal structure identified a peptide-binding site that resembles the site found in MHCI; mutations in this site reduce Ire1-dependent signal transduction [66]. Nevertheless, the structure of this domain in human IRE1 is collapsed, making it less obvious whether peptides fit into this site [67]. The solution to this controversy might be explained by the fact that yeast Ire1 binds unfolded proteins and BiP prevents the formation of Ire1 oligomers, which when activated primes Ire1 to initiate the UPR [68]. In mammals, BiP may provide both a threshold and dictate UPR induction kinetics [69].
ER Associated Degradation: A Quick-Fix for Misfolded Proteins in the ER
The ERAD pathway identifies and destroys individual proteins unable to pass ERQC and can be subdivided into unique steps. First, an ERAD substrate must be recognized. Molecular chaperones were first shown to play this role, but over time it has become clear that ER-resident lectins, discussed above, aid in substrate recognition. Second, the substrate must be presented to a protease, which was initially thought to reside in the ER. Over time it became clear that the cytoplasmic proteasome degrades misfolded proteins, improperly modified proteins, and orphaned subunits of multimeric complexes in the ER [70-72]. Thus, lumenal domains of membrane proteins and soluble protein substrates must be “retrotranslocated” from the ER. Third, proteasome delivery requires ubiquitination [73], and not surprisingly, enzymes required for ubiquitin conjugation and ligation are critical for ERAD. And fourth, the proteasome recognizes and destroys the retrotranslocated substrate.
Based on the many topologies of ERAD substrates, factors required for substrate recognition were proposed to reside in the ER lumen, cytoplasm, and membrane. Substrates with folding lesions in each compartment were then analyzed to reveal distinctions in the degradation requirements for ERAD-“M” (membrane), ERAD-“L” (lumenal), and ERAD-“C” (cytosolic) substrates [74] (Figure 3). Moreover, E3 ubiquitin ligases that append ubiquitin onto ERAD-L/M versus ERAD-C substrates reside in distinct multiprotein complexes [75-77]. One caveat to these designations is that a substrate—especially those with complex topologies—might expose misfolded domains in more than one compartment or might prove too hard to handle by one pathway. Indeed, some substrates in yeast do not fall cleanly within these designations [78-80], and in mammals the membrane tethering of a soluble ERAD-L substrate alters the degradation requirements [81].
Figure 3. ERAD substrates can be classified into those with misfolded protein lesions either in the ER lumen (ERAD-L), the cytoplasm (ERAD-C), or the membrane (ERAD-M).

The site of the misfolding lesion is depicted by a
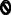 . Select yeast factors (in parentheses) required for the degradation of each class are listed, and mammalian homologs are indicated in cases in which these have also been shown to function during ERAD, although not necessarily in a class-specific manner. Note: Many mammalian E3s have been identified as contributing to the ERAD of select factors (e.g., CHIP, HRD1, RMA1, gp78, TEB4, and Parkin), but for simplicity these are not shown. In addition, there is evidence that Hsp70 and its constitutively expressed isoform, Hsc70, may play unique roles during protein folding and ERAD, respectively [153].
. Select yeast factors (in parentheses) required for the degradation of each class are listed, and mammalian homologs are indicated in cases in which these have also been shown to function during ERAD, although not necessarily in a class-specific manner. Note: Many mammalian E3s have been identified as contributing to the ERAD of select factors (e.g., CHIP, HRD1, RMA1, gp78, TEB4, and Parkin), but for simplicity these are not shown. In addition, there is evidence that Hsp70 and its constitutively expressed isoform, Hsc70, may play unique roles during protein folding and ERAD, respectively [153].
As discussed above, chaperone- and lectin-containing complexes in the ER recognize ERAD substrates. Perhaps given the evolution of diverse substrates, some proteins involved in substrate selection are unique to mammals [82,83]. Although the enzymes required for glycan quality control were characterized several years ago using overexpressed proteins, the first endogenous substrate for UGT was only recently identified [84]. There is also an appreciation that UGT and CNX/CRT are not alone in being both lectins and peptide-binding chaperones; EDEM also recognizes non-glycosylated substrates [85].
Mannose trimmed substrates are recognized by the homologous Yos9 or OS-9/XTP3-B proteins (Figure 1), which also associate with components that recognize, ferry, and ubiquitinate ERAD substrates [33,75-77,86-88]. A critical event for retrotranslocation may be disulfide bond reduction, which is catalyzed by a J-domain and thioredoxin-domain containing protein, ERdj5 [89]. The redox potential of ERdj5 positions the enzyme as the most reducing PDI. Moreover, ERdj5 binds EDEM. Recently, an ERdj5 flavoprotein partner was discovered, ERFAD [90], but it is unknown whether ERdj5 and ERFAD form an electron relay. It also remains mysterious how yeast, which lack ERdj5, reduce ERAD substrates, or whether ERdj5 is required for all mammalian substrates. Indeed, aggregated ERAD substrates can be removed by other pathways, such as autophagy [91-93].
How are soluble proteins retrotranslocated? One candidate for a retrotranslocation channel is Sec61 [94-98]. Another candidate is a polytopic E3 ubiquitin ligase that forms multimers, namely Hrd1 [99]. Hrd1, which is required for the degradation of ERAD-L and ERAD-M substrates (Figure 3), crosslinks to a lumenally trapped ERAD substrate [100]. Crosslinking requires Cdc48, a AAA-ATPase that extracts substrates from the ER (see below). Consistent with channel-like activity, Hrd1 partners determine the specificity for diverse Hrd1 substrates [101], and Hrd1 may directly recognize polar and charged residues in ERAD-M substrates that are exposed when membrane segments fail to pack properly in the bilayer [102]. In mammalian cells, the exposure of a dibasic motif in opsin's transmembrane domain triggered Hrd1-dependent ERAD [103], consistent with Hrd1 acting as both a ubiquitin ligase and a chaperone for defective transmembrane segments. Hrd1 may even be a component of or constitute the retrotranslocation channel for soluble proteins. But, whether a channel is needed for membrane proteins remains unclear, nor is the case closed on whether alternate factors—such as Sec61 or the Derlins, membrane proteins that are required for the ERAD of some substrates [104-107]—form part of the channel or are substrate-specific “retrotranslocons”.
Doa10 is another integral membrane ubiquitin ligase and is required for ERAD-C in yeast [108]. In higher cells, the list of E3s is expanded, and examples of the sequential action of unique ligases have been observed [109]. Although it was previously thought that ERAD substrates possessed Lys48-derived polyubiquitin chains, recent proteomic analyses indicate that ERAD substrates contain Lys48- and Lys11-linkages [110]. Modification on residues other than Lys, particularly Ser and Thr, during ERAD has also been observed [111-113]. In any event, once a protein is polyubiquitinated, the AAA-ATPase, Cdc48 (in yeast) or p97 (in mammals) bind the substrate by virtue of its associated Npl4-Ufd1 cofactors, and Cdc48/p97-mediated ATP hydrolysis is used to facilitate membrane extraction. Proteasome adaptors then serve as intermediaries during ubiquitinated protein targeting to the proteasome [114].
Given that AAA-ATPases function as force-generating engines, it was anticipated that p97/Cdc48-dependence is dictated by the relative difficulty in retrotranslocating a substrate. In fact, genetic evidence indicates that ERAD-M substrates are most sensitive to Cdc48 depletion [115], and p97 dependence correlates with the relative stability of a polytopic membrane protein in the bilayer [116]. In contrast, some ERAD substrates are retrotranslocated in a Cdc48/p97-independent fashion and instead use an analogous AAA complex in the proteasome cap [117-120]. One substrate even retrotranslocates in a p97-dependent but Ufd1-Npl4-independent manner [121]. Regardless, p97/Cdc48 tethering to the ER is critical for maximal ERAD, and in mammals a p97 organizing protein, erasin, may fulfill this role [122]. In yeast, Ubx2 might play this role, or it could help recruit ubiquitinated ERAD substrates to Cdc48 [123-125].
Because a significant population of proteasomes resides at the ER membrane, retrotranslocation and degradation should be coupled. In most cases this is true, but ubiquitinated, integral membrane proteins have been observed in solution after retrotranslocation from the yeast [126,127] and mammalian ER [128-130]. How these proteins are stabilized is unclear, but the characterization of cytoplasmic inclusions may help answer this question [131,132].
Before degradation, the polyubiquitin chain on ERAD substrates is removed, presumably by the same deubiquitinating enzymes (Dubs) required for most proteasome substrates [73]. Thus, it was surprising that a p97-associated Dub in mammals contributed to the retrotranslocation of a soluble and membrane substrate [133]. It was proposed that ubiquitin removal helps ERAD substrates enter the p97 hexamer, but presumably sufficient ubiquitin remains to ensure proteasome-capture. There is also continued interest in identifying proteases that aid the proteasome during ERAD. One enzyme that fills this role is the signal peptide peptidase, which is ideally positioned to clip residual transmembrane segments [134,135]. Nevertheless, the hunt for other ER proteases is ongoing.
Conclusions
There has been intense interest in defining the factors that facilitate ER protein folding, but it must be appreciated that only a small fraction of all secreted proteins have been analyzed. Undoubtedly, new paradigms will emerge as additional substrates are examined. It is also clear that the UPR and ERAD have been co-opted to facilitate non-quality control activities. For example, the UPR is critical for homeostasis during unstressed conditions [136], and can initiate specific cellular responses or developmental decisions [137]; conversely, preparing the ER for a subsequent onslaught of secreted proteins—by upregulating the synthesis of ER chaperones—prevents UPR induction [138]. The ERAD pathway is also used to regulate metabolic processes [139] and can modulate cadmium detoxification in yeast [140] and proteasome levels in mammals [141]. The retrotranslocation of bacterial toxins and the mechanisms underlying cytomegalovirus pathogenesis represent other examples in which this house-keeping pathway has been usurped for defined purposes [142]. Recently, components of the ERAD machinery (e.g., Cdc48/p97) have even been shown to facilitate the extraction and proteasome-mediated degradation of ubiquitin-conjugated components in the outer mitochondrial membrane [143-145].
Perhaps misfolded proteins in other intracellular compartments also utilize this pathway.
Finally, the connection between the ER and human disease has generated interest in exploiting ERQC pathways for therapeutics. More than 100 disorders ranging from cystic fibrosis, liver disease, epilepsy, cardiac arrythmias, blindness, and Alzheimer's disease, share conserved cellular pathology, in which misfolded ER substrates are prematurely degraded or accumulate as toxic aggregates. This has prompted three strategies for intervention that are directed at either the substrate itself, specific cellular factors, or the global folding environment. The first approach arose from observations that mutant proteins retain biological function when rescued by osmolytes [146,147]. Subsequent findings revealed that protein stability was improved by specific ligands, i.e., pharmacologic chaperones [148]. Efforts to specifically correct disease-related mutants are encouraging, and the first agents that restore misfolding protein trafficking are entering the clinic. The second approach targets the ERQC machinery. Compounds that modulate Hsp70 and Hsp90 chaperones, regulatory cochaperones, and lectins are of significant interest, and investigations to establish clinical utility are underway. Interestingly, proteasome inhibition has proven effective in treating refractory multiple myeloma because the synthetic load in malignant plasma cells sensitizes the cells to proteasome loss [149]. Proteasome stimulation by inhibiting a Dub may also improve clearance of toxic aggregates [150]. Third, the fate of ER substrates is determined by their interactions with solutes, metabolites, and macromolecules that comprise the proteostatic environment, which is in turn sensitive to stress, age and biosynthetic load [2]. Thus, manipulating proteostatic regulators, such as transcription factors, chaperone systems, or epigenetic modifiers may provide a means to treat these disorders. Similarly, upregulation of ER chaperones, such as BiP, corrects specific diseases in animal models [151,152]. The implications of these findings are profound, as proteostatic pathways are connected to pathologic processes that range from metabolic syndrome, to cancer, longevity and degenerative disease. This is clearly an exciting time to be in this field.
Acknowledgments
The authors acknowledge support from grants from the National Institutes of Health (GM75061 and DK79307, “The Pittsburgh Center for Kidney Research”, to J.L.B, and GM53457 and DK51818 to W.R.S.) and from Cystic Fibrosis Foundation Therapeutics.
Contributor Information
Jeffrey L. Brodsky, Email: jbrodsky@pitt.edu.
William R. Skach, Email: skachw@ohsu.edu.
References
- 1.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 2.Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009;78:959–991. doi: 10.1146/annurev.biochem.052308.114844. [DOI] [PubMed] [Google Scholar]
- 3.Crowley KS, Liao S, Worrell VE, Reinhart GD, Johnson AE. Secretory proteins move through the endoplasmic reticulum membrane via an aqueous, gated pore. Cell. 1994;78:461–471. doi: 10.1016/0092-8674(94)90424-3. [DOI] [PubMed] [Google Scholar]
- 4.Becker T, Bhushan S, Jarasch A, Armache JP, Funes S, Jossinet F, Gumbart J, Mielke T, Berninghausen O, Schulten K, et al. Structure of monomeric yeast and mammalian Sec61 complexes interacting with the translating ribosome. Science. 2009;326:1369–1373. doi: 10.1126/science.1178535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Woolhead CA, McCormick PJ, Johnson AE. Nascent membrane and secretory proteins differ in FRET-detected folding far inside the ribosome and in their exposure to ribosomal proteins. Cell. 2004;116:725–736. doi: 10.1016/s0092-8674(04)00169-2. [DOI] [PubMed] [Google Scholar]
- 6.Daniel CJ, Conti B, Johnson AE, Skach WR. Control of translocation through the Sec61 translocon by nascent polypeptide structure within the ribosome. J Biol Chem. 2008;283:20864–20873. doi: 10.1074/jbc.M803517200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liao S, Lin J, Do H, Johnson AE. Both lumenal and cytosolic gating of the aqueous ER translocon pore are regulated from inside the ribosome during membrane protein integration. Cell. 1997;90:31–41. doi: 10.1016/s0092-8674(00)80311-6. [DOI] [PubMed] [Google Scholar]
- 8.Alder NN, Shen Y, Brodsky JL, Hendershot LM, Johnson AE. The molecular mechanisms underlying BiP-mediated gating of the Sec61 translocon of the endoplasmic reticulum. J Cell Biol. 2005;168:389–399. doi: 10.1083/jcb.200409174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Skach WR. Cellular mechanisms of membrane protein folding. Nat Struct Mol Biol. 2009;16:606–612. doi: 10.1038/nsmb.1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buck TM, Wagner J, Grund S, Skach WR. A novel tripartite motif involved in aquaporin topogenesis, monomer folding and tetramerization. Nat Struct Mol Biol. 2007;14:762–769. doi: 10.1038/nsmb1275. [DOI] [PubMed] [Google Scholar]
- 11.Cheng Z, Gilmore R. Slow translocon gating causes cytosolic exposure of transmembrane and lumenal domains during membrane protein integration. Nat Struct Mol Biol. 2006;13:930–936. doi: 10.1038/nsmb1146. [DOI] [PubMed] [Google Scholar]
- ••12.Jonikas MC, Collins SR, Denic V, Oh E, Quan EM, Schmid V, Weibezahn J, Schwappach B, Walter P, Weissman JS, et al. Comprehensive characterization of genes required for protein folding in the endoplasmic reticulum. Science. 2009;323:1693–1697. doi: 10.1126/science.1167983. A non-biased systems approach to UPR signaling identified integrated components of pathways involving ER targeting, protein folding, processing, degradation, and trafficking. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kowarik M, Kung S, Martoglio B, Helenius A. Protein folding during cotranslational translocation in the endoplasmic reticulum. Mol Cell. 2002;10:769–778. doi: 10.1016/s1097-2765(02)00685-8. [DOI] [PubMed] [Google Scholar]
- 14.Dudek J, Greiner M, Muller A, Hendershot LM, Kopsch K, Nastainczyk W, Zimmermann R. ERj1p has a basic role in protein biogenesis at the endoplasmic reticulum. Nat Struct Mol Biol. 2005;12:1008–1014. doi: 10.1038/nsmb1007. [DOI] [PubMed] [Google Scholar]
- 15.Blau M, Mullapudi S, Becker T, Dudek J, Zimmermann R, Penczek PA, Beckmann R. ERj1p uses a universal ribosomal adaptor site to coordinate the 80S ribosome at the membrane. Nat Struct Mol Biol. 2005;12:1015–1016. doi: 10.1038/nsmb998. [DOI] [PubMed] [Google Scholar]
- •16.Benedix J, Lajoie P, Jaiswal H, Burgard C, Greiner M, Zimmermann R, Rospert S, Snapp EL, Dudek J. BiP modulates the affinity of its co-chaperone ERj1 for ribosomes. J Biol Chem. 2010;285:36427–36433. doi: 10.1074/jbc.M110.143263. The affinity of ERj1 for ribosomes at the cytosolic face of the ER is decreased upon BiP binding to its lumenal domain. This activity couples translation to to the local availability of chaperones in the lumenal compartment, a property shared by Sec62/63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muller L, de Escauriaza MD, Lajoie P, Theis M, Jung M, Muller A, Burgard C, Greiner M, Snapp EL, Dudek J, et al. Evolutionary gain of function for the ER membrane protein Sec62 from yeast to humans. Mol Biol Cell. 2010;21:691–703. doi: 10.1091/mbc.E09-08-0730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aebi M, Bernasconi R, Clerc S, Molinari M. N-glycan structures: recognition and processing in the ER. Trends Biochem Sci. 2010;35:74–82. doi: 10.1016/j.tibs.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 19.Nilsson I, Kelleher DJ, Miao Y, Shao Y, Kreibich G, Gilmore R, von Heijne G, Johnson AE. Photocross-linking of nascent chains to the STT3 subunit of the oligosaccharyltransferase complex. J Cell Biol. 2003;161:715–725. doi: 10.1083/jcb.200301043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gorlich D, Hartmann E, Prehn S, Rapoport TA. A protein of the endoplasmic reticulum involved early in polypeptide translocation. Nature. 1992;357:47–52. doi: 10.1038/357047a0. [DOI] [PubMed] [Google Scholar]
- 21.Shibatani T, David LL, McCormack AL, Frueh K, Skach WR. Proteomic analysis of mammalian oligosaccharyltransferase reveals multiple subcomplexes that contain Sec61, TRAP, and two potential new subunits. Biochemistry. 2005;44:5982–5992. doi: 10.1021/bi047328f. [DOI] [PubMed] [Google Scholar]
- •22.Ruiz-Canada C, Kelleher DJ, Gilmore R. Cotranslational and posttranslational N-glycosylation of polypeptides by distinct mammalian OST isoforms. Cell. 2009;136:272–283. doi: 10.1016/j.cell.2008.11.047. This study shows that two isoforms of OST containing either STT3A or STT3B perform overlapping functions in cotranslational glycosylation, while an separate pool of STT3 , not associated with the translocon, posttranslationally glycosylates (C-terminal) consensus sites in unfolded proteins after translocation has been completed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •23.Schulz BL, Stirnimann CU, Grimshaw JP, Brozzo MS, Fritsch F, Mohorko E, Capitani G, Glockshuber R, Grutter MG, Aebi M. Oxidoreductase activity of oligosaccharyltransferase subunits Ost3p and Ost6p defines site-specific glycosylation efficiency. Proc Natl Acad Sci U S A. 2009;106:11061–11066. doi: 10.1073/pnas.0812515106. This study shows that CXXC motifs in oxidoreductase domains of Ost6p and Ost3P are important for attachment of N-linked glycans to certain ER substrates. Thus, OST function in glycosylation is directly tied to an additional role in disulfide bond formation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schrag JD, Bergeron JJ, Li Y, Borisova S, Hahn M, Thomas DY, Cygler M. The Structure of calnexin, an ER chaperone involved in quality control of protein folding. Mol Cell. 2001;8:633–644. doi: 10.1016/s1097-2765(01)00318-5. [DOI] [PubMed] [Google Scholar]
- 25.Thomson SP, Williams DB. Delineation of the lectin site of the molecular chaperone calreticulin. Cell Stress Chaperones. 2005;10:242–251. doi: 10.1379/CSC-126.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kapoor M, Ellgaard L, Gopalakrishnapai J, Schirra C, Gemma E, Oscarson S, Helenius A, Surolia A. Mutational analysis provides molecular insight into the carbohydrate-binding region of calreticulin: pivotal roles of tyrosine-109 and aspartate-135 in carbohydrate recognition. Biochemistry. 2004;43:97–106. doi: 10.1021/bi0355286. [DOI] [PubMed] [Google Scholar]
- •27.Kozlov G, Pocanschi CL, Rosenauer A, Bastos-Aristizabal S, Gorelik A, Williams DB, Gehring K. Structural basis of carbohydrate recognition by calreticulin. J Biol Chem. 2010;285:38612–38620. doi: 10.1074/jbc.M110.168294. The 1.95 Å crystal structure of CRT bound to the tetrasaccharide Man3Glc1 revealed that all four sugars fit precisely into a long binding pocket with an extensive network of hydrogen bonds, particularly to the terminal glucose residue, thus explaining the exquisite binding specificity of CRT (and likely CNX) for monoglycosylated over di- and tri-glucosylated N-linked glycans. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Williams DB. Beyond lectins: the calnexin/calreticulin chaperone system of the endoplasmic reticulum. J Cell Sci. 2006;119:615–623. doi: 10.1242/jcs.02856. [DOI] [PubMed] [Google Scholar]
- 29.Quan EM, Kamiya Y, Kamiya D, Denic V, Weibezahn J, Kato K, Weissman JS. Defining the glycan destruction signal for endoplasmic reticulum-associated degradation. Mol Cell. 2008;32:870–877. doi: 10.1016/j.molcel.2008.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clerc S, Hirsch C, Oggier DM, Deprez P, Jakob C, Sommer T, Aebi M. Htm1 protein generates the N-glycan signal for glycoprotein degradation in the endoplasmic reticulum. J Cell Biol. 2009;184:159–172. doi: 10.1083/jcb.200809198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hosokawa N, Tremblay LO, Sleno B, Kamiya Y, Wada I, Nagata K, Kato K, Herscovics A. EDEM1 accelerates the trimming of alpha1,2-linked mannose on the C branch of N-glycans. Glycobiology. 2010;20:567–575. doi: 10.1093/glycob/cwq001. [DOI] [PubMed] [Google Scholar]
- 32.Cormier JH, Pearse BR, Hebert DN. Yos9p: a sweet-toothed bouncer of the secretory pathway. Mol Cell. 2005;19:717–719. doi: 10.1016/j.molcel.2005.08.029. [DOI] [PubMed] [Google Scholar]
- 33.Christianson JC, Shaler TA, Tyler RE, Kopito RR. OS-9 and GRP94 deliver mutant alpha1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat Cell Biol. 2008;10:272–282. doi: 10.1038/ncb1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •34.Groisman B, Shenkman M, Ron E, Lederkremer GZ. Mannose trimming is required for delivery of a glycoprotein from EDEM1 to XTP3-B and to late ER-associated degradation steps. J Biol Chem. 2011;286:1292–1300. doi: 10.1074/jbc.M110.154849. This study demonstates that XTP3-B acts as a lectin acceptor in the ERQC compartment that accepts ERAD substrates only after mannose trimming by ER ManI and EDEM and facilitates delivery to E3 ubiquitin ligases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fass D. Hunting for alternative disulfide bond formation pathways: endoplasmic reticulum janitor turns professor and teaches a lesson. Mol Cell. 2010;40:685–686. doi: 10.1016/j.molcel.2010.11.034. [DOI] [PubMed] [Google Scholar]
- 36.Hatahet F, Ruddock LW. Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxid Redox Signal. 2009;11:2807–2850. doi: 10.1089/ars.2009.2466. [DOI] [PubMed] [Google Scholar]
- 37.Park SW, Zhen G, Verhaeghe C, Nakagami Y, Nguyenvu LT, Barczak AJ, Killeen N, Erle DJ. The protein disulfide isomerase AGR2 is essential for production of intestinal mucus. Proc Natl Acad Sci U S A. 2009;106:6950–6955. doi: 10.1073/pnas.0808722106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •38.Jessop CE, Watkins RH, Simmons JJ, Tasab M, Bulleid NJ. Protein disulphide isomerase family members show distinct substrate specificity: P5 is targeted to BiP client proteins. J Cell Sci. 2009;122:4287–4295. doi: 10.1242/jcs.059154. Mixed disulfide trapping mutants (CXXA) of multiple ER PDIs revealed common interactions with Ero1 and overlapping yet distinct substrate specificites that may be partially dependent upon interactions with either lectin-based or BiP chaperone systems. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •39.Rutkevich LA, Cohen-Doyle MF, Brockmeier U, Williams DB. Functional relationship between protein disulfide isomerase family members during the oxidative folding of human secretory proteins. Mol Biol Cell. 2010;21:3093–3105. doi: 10.1091/mbc.E10-04-0356. SiRNA depletion of PDI's reveal that both ERp57 and mammalian PDI show broad substrate specificty, with ERp57 prefering glycosylated substrates, and weak phenotype for ERp72 and P5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sakoh-Nakatogawa M, Nishikawa S, Endo T. Roles of protein-disulfide isomerase-mediated disulfide bond formation of yeast Mnl1p in endoplasmic reticulum-associated degradation. J Biol Chem. 2009;284:11815–11825. doi: 10.1074/jbc.M900813200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tian G, Xiang S, Noiva R, Lennarz WJ, Schindelin H. The crystal structure of yeast protein disulfide isomerase suggests cooperativity between its active sites. Cell. 2006;124:61–73. doi: 10.1016/j.cell.2005.10.044. [DOI] [PubMed] [Google Scholar]
- 42.Byrne LJ, Sidhu A, Wallis AK, Ruddock LW, Freedman RB, Howard MJ, Williamson RA. Mapping of the ligand-binding site on the b' domain of human PDI: interaction with peptide ligands and the x-linker region. Biochem J. 2009;423:209–217. doi: 10.1042/BJ20090565. [DOI] [PubMed] [Google Scholar]
- 43.Kozlov G, Maattanen P, Schrag JD, Pollock S, Cygler M, Nagar B, Thomas DY, Gehring K. Crystal structure of the bb' domains of the protein disulfide isomerase ERp57. Structure. 2006;14:1331–1339. doi: 10.1016/j.str.2006.06.019. [DOI] [PubMed] [Google Scholar]
- •44.Kozlov G, Maattanen P, Schrag JD, Hura GL, Gabrielli L, Cygler M, Thomas DY, Gehring K. Structure of the noncatalytic domains and global fold of the protein disulfide isomerase ERp72. Structure. 2009;17:651–659. doi: 10.1016/j.str.2009.02.016. The crystal structure of ERp72, while stucturally simialr to ERp57, lacks key components in the noncatalytic b' domain necessary CNX/CRT binding, thereby explaining the structural basis for substrate specificity of these related disulfide isomerases. [DOI] [PubMed] [Google Scholar]
- 45.Kincaid MM, Cooper AA. Misfolded proteins traffic from the endoplasmic reticulum (ER) due to ER export signals. Mol Biol Cell. 2007;18:455–463. doi: 10.1091/mbc.E06-08-0696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •46.Ashok A, Hegde RS. Selective processing and metabolism of disease-causing mutant prion proteins. PLoS Pathog. 2009;5:e1000479. doi: 10.1371/journal.ppat.1000479. This study identifies clearly misfolded forms of prion protein PrP, that fail to be recognized or degraded by ERAD components, but rather exit to the Golgi and are selectively trafficked and degraded in a lysosomal compartment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xie W, Kanehara K, Sayeed A, Ng DT. Intrinsic conformational determinants signal protein misfolding to the Hrd1/Htm1 endoplasmic reticulum-associated degradation system. Mol Biol Cell. 2009;20:3317–3329. doi: 10.1091/mbc.E09-03-0231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang S, Ng DT. Evasion of endoplasmic reticulum surveillance makes Wsc1p an obligate substrate of Golgi quality control. Mol Biol Cell. 2010;21:1153–1165. doi: 10.1091/mbc.E09-10-0910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •49.Kawaguchi S, Hsu CL, Ng DT. Interplay of substrate retention and export signals in endoplasmic reticulum quality control. PLoS One. 2010;5:e15532. doi: 10.1371/journal.pone.0015532. This study shows that functional ER export signals are in kinetic competition with recognition of misfolded proeins by ERAD comopnents. The resulting substate export may protect the ER in times of severe stress by utilizing post-ERQC mechanisms that involve direct transport to lysosomes or subsequent retrograde transport to the ER and degradation by ERAD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kawahara T, Yanagi H, Yura T, Mori K. Endoplasmic reticulum stress-induced mRNA splicing permits synthesis of transcription factor Hac1p/Ern4p that activates the unfolded protein response. Mol Biol Cell. 1997;8:1845–1862. doi: 10.1091/mbc.8.10.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Patil CK, Li H, Walter P. Gcn4p and novel upstream activating sequences regulate targets of the unfolded protein response. PLoS Biol. 2004;2:E246. doi: 10.1371/journal.pbio.0020246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •52.Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. doi: 10.1016/s0092-8674(00)80835-1. Transcriptional profiling of the UPR in yeast for the first time uncovered the diverse pathways that are utilized to off-set ER stress. [DOI] [PubMed] [Google Scholar]
- 53.Schuck S, Prinz WA, Thorn KS, Voss C, Walter P. Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J Cell Biol. 2009;187:525–536. doi: 10.1083/jcb.200907074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li H, Korennykh AV, Behrman SL, Walter P. Mammalian endoplasmic reticulum stress sensor IRE1 signals by dynamic clustering. Proc Natl Acad Sci U S A. 2010;107:16113–16118. doi: 10.1073/pnas.1010580107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Korennykh AV, Egea PF, Korostelev AA, Finer-Moore J, Zhang C, Shokat KM, Stroud RM, Walter P. The unfolded protein response signals through high-order assembly of Ire1. Nature. 2009;457:687–693. doi: 10.1038/nature07661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Merksamer PI, Trusina A, Papa FR. Real-time redox measurements during endoplasmic reticulum stress reveal interlinked protein folding functions. Cell. 2008;135:933–947. doi: 10.1016/j.cell.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Haynes CM, Titus EA, Cooper AA. Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol Cell. 2004;15:767–776. doi: 10.1016/j.molcel.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 58.Babour A, Bicknell AA, Tourtellotte J, Niwa M. A surveillance pathway monitors the fitness of the endoplasmic reticulum to control its inheritance. Cell. 2010;142:256–269. doi: 10.1016/j.cell.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •59.Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Gorgun CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–1140. doi: 10.1126/science.1128294. The concept that small molecule modulators of ER stress might ameliorate a common protein misfolding disease was established using various experimental models. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Malhotra JD, Miao H, Zhang K, Wolfson A, Pennathur S, Pipe SW, Kaufman RJ. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc Natl Acad Sci U S A. 2008;105:18525–18530. doi: 10.1073/pnas.0809677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••61.Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313:104–107. doi: 10.1126/science.1129631. Although IRE1 was first thought to act as a specific splicing factor--one that activated a downstream transcriptional factor--this study established a more general role for the RNase activity of the protein: ridding the ER of messages encoding potential ER cargo. [DOI] [PubMed] [Google Scholar]
- 62.Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol. 2009;186:323–331. doi: 10.1083/jcb.200903014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Han D, Lerner AG, Vande Walle L, Upton JP, Xu W, Hagen A, Backes BJ, Oakes SA, Papa FR. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell. 2009;138:562–575. doi: 10.1016/j.cell.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 65.Lai CW, Aronson DE, Snapp EL. BiP availability distinguishes states of homeostasis and stress in the endoplasmic reticulum of living cells. Mol Biol Cell. 2010;21:1909–1921. doi: 10.1091/mbc.E09-12-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••66.Credle JJ, Finer-Moore JS, Papa FR, Stroud RM, Walter P. On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc Natl Acad Sci U S A. 2005;102:18773–18784. doi: 10.1073/pnas.0509487102. A model to describe the molecular mechanism of UPR induction became immediately evident once the structure of the lumenal domain of Ire1 revealed the potential for direct interaction with unfolded polypeptides. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhou J, Liu CY, Back SH, Clark RL, Peisach D, Xu Z, Kaufman RJ. The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proc Natl Acad Sci U S A. 2006;103:14343–14348. doi: 10.1073/pnas.0606480103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kimata Y, Ishiwata-Kimata Y, Ito T, Hirata A, Suzuki T, Oikawa D, Takeuchi M, Kohno K. Two regulatory steps of ER-stress sensor Ire1 involving its cluster formation and interaction with unfolded proteins. J Cell Biol. 2007;179:75–86. doi: 10.1083/jcb.200704166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Onn A, Ron D. Modeling the endoplasmic reticulum unfolded protein response. Nat Struct Mol Biol. 2010;17:924–925. doi: 10.1038/nsmb0810-924. [DOI] [PubMed] [Google Scholar]
- 70.Vembar SS, Brodsky JL. One step at a time: endoplasmic reticulum-associated degradation. Nat Rev Mol Cell Biol. 2008;9:944–957. doi: 10.1038/nrm2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Buchberger A, Bukau B, Sommer T. Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Mol Cell. 2010;40:238–252. doi: 10.1016/j.molcel.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 72.Hoseki J, Ushioda R, Nagata K. Mechanism and components of endoplasmic reticulum-associated degradation. J Biochem. 2010;147:19–25. doi: 10.1093/jb/mvp194. [DOI] [PubMed] [Google Scholar]
- 73.Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem. 2009;78:477–513. doi: 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •74.Vashist S, Ng DT. Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J Cell Biol. 2004;165:41–52. doi: 10.1083/jcb.200309132. The binning of ERAD substrates, and some of the first “rules” that allowed the classification of these substrates, became apparent when model secreted polypeptides were created and examined in yeast mutated for a collection of genes known to impact ERAD efficiency. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •75.Gauss R, Jarosch E, Sommer T, Hirsch C. A complex of Yos9p and the HRD ligase integrates endoplasmic reticulum quality control into the degradation machinery. Nat Cell Biol. 2006;8:849–854. doi: 10.1038/ncb1445. A separation-of-functions for the Hrd3 component of the ERAD-L machinery and its partner, the lectin, Yos9, during degradation was elucidated. [DOI] [PubMed] [Google Scholar]
- •76.Denic V, Quan EM, Weissman JS. A luminal surveillance complex that selects misfolded glycoproteins for ER-associated degradation. Cell. 2006;126:349–359. doi: 10.1016/j.cell.2006.05.045. [DOI] [PubMed] [Google Scholar]
- ••77.Carvalho P, Goder V, Rapoport TA. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell. 2006;126:361–373. doi: 10.1016/j.cell.2006.05.043. A proteomic attack was used in these papers to determine the unique components that underlie events during the degradation of ERAD-L, ERAD-M, and ERAD-C type substrates. [DOI] [PubMed] [Google Scholar]
- 78.Buck TM, Kolb AR, Boyd CR, Kleyman TR, Brodsky JL. The endoplasmic reticulum-associated degradation of the epithelial sodium channel requires a unique complement of molecular chaperones. Mol Biol Cell. 2010;21:1047–1058. doi: 10.1091/mbc.E09-11-0944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kota J, Gilstring CF, Ljungdahl PO. Membrane chaperone Shr3 assists in folding amino acid permeases preventing precocious ERAD. J Cell Biol. 2007;176:617–628. doi: 10.1083/jcb.200612100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gnann A, Riordan JR, Wolf DH. Cystic fibrosis transmembrane conductance regulator degradation depends on the lectins Htm1p/EDEM and the Cdc48 protein complex in yeast. Mol Biol Cell. 2004;15:4125–4135. doi: 10.1091/mbc.E04-01-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bernasconi R, Galli C, Calanca V, Nakajima T, Molinari M. Stringent requirement for HRD1, SEL1L, and OS-9/XTP3-B for disposal of ERAD-LS substrates. J Cell Biol. 2010;188:223–235. doi: 10.1083/jcb.200910042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ostrovsky O, Ahmed NT, Argon Y. The chaperone activity of GRP94 toward insulin-like growth factor II is necessary for the stress response to serum deprivation. Mol Biol Cell. 2009;20:1855–1864. doi: 10.1091/mbc.E08-04-0346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Okuda-Shimizu Y, Hendershot LM. Characterization of an ERAD pathway for nonglycosylated BiP substrates, which require Herp. Mol Cell. 2007;28:544–554. doi: 10.1016/j.molcel.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pearse BR, Tamura T, Sunryd JC, Grabowski GA, Kaufman RJ, Hebert DN. The role of UDP-Glc:glycoprotein glucosyltransferase 1 in the maturation of an obligate substrate prosaposin. J Cell Biol. 2010;189:829–841. doi: 10.1083/jcb.200912105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cormier JH, Tamura T, Sunryd JC, Hebert DN. EDEM1 recognition and delivery of misfolded proteins to the SEL1L-containing ERAD complex. Mol Cell. 2009;34:627–633. doi: 10.1016/j.molcel.2009.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hosokawa N, Wada I, Nagasawa K, Moriyama T, Okawa K, Nagata K. Human XTP3-B forms an endoplasmic reticulum quality control scaffold with the HRD1-SEL1L ubiquitin ligase complex and BiP. J Biol Chem. 2008;283:20914–20924. doi: 10.1074/jbc.M709336200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mueller B, Klemm EJ, Spooner E, Claessen JH, Ploegh HL. SEL1L nucleates a protein complex required for dislocation of misfolded glycoproteins. Proc Natl Acad Sci U S A. 2008;105:12325–12330. doi: 10.1073/pnas.0805371105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Maattanen P, Gehring K, Bergeron JJ, Thomas DY. Protein quality control in the ER: the recognition of misfolded proteins. Semin Cell Dev Biol. 2010;21:500–511. doi: 10.1016/j.semcdb.2010.03.006. [DOI] [PubMed] [Google Scholar]
- •89.Ushioda R, Hoseki J, Araki K, Jansen G, Thomas DY, Nagata K. ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science. 2008;321:569–572. doi: 10.1126/science.1159293. The role of this J-domain and PDI-like protein during ERAD was examined, and evidence was provided indicating a specific effect on the degradation of disulfide bonded substrates, but not on those lacking disulfide bonds. ERdj5 also interacts with several components required for ERAD, further suggesting that this protein reduces disulfide bonds prior to substrate retrotranslocation. [DOI] [PubMed] [Google Scholar]
- 90.Riemer J, Appenzeller-Herzog C, Johansson L, Bodenmiller B, Hartmann-Petersen R, Ellgaard L. A luminal flavoprotein in endoplasmic reticulum-associated degradation. Proc Natl Acad Sci U S A. 2009;106:14831–14836. doi: 10.1073/pnas.0900742106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kruse KB, Brodsky JL, McCracken AA. Characterization of an ERAD gene as VPS30/ATG6 reveals two alternative and functionally distinct protein quality control pathways: one for soluble Z variant of human alpha-1 proteinase inhibitor (A1PiZ) and another for aggregates of A1PiZ. Mol Biol Cell. 2006;17:203–212. doi: 10.1091/mbc.E04-09-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kamimoto T, Shoji S, Hidvegi T, Mizushima N, Umebayashi K, Perlmutter DH, Yoshimori T. Intracellular inclusions containing mutant alpha1-antitrypsin Z are propagated in the absence of autophagic activity. J Biol Chem. 2006;281:4467–4476. doi: 10.1074/jbc.M509409200. [DOI] [PubMed] [Google Scholar]
- 93.Ishida Y, Nagata K. Autophagy eliminates a specific species of misfolded procollagen and plays a protective role in cell survival against ER stress. Autophagy. 2009;5:1217–1219. doi: 10.4161/auto.5.8.10168. [DOI] [PubMed] [Google Scholar]
- 94.Wiertz EJ, Tortorella D, Bogyo M, Yu J, Mothes W, Jones TR, Rapoport TA, Ploegh HL. Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature. 1996;384:432–438. doi: 10.1038/384432a0. [DOI] [PubMed] [Google Scholar]
- 95.Xiong X, Chong E, Skach WR. Evidence that endoplasmic reticulum (ER)-associated degradation of cystic fibrosis transmembrane conductance regulator is linked to retrograde translocation from the ER membrane. J Biol Chem. 1999;274:2616–2624. doi: 10.1074/jbc.274.5.2616. [DOI] [PubMed] [Google Scholar]
- 96.Willer M, Forte GM, Stirling CJ. Sec61p is required for ERAD-L: genetic dissection of the translocation and ERAD-L functions of Sec61P using novel derivatives of CPY. J Biol Chem. 2008;283:33883–33888. doi: 10.1074/jbc.M803054200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Scott DC, Schekman R. Role of Sec61p in the ER-associated degradation of short-lived transmembrane proteins. J Cell Biol. 2008;181:1095–1105. doi: 10.1083/jcb.200804053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schafer A, Wolf DH. Sec61p is part of the endoplasmic reticulum-associated degradation machinery. EMBO J. 2009;28:2874–2884. doi: 10.1038/emboj.2009.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Horn SC, Hanna J, Hirsch C, Volkwein C, Schutz A, Heinemann U, Sommer T, Jarosch E. Usa1 functions as a scaffold of the HRD-ubiquitin ligase. Mol Cell. 2009;36:782–793. doi: 10.1016/j.molcel.2009.10.015. [DOI] [PubMed] [Google Scholar]
- 100.Carvalho P, Stanley AM, Rapoport TA. Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase Hrd1p. Cell. 2010;143:579–591. doi: 10.1016/j.cell.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kanehara K, Xie W, Ng DT. Modularity of the Hrd1 ERAD complex underlies its diverse client range. J Cell Biol. 2010;188:707–716. doi: 10.1083/jcb.200907055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••102.Sato BK, Schulz D, Do PH, Hampton RY. Misfolded membrane proteins are specifically recognized by the transmembrane domain of the Hrd1p ubiquitin ligase. Mol Cell. 2009;34:212–222. doi: 10.1016/j.molcel.2009.03.010. Evidence was obtained to suggest that the Hrd1 ubiquitin ligase directly recognizes misfolded, transmembrane segments in ERAD substrates. Thus, this protein functions as both a recognition element during ERAD and can ubiquitin identified substrates. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ray-Sinha A, Cross BC, Mironov A, Wiertz E, High S. Endoplasmic reticulum-associated degradation of a degron-containing polytopic membrane protein. Mol Membr Biol. 2009;26:448–464. doi: 10.3109/09687680903333839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •104.Knop M, Finger A, Braun T, Hellmuth K, Wolf DH. Der1, a novel protein specifically required for endoplasmic reticulum degradation in yeast. Embo J. 1996;15:753–763. The first gene specifically shown to be required for ERAD was isolated. Der1 is an integral membrane protein and is homologous to the mammalian Derlins, which have been suggested to act as a component of the retrotranslocon. [PMC free article] [PubMed] [Google Scholar]
- 105.Lilley BN, Ploegh HL. A membrane protein required for dislocation of misfolded proteins from the ER. Nature. 2004;429:834–840. doi: 10.1038/nature02592. [DOI] [PubMed] [Google Scholar]
- 106.Ye Y, Shibata Y, Yun C, Ron D, Rapoport TA. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature. 2004;429:841–847. doi: 10.1038/nature02656. [DOI] [PubMed] [Google Scholar]
- 107.Wahlman J, DeMartino GN, Skach WR, Bulleid NJ, Brodsky JL, Johnson AE. Real-time fluorescence detection of ERAD substrate retrotranslocation in a mammalian in vitro system. Cell. 2007;129:943–955. doi: 10.1016/j.cell.2007.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ravid T, Kreft SG, Hochstrasser M. Membrane and soluble substrates of the Doa10 ubiquitin ligase are degraded by distinct pathways. EMBO J. 2006;25:533–543. doi: 10.1038/sj.emboj.7600946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Younger JM, Chen L, Ren HY, Rosser MF, Turnbull EL, Fan CY, Patterson C, Cyr DM. Sequential quality-control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell. 2006;126:571–582. doi: 10.1016/j.cell.2006.06.041. [DOI] [PubMed] [Google Scholar]
- 110.Xu P, Duong DM, Seyfried NT, Cheng D, Xie Y, Robert J, Rush J, Hochstrasser M, Finley D, Peng J. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell. 2009;137:133–145. doi: 10.1016/j.cell.2009.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang X, Herr RA, Rabelink M, Hoeben RC, Wiertz EJ, Hansen TH. Ube2j2 ubiquitinates hydroxylated amino acids on ER-associated degradation substrates. J Cell Biol. 2009;187:655–668. doi: 10.1083/jcb.200908036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ishikura S, Weissman AM, Bonifacino JS. Serine residues in the cytosolic tail of the T-cell antigen receptor alpha-chain mediate ubiquitination and endoplasmic reticulum-associated degradation of the unassembled protein. J Biol Chem. 2010;285:23916–23924. doi: 10.1074/jbc.M110.127936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shimizu Y, Okuda-Shimizu Y, Hendershot LM. Ubiquitylation of an ERAD substrate occurs on multiple types of amino acids. Mol Cell. 2010;40:917–926. doi: 10.1016/j.molcel.2010.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Raasi S, Wolf DH. Ubiquitin receptors and ERAD: a network of pathways to the proteasome. Semin Cell Dev Biol. 2007;18:780–791. doi: 10.1016/j.semcdb.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 115.Bosis E, Salomon D, Ohayon O, Sivan G, Bar-Nun S, Rabinovich E. Ssz1 restores endoplasmic reticulum-associated protein degradation in cells expressing defective cdc48-ufd1-npl4 complex by upregulating cdc48. Genetics. 2010;184:695–706. doi: 10.1534/genetics.109.111419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Carlson EJ, Pitonzo D, Skach WR. p97 functions as an auxiliary factor to facilitate TM domain extraction during CFTR ER-associated degradation. EMBO J. 2006;25:4557–4566. doi: 10.1038/sj.emboj.7601307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lee RJ, Liu CW, Harty C, McCracken AA, Latterich M, Romisch K, DeMartino GN, Thomas PJ, Brodsky JL. Uncoupling retro-translocation and degradation in the ER-associated degradation of a soluble protein. Embo J. 2004;23:2206–2215. doi: 10.1038/sj.emboj.7600232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kothe M, Ye Y, Wagner JS, De Luca HE, Kern E, Rapoport TA, Lencer WI. Role of p97 AAA-ATPase in the retrotranslocation of the cholera toxin A1 chain, a non-ubiquitinated substrate. J Biol Chem. 2005;280:28127–28132. doi: 10.1074/jbc.M503138200. [DOI] [PubMed] [Google Scholar]
- 119.Wojcik C, Rowicka M, Kudlicki A, Nowis D, McConnell E, Kujawa M, DeMartino GN. Valosin-containing protein (p97) is a regulator of endoplasmic reticulum stress and of the degradation of N-end rule and ubiquitin-fusion degradation pathway substrates in mammalian cells. Mol Biol Cell. 2006;17:4606–4618. doi: 10.1091/mbc.E06-05-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lipson C, Alalouf G, Bajorek M, Rabinovich E, Atir-Lande A, Glickman M, Bar-Nun S. A proteasomal ATPase contributes to dislocation of endoplasmic reticulum-associated degradation (ERAD) substrates. J Biol Chem. 2008;283:7166–7175. doi: 10.1074/jbc.M705893200. [DOI] [PubMed] [Google Scholar]
- 121.Soetandyo N, Ye Y. The p97 ATPase dislocates MHC class I heavy chain in US2-expressing cells via a Ufd1-Npl4-independent mechanism. J Biol Chem. 2010;285:32352–32359. doi: 10.1074/jbc.M110.131649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lim PJ, Danner R, Liang J, Doong H, Srinivasan D, Rothenberg C, Wang H, Ye Y, Fang S, Monteiro MJ. Ubiquilin and p97/VCP bind erasin, forming a complex involved in ERAD. J Cell Biol. 2010;187:201–217. doi: 10.1083/jcb.200903024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Neuber O, Jarosch E, Volkwein C, Walter J, Sommer T. Ubx2 links the Cdc48 complex to ER-associated protein degradation. Nat Cell Biol. 2005;7:993–998. doi: 10.1038/ncb1298. [DOI] [PubMed] [Google Scholar]
- 124.Schuberth C, Buchberger A. Membrane-bound Ubx2 recruits Cdc48 to ubiquitin ligases and their substrates to ensure efficient ER-associated protein degradation. Nat Cell Biol. 2005;7:999–1006. doi: 10.1038/ncb1299. [DOI] [PubMed] [Google Scholar]
- 125.Wilson JD, Liu Y, Bentivoglio CM, Barlowe C. Sel1p/Ubx2p participates in a distinct Cdc48p-dependent endoplasmic reticulum-associated degradation pathway. Traffic. 2006;7:1213–1223. doi: 10.1111/j.1600-0854.2006.00460.x. [DOI] [PubMed] [Google Scholar]
- 126.Nakatsukasa K, Huyer G, Michaelis S, Brodsky JL. Dissecting the ER-associated degradation of a misfolded polytopic membrane protein. Cell. 2008;132:101–112. doi: 10.1016/j.cell.2007.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Garza RM, Sato BK, Hampton RY. In vitro analysis of Hrd1p-mediated retrotranslocation of its multispanning membrane substrate 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase. J Biol Chem. 2009;284:14710–14722. doi: 10.1074/jbc.M809607200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hartman IZ, Liu P, Zehmer JK, Luby-Phelps K, Jo Y, Anderson RG, DeBose-Boyd RA. Sterol-induced dislocation of 3-hydroxy-3-methylglutaryl coenzyme A reductase from endoplasmic reticulum membranes into the cytosol through a subcellular compartment resembling lipid droplets. J Biol Chem. 2010;285:19288–19298. doi: 10.1074/jbc.M110.134213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Leichner GS, Avner R, Harats D, Roitelman J. Dislocation of HMG-CoA reductase and Insig-1, two polytopic endoplasmic reticulum proteins, en route to proteasomal degradation. Mol Biol Cell. 2009;20:3330–3341. doi: 10.1091/mbc.E08-09-0953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Acharya P, Liao M, Engel JC, Correia MA. Liver cytochrome P450 3A endoplasmic reticulum-associated degradation (ERAD): A major role for the p97 AAA ATPase in cytochrome P450 3A extraction into the cytosol. J Biol Chem. 2011 doi: 10.1074/jbc.M110.186981. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •131.Kaganovich D, Kopito R, Frydman J. Misfolded proteins partition between two distinct quality control compartments. Nature. 2008;454:1088–1095. doi: 10.1038/nature07195. Different classes of misfolded proteins in the cytoplasm can reside in distinct locales, which ultimately determines their fate. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Shibatani T, Carlson EJ, Larabee F, McCormack AL, Frueh K, Skach WR. Global organization and function of mammalian cytosolic proteasome pools: implications for PA28 an 19S regulatory complexes. Mol Biol Cell. 2006;17:4962–4971. doi: 10.1091/mbc.E06-04-0311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ernst R, Mueller B, Ploegh HL, Schlieker C. The otubain YOD1 is a deubiquitinating enzyme that associates with p97 to facilitate protein dislocation from the ER. Mol Cell. 2009;36:28–38. doi: 10.1016/j.molcel.2009.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Stagg HR, Thomas M, van den Boomen D, Wiertz EJ, Drabkin HA, Gemmill RM, Lehner PJ. The TRC8 E3 ligase ubiquitinates MHC class I molecules before dislocation from the ER. J Cell Biol. 2009;186:685–692. doi: 10.1083/jcb.200906110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Loureiro J, Lilley BN, Spooner E, Noriega V, Tortorella D, Ploegh HL. Signal peptide peptidase is required for dislocation from the endoplasmic reticulum. Nature. 2006;441:894–897. doi: 10.1038/nature04830. [DOI] [PubMed] [Google Scholar]
- 136.Rutkowski DT, Hegde RS. Regulation of basal cellular physiology by the homeostatic unfolded protein response. J Cell Biol. 2010;189:783–794. doi: 10.1083/jcb.201003138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hetz C, Glimcher LH. Fine-tuning of the unfolded protein response: Assembling the IRE1alpha interactome. Mol Cell. 2009;35:551–561. doi: 10.1016/j.molcel.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Christis C, Fullaondo A, Schildknegt D, Mkrtchian S, Heck AJ, Braakman I. Regulated increase in folding capacity prevents unfolded protein stress in the ER. J Cell Sci. 2010;123:787–794. doi: 10.1242/jcs.041111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hampton RY, Garza RM. Protein quality control as a strategy for cellular regulation: lessons from ubiquitin-mediated regulation of the sterol pathway. Chem Rev. 2009;109:1561–1574. doi: 10.1021/cr800544v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Adle DJ, Wei W, Smith N, Bies JJ, Lee J. Cadmium-mediated rescue from ER-associated degradation induces expression of its exporter. Proc Natl Acad Sci U S A. 2009;106:10189–10194. doi: 10.1073/pnas.0812114106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Steffen J, Seeger M, Koch A, Kruger E. Proteasomal degradation is transcriptionally controlled by TCF11 via an ERAD-dependent feedback loop. Mol Cell. 40:147–158. doi: 10.1016/j.molcel.2010.09.012. [DOI] [PubMed] [Google Scholar]
- 142.Lord JM, Roberts LM, Lencer WI. Entry of protein toxins into mammalian cells by crossing the endoplasmic reticulum membrane: co-opting basic mechanisms of endoplasmic reticulum-associated degradation. Curr Top Microbiol Immunol. 2005;300:149–168. doi: 10.1007/3-540-28007-3_7. [DOI] [PubMed] [Google Scholar]
- 143.Cohen MM, Leboucher GP, Livnat-Levanon N, Glickman MH, Weissman AM. Ubiquitin-proteasome-dependent degradation of a mitofusin, a critical regulator of mitochondrial fusion. Mol Biol Cell. 2008;19:2457–2464. doi: 10.1091/mbc.E08-02-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Heo JM, Livnat-Levanon N, Taylor EB, Jones KT, Dephoure N, Ring J, Xie J, Brodsky JL, Madeo F, Gygi SP, Ashrafi K, Glickman MH, Rutter J. A stress-responsive system for mitochondrial protein degradation. Mol Cell. 2010;40:465–480. doi: 10.1016/j.molcel.2010.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Xu S, Peng G, Wang Y, Fang S, Karbowski M. The AAA-ATPase p97 is essential for outer mitochondrial membrane protein turnover. Mol Biol Cell. 2011;22:291–300. doi: 10.1091/mbc.E10-09-0748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Sato S, Ward CL, Krouse ME, Wine JJ, Kopito RR. Glycerol reverses the misfolding phenotype of the most common cystic fibrosis mutation. J Biol Chem. 1996;271:635–638. doi: 10.1074/jbc.271.2.635. [DOI] [PubMed] [Google Scholar]
- 147.Brown CR, Hong-Brown LQ, Biwersi J, Verkman AS, Welch WJ. Chemical chaperones correct the mutant phenotype of the delta F508 cystic fibrosis transmembrane conductance regulator protein. Cell Stress Chaperones. 1996;1:117–125. doi: 10.1379/1466-1268(1996)001<0117:ccctmp>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hammarstrom P, Wiseman RL, Powers ET, Kelly JW. Prevention of transthyretin amyloid disease by changing protein misfolding energetics. Science. 2003;299:713–716. doi: 10.1126/science.1079589. [DOI] [PubMed] [Google Scholar]
- •149.Bianchi G, Oliva L, Cascio P, Pengo N, Fontana F, Cerruti F, Orsi A, Pasqualetto E, Mezghrani A, Calbi V, et al. The proteasome load versus capacity balance determines apoptotic sensitivity of multiple myeloma cells to proteasome inhibition. Blood. 2009;113:3040–3049. doi: 10.1182/blood-2008-08-172734. An elegant study revealing that sensitivity of multiple myeloma cells to Bortezomib, a proteasome inhibitor, is directly proportional to the biosynthetic workload of the ER and proteasome. This raises the potential to predict clinical responses in specific subsets of tumors. [DOI] [PubMed] [Google Scholar]
- ••150.Lee BH, Lee MJ, Park S, Oh DC, Elsasser S, Chen PC, Gartner C, Dimova N, Hanna J, Gygi SP, et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature. 2010;467:179–184. doi: 10.1038/nature09299. This study demonstrates that a small molecule inhibitor of the proteasome-bound Dub, USP14, increases proteasome activity, thereby accelerating degradation of toxic aggregate-prone substrates that cause neurodegenerative disorders. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Gorbatyuk MS, Knox T, LaVail MM, Gorbatyuk OS, Noorwez SM, Hauswirth WW, Lin JH, Muzyczka N, Lewin AS. Restoration of visual function in P23H rhodopsin transgenic rats by gene delivery of BiP/Grp78. Proc Natl Acad Sci U S A. 2010;107:5961–5966. doi: 10.1073/pnas.0911991107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kammoun HL, Chabanon H, Hainault I, Luquet S, Magnan C, Koike T, Ferre P, Foufelle F. GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J Clin Invest. 2009;119:1201–1215. doi: 10.1172/JCI37007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Goldfarb SB, Kashlan OB, Watkins JN, Suaud L, Yan W, Kleyman TR, Rubenstein RC. Differential effects of Hsc70 and Hsp70 on the intracellular trafficking and functional expression of epithelial sodium channels. Proc Natl Acad Sci U S A. 2006;103:5817–5822. doi: 10.1073/pnas.0507903103. [DOI] [PMC free article] [PubMed] [Google Scholar]