

Preface
The mitotic checkpoint guards against chromosome missegregation and the production of aneuploid daughter cells. Aneuploidy is a common characteristic of tumor cells and has been proposed for over a century to drive tumor progression. However, recent evidence has revealed that although aneuploidy can increase the potential for cellular transformation, it also acts to antagonize tumorigenesis in certain genetic contexts. A clearer understanding of the tumor suppressive function of aneuploidy may reveal new avenues for anticancer therapy.
Introduction
Each time a cell divides it must accurately duplicate its genome and faithfully partition it into the daughter cells. If this process fails to occur accurately then the resulting daughters may inherit too many or too few chromosomes, a condition known as aneuploidy. Over a century ago the German zoologist Theodor Boveri described the effect of aneuploidy on organism development. Studying sea urchin embryos undergoing abnormal mitotic divisions, Boveri demonstrated that aneuploidy has a detrimental effect on cell and organism physiology1. Drawing on this discovery and von Hansemann's observations of abnormal mitotic figures in tumor cells2, Boveri proposed that an abnormal chromosome constitution may promote cancer3. Today it is clear that aneuploidy is a common genetic feature of solid human tumors4. However, whether aneuploidy is a cause or a consequence of malignant transformation remains hotly debated.
Part of the difficulty in studying the role of aneuploidy in cancer has stemmed from the complex and diverse array of chromosomal abnormalities found even among clinically similar tumors. Indeed, coupled with numerical changes in whole chromosomes, cancer cells also often display structural chromosomal alterations, including deletions, amplifications and translocations. Structural alterations of chromosomes are a well-established cause of cancer and thus, for the purpose of this Opinion article, we use aneuploidy to describe numerical alterations in whole chromosomes. Here, we review the pathways by which aneuploidy arises and consider the evidence suggesting a causative role for aneuploidy in the development of tumors, as well as surprising new evidence which shows aneuploidy can suppresses tumorigenesis in certain genetic contexts and cell types5.
The roads to aneuploidy
Weakening mitotic checkpoint signaling
Aneuploidy is often caused by errors in chromosome partitioning during mitosis. A surveillance mechanism known as the mitotic checkpoint (also known as spindle assembly checkpoint) is a primary guard against chromosome missegregation (Box 1)6, 7. Under normal circumstances the mitotic checkpoint delays mitotic progression in response to a single unattached kinetochore8. However, if checkpoint signaling is compromised, cells can initiate anaphase before all chromosomes have established proper spindle attachments, leading to chromosome missegregation and subsequent aneuploidy (Figure 1A). An extensive search has uncovered altered expression or mutation of mitotic checkpoint components in a subset of aneuploid human cancers including, leukemias, breast, colorectal, ovarian and lung cancer4. In addition, germline mutations in the mitotic checkpoint component BubR1 have been identified in patients suffering from the rare genetic disorder Mosaic Variegated Aneuploidy (MVA), in which as many as 25% of cells in multiple tissues are aneuploid9, 10. Nevertheless, mutated or altered expression of mitotic checkpoint genes account for, at most, a minor proportion of the aneuploidy observed in human tumors.
Box 1. The mitotic checkpoint: a safeguard to protect against aneuploidy.
The microtubule organizing center of the cell, the centrosome, is duplicated during S phase and separates at the beginning of mitosis. Microtubules nucleated by the centrosomes overlap to form a bilaterally symmetrical mitotic spindle, with each of the spindle poles organized around a single centrosome. Chromosomes attach to spindle microtubules at specialized proteinaceous structures known as kinetochores, which are assembled during mitosis upon centromeric chromatin. To ensure microtubules will pull sister chromatids to opposite sides of the cell, kinetochores of duplicated chromosomes must attach to microtubules emanating from opposite spindle poles, a state known as bi-orientation. Errors in this process lead to chromosome missegregation and the production of aneuploid daughter cells.
To guard against chromosome missegregation cells have evolved a surveillance mechanism known as the mitotic checkpoint (also known as spindle assembly checkpoint)7, which acts to delay the onset of anaphase until all chromosomes are properly attached and bi-oriented on the microtubule spindle6. In some organisms, such as yeast and flies, the mitotic checkpoint is not essential for viability64-66. In mammals, however, inactivation of the mitotic checkpoint leads to massive chromosome missegregation, cell autonomous killing and early embryonic lethality28, 30, 67-69.
Core components of the mitotic checkpoint machinery include MAD1, MAD2, BUB1, BUBR1, BUB3 and CENP-E. These proteins localize to unattached or maloriented kinetochores, which in turn catalytically generate a diffusible signal70 that inhibits CDC20 mediated activation of an E3 ubiquitin ligase, the Anpahase Promoting Complex/Cyclosome (APC/C). SEPARASE, the protease that cleaves the cohesins holding sister chromatids together, is inhibited by the binding of SECURIN or CYCLIN B71. Following attachment and alignment of all the chromosomes at metaphase, the checkpoint signal is silenced and the APC/C ubiquitylates and targets SECURIN and CYCLIN B for proteasome mediated destruction, thereby initiating anaphase. At the same time, the degradation of CYCLIN B inactivates CDK1 promoting mitotic exit.
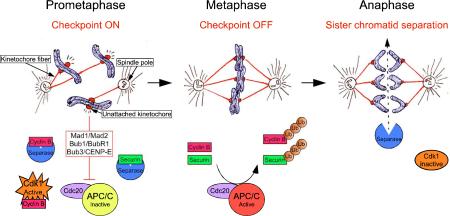
Figure 1. Pathways to aneuploidization.

There are several pathways by which cells may gain or lose chromosomes during mitosis. A. Defects in mitotic checkpoint signaling. A weakened mitotic checkpoint may allow cells to enter anaphase in the presence of unattached or misaligned chromosomes. As a consequence, both copies of one chromosome may be deposited into a single daughter cell. B. Cohesion defects. If sister chromatid cohesion is lost prematurely or persists during anaphase then chromosomes may be missegregated. C. Merotelic attachment. One kinetochore may attach to microtubules from both poles of the spindle. If these attachments persist into anaphase then lagging chromatid pairs may be missegregated or excluded from both daughter cells during cytokinesis. D. Multipolar mitotic divisions. Cells possessing more than two centrosomes may form multiple spindle poles during mitosis. If this defect is not corrected, then a multipolar division will occur resulting in the production of highly aneuploid and often inviable daughter cells. Often, however, centrosomes in multipolar spindles cluster into two groups to allow cells to divide in a bipolar fashion. Centrosome clustering will increase the formation of incorrect kinetocore-microtubule attachments (such as merotelic attachments). Extra centrosomes are thus, capable of driving low rates of chromosome missegregation through a mechanism independent of multipolar divisions.
Defects in chromosome cohesion or attachment
To identify other mechanisms leading to aneuploidization, genes with putative functions in guarding against chromosome missegregation were systematically sequenced in a panel of aneuploid colorectal cancers11. Surprisingly, 10 of the 11 mutations identified were in genes that directly contribute to sister chromatid cohesion, indicating that defects in the machinery-controlling sister chromatid cohesion may promote aneuploidy (Figure 1B). Consistently, overexpression of SEPARSE or SECURIN, two key regulators controlling the loss of chromatid cohesion, promotes aneuploidy and cellular transformation12-15. Chromosome missegregation may also arise from the improper attachment of kinetochores to spindle microtubules. This can occur when a single kinetochore attaches to microtubules emanating from both poles of the spindle, a situation known as merotelic attachment (Figure 1C)16. Since merotelically oriented kinetochores are attached and under tension, their presence does not continue to activate mitotic checkpoint signaling. Although merotelic attachments are usually corrected before entry into anaphase17, if they persist both sister chromatids may be missegreagted towards the same pole (called chromosome non-disjunction) or lagging chromosomes may be left in the spindle midzone and excluded from both daughter nuclei18, 19.
Multipolar spindles
A final source of aneuploidy arises when a cell containing more than two centrosomes enters mitosis (Figure 2A). The centrosome forms the poles of the mitotic spindle and cells possessing extra centrosomes can form multipolar spindles. If not corrected, a multipolar anaphase can produce three or more highly aneuploid daughter cells that are likely to be inviable (Figure 1D). Multipolar mitotic divisions are rare because in most cases extra centrosomes are clustered into two groups allowing bipolar spindles to form20, 21 ((Box 2). While the progeny of these divisions are likely to survive, the passage through a multipolar intermediate prior to centrosomal clustering will increase the frequency of improper kinetocore-microtubule attachments, including merotelic orientations (Figure 1D), thereby promoting chromosome missegregation through a mechanism independent of multipolar divisions.
Figure 2. Pathways to acquiring extra centrosomes.

The centrosome consists of a pair of connected centrioles surrounded by the pericentriolar material (PCM). There are two major mechanisms by which cells can gain extra centrosomes. A. Centrosome amplification. Defects in the processes controlling centriole replication can lead to centriole overduplication, which results in multiple centrosomes in the next cell cycle. This process can occur when PLK4, a regulator of centriole biogenesis, is overexpressed72, 77. Impairment of centrosome structure can cause fragmentation of the pericentriolar material. The acentriolar fragments can then serve to nucleate microtubules and create multipolar spindles. This has been found to occur following cellular infection with the human T cell lymphotrophic virus Type 1 (HTLV-1)78. Finally, defects in centriole cohesion can lead to the separation of paired centrioles before the completion of chromosome segregation, creating multiple microtubule nucleating foci. Cells with reduced levels of sSgo1 have been shown to loose centriole cohesion prematurely79. B. Cells become tetraploid. This can occur following cell-cell fusion or after cytokinesis failure. Alternatively, cells may skip mitosis altogether and endoreduplicate or “slip” out of mitosis and progress into the next cell cycle without undergoing anaphase or cytokinesis. In all these situations G1 tetraploid cells are created with two centrosomes that are duplicated during the next cell cycle.
Box 2. Centrosome amplification in cancer.
In addition to numerical alterations in chromosomes, cancer cells frequently exhibit an amplification of centrosome number21. Extra centrosomes can lead to the formation of multiple spindle poles during mitosis resulting in the unequal distribution of chromosomes and the production of aneuploid daughter cells. This has led to the proposal that centrosome amplification may be a driving force behind genomic instability and tumorigenesis3. A direct test of the role of centrosome amplification in cancer was recently performed in the fly72. Remarkably, flies possessing extra centrosomes in ~60% of somatic cells were overtly normal and exhibited no dramatic increase in genetic instability. Nevertheless, larval brain cells with extra centrosomes generated metastatic tumors when transplanted into the abdomen of host flies, demonstrating that centrosome amplification can initiate tumorigenesis72.
The observation that cells from flies and human cancers proliferate normally in the presence of extra centrosomes is consistent with previous studies indicating cells have evolved pathways to minimize the damaging effect of centrosome amplification20. At least three mechanisms are known to exist. First, centrosomes are clustered into two groups to allow division to occur in a bipolar fashion73-75. Second, centrosomes are inactivated such that they no longer nucleate microtubules and participate in spindle formation72. Finally, the mitotic checkpoint is activated by the unstable/incorrect microtubule attachments formed in multipolar mitotic spindles72, 73, 76. This acts to delay cells in mitosis and provide additional time to cluster and inactivate centrosomes to enable a bipolar spindle to form. Recently, it has been shown that blocking processes that suppress the formation of multipolar spindles can selectively kill cells with amplified centrosomes, providing a possible therapeutic avenue for the treatment of cancer cells with supernumerary centrosomes76.
Aneuploidy and CIN
Some tumor cells are stably aneuploid, reflecting a transient chromosome missegregation event at some point in the development of the tumor leading to an abnormal karyotype that is stably propagated and inherited. More often, however, aneuploidy is a result of an underlying chromosomal instability (CIN), characterized by an increase in the rate of gains and losses of whole chromosomes during division22. Aneuploidy and CIN are not synonymous: while aneuploidy describes the state of having an abnormal chromosome number, CIN refers to an elevated rate of chromosome gain or loss. This is exemplified in Down's syndrome, a condition associated with widespread aneuploidy but not CIN.
For more than a decade the molecular mechanisms underlying CIN have remained unclear. Cells possessing CIN were originally reported to have an impaired ability to sustain a mitotic arrest in response to spindle toxins23, leading to wide spread acceptance of the proposal that an attenuated mitotic checkpoint may be the primary cause of CIN6. This view is probably wrong. Direct measurements using live cell imaging to visualize mitosis have revealed that in response to spindle toxins, the duration of mitosis in CIN cells is at least as long as in chromosomally stable diploid cells24. Moreover, CIN cells were not found to enter anaphase in the presence of misaligned chromosomes, demonstrating that at least in these cells, mitotic checkpoint dysfunction is not a primary cause of CIN24, 25. Although CIN cells did not enter anaphase precociously, they did exhibit an increase in the incidence of lagging anaphase chromosomes, caused at least in part by unresolved merotelic attachments.
The underlying cause of increased malorientations in CIN cells has not been determined, but could be caused by an acquired defect in resolving merotelic orientations prior to anaphase or in spindle assembly. For example, the clustering of centrosomes in a multipolar mitotic spindle may increase the number of kinetochore mal-orientations ((Box 2 and Figure 1D). Recently, it was shown that reductions in kinetochore-microtubule turnover in early mitosis increase the frequency of kinetochore mal-orientations and chromosome missegregation26. Remarkably, a modest increase in expression of either of a pair of kinetochore localized microtubule-depolymerizing enzymes substantially reduced the occurrence of lagging anaphase chromosomes and chromosome missegregation in CIN cells, suggesting a causal relationship between kinetochore-microtubule dynamics and CIN26.
Aneuploidy facilitates tumor formation
The role of aneuploidy in tumorigenesis has been extensively studied in mouse models of mitotic checkpoint dysfunction. So far, conventional gene knockouts have been constructed for almost all known mitotic checkpoint genes, including Mad1, Mad2, Bub1, Bub3, BubR1, Rae1 and CENP-E27-33. In addition, hypomorphic alleles expressing dramatically reduced levels of BUB1 and BUBR1 have also been generated34, 35. While complete loss of these gene products results in early embryonic lethality, heterozygous and hypomorphic mice are viable and fertile. In all cases, mice with genetically reduced levels of mitotic checkpoint components display an increased level of aneuploidy and CIN in mouse embryonic fibroblasts (MEFs) and tissues27-30, 32, 34-37. However, the degree of aneuploidy, including the proportion of aneuploid cells and the spectrum of chromosome losses and gains, varies depending on which gene product has been reduced and to what level (Table 1).
Table I.
Context dependent roles of aneuploidy in tumorigenesis
| Mouse Genotype | % Aneuploidy | Tumorigenesis | Comments/Ref | ||
|---|---|---|---|---|---|
| MEFs (Cont) | Spleen (Cont) | Spontaneous | Carcinogen-induced | ||
|
Contexts in which aneuploidy promotes tumorigenesis | |||||
| Aneuploid mouse models showing an increase in spontaneous tumorigenesis | |||||
| Mad1+/- | ND | ND | 24% develop tumors | ND | 27 |
| Mad2+/- | 57(16) | ND | 28% develop lung tumors | ND | 28 |
| Bub1H/H | 35(7) | 35(1) | 48% develop lethal tumors | ND | 34 |
| CENP-E+/- | 36(18) | 35(10) | 20% develop spleen and lung tumors | See Below | 38 |
| BubR1+/-;APCMin/+ | 65(14) | ND | BubR1+/-;APCMin/+ show a 10 fold increase in colon tumors relative to APCMin/+ | ND | 37 |
| BubR1H/H;p16-/- | ND | ND | BubR1H/H;p16-/- show increased lung tumors relative to p16-/- | ND | 44 |
| Aneuploid mouse models showing an increase in carcinogen induced tumors | |||||
| Bub1+/- | 14(7) | 16(1) | Normal | DMBA-induced | 35 |
| Bub3+/- | 19(9) | 9(0) | Normal | DMBA-induced | 29 |
| 42(35) | ND | Normal | ND | 39 | |
| Rae1+/- | 19(9) | 9(0) | Normal | DMBA-induced | 29 |
| Bub3+/-;Rae1+/- | 41(9) | 37(0) | Normal | DMBA-induced | Premature aging29, 40 |
| Rae1+/-;Nup98+/- | 37(9) | 32(0) | Normal | DMBA-induced | 42, 43 |
| BubR1+/- | 14(9) | 0(0) | Normal | ND | 34, 40, 33, 37 |
| 61(14) | ND | ND | Azoxymethane-induced | ||
| BubR1H/H | 36(9) | 15(0) | Normal | DMBA-induced | Premature aging38 |
| Aneuploid mouse models with overexpressed mitotic components | |||||
| Mad2 OE | 53(5) | ND | 50% develop tumors | ND | Increase in structural chromosomal alterations and the proportion of tetraploid cells46 |
| Hec1 OE | 31(16) | ND | 40% develop tumors | ND | 45 |
| Aurora A OE | ND | ND | 40% developed mammary tumors | ND | Aurora A is overexpressed specifically in the mammary gland80 |
|
Contexts in which aneuploidy suppresses tumorigenesis | |||||
| CENP-E+/- | 36(18) | 35(10) | 50% decrease in Liver tumors | DMBA-reduced | 38 |
| CENP-E+/;p19ARF-/- | ND | ND | CENP-E+/;p19ARF-/- have a 93 day increase in survival relative to p19ARF-/- | ND | 38 |
| BubR1+/-;APCMin/+ | 65(14) | ND | BubR1+/-;APCMin/+ show a 50% decrease in small intestine tumors relative to APCMin/+ | ND | 37 |
| Ts65Dn;APCMin/+ | 100%* | 100%* | Ts65Dn;APCMin/+ show a 44% decrease in small intestine tumors relative to APCMin/+ | ND | *Ts65Dn mice are trismoic for ~50% of the orthologous genes on human chr2156 |
| Pttg-/-;Rb+/- | ND | ND | Pttg-/-;Rb+/- mice have a 56% decrease in pituitary tumors relative to Rb+/- | ND | 55 |
H = hypomophic allele; - = null allele; + = wild type allele; Cont = Control; OE = over-expression; ND = Not determined; DMBA = 7,12-Dimethylbenz[α]anthracene
Downregulation of mitotic checkpoint components
In some instances, reduced expression of mitotic checkpoint components is associated with an increase in spontaneous cancer (Table 1). Specifically, Mad1 and Mad2 heterozygous mice develop lung tumors while CENP-E heterozygous animals show an increased incidence of lung tumors and splenic lymphomas27, 28, 38. The cancers formed in these animals are benign and occur late in life (<18 months), suggesting the acquisition of a transformed karyotype is a rare event that requires many consecutive generations of chromosome missegregation. In contrast, Bub1 hypomorphic mice develop a wide array of lethal cancers including, lymphomas, lung and liver tumors35. Nevertheless, in all situations where aneuploidy has been found to promote spontaneous tumorigenesis, tumors form in only a fraction of animals (Table 1), suggesting that the transformation of aneuploid cells relies upon the chance acquisition of additional, cooperating mutations in regulatory genes.
Several mitotic checkpoint deficient mice display a significantly elevated level of aneuploidy without an increase in spontaneous tumorigenesis, demonstrating that cancer is not an inescapable fate of aneuploidy29, 34, 35, 39-42. Indeed, to date there is no direct correlation between the level of aneuploidy and the incidence of spontaneous tumor development. Indeed, Bub3;Rae1 and Rae1;Nup98 compound heterozygotes possess similar levels of aneuploidy to Bub1 hypomorphic mice; however, unlike Bub1 hypomoprhs, Bub3;Rae1 and Rae1;Nup98 exhibit no increase in spontaneous tumor development (Table 1)35, 40, 42, 43.
It remains unclear why a reduction in some mitotic checkpoint components drives spontaneous tumorigenesis while others do not. One possibility is that in addition to guarding against aneuploidy, some mitotic checkpoint proteins have other tumor suppressive roles. For example, BUB1 has recently been proposed to play a role in eliminating aneuploid cells from the population, which may explain the high tumor susceptibility of Bub1 hypomorphic mice35. Alternatively, loss of different mitotic checkpoint components may give rise to distinct types of aneuploidy that could have different effects on tumorigenesis. For instance, aneuploid splenocytes from mice with reduced levels of BUB1, BUBR1, BUB3 and RAE1 show both gain and losses of whole chromosomes29, 34, 35, while CENP-E heterozygous animals show almost exclusive chromosome loss38.
Although aneuploid Bub1, BubR1, Bub3, Rae1 and Rae1;Nup98 heterozygous animals fail to display an increase in spontaneous tumorigenesis, these mice are prone to carcinogen-induced tumors (Table 1)29, 35, 41. This suggests that in these mouse models aneuploidy does not initiate cancer, but rather drives tumor formation in cases where mutations at oncogenic or tumor suppressor loci have already increased the potential for cellular transformation. Consistently, mutations in some tumor suppressor genes cooperate with aneuploidy to promote tumor progression. For example, reduced levels of BUBR1 promote an increase in lung tumors in mice lacking the p16 tumor suppressor44 and a 10-fold increase in colon tumors in sensitized APCMin/+ animals37 (APCMin/+ mice carry a heterozygous truncating mutation in the APC tumor suppressor resulting in the development of benign colon and intestinal tumors at an early age). Together, these data suggest that the mutations that co-operate with aneuploidy to promote tumor formation do not occur at a significantly frequency during the lifetime of laboratory mice.
Upregulation of mitotic checkpoint components
Paradoxically, inactivating mutations in mitotic checkpoint genes are rarely observed in human cancer, however, abnormally high expression is much more frequent4. Indeed, overexpression of MAD2 and the kinetochore component HEC1 are common in human tumors and elevated levels of these proteins are often associated with a poor prognosis. Increased expression of HEC1 has been shown to drive aneuploidy and an elevation in spontaneous lung and liver tumors in mice45. In addition, conditional overexpression of MAD2 predisposes animals to a wide spectrum of early-onset, lethal tumors46. Importantly, continued tumor growth does not remain dependent on expression of the MAD2 transgene, suggesting that once neoplastic transformation has occurred, excessive MAD2 is not required for tumor maintenance. Surprisingly, MAD2 transgenic mice are considerably more tumor prone than mice with reduced levels of MAD228, 46. However, in addition to rampant aneuploidy, mice overexpressing MAD2 also show large-scale structural defects, including chromosomal breaks, fusions amplifications and interstitial deletions. Thus, it remains unclear whether it is aneuploidy or structural defects that are the primary cause of tumorigenesis in these animals.
Taken together, mouse models have unequivocally demonstrated that aneuploidy is capable of increasing the risk of neoplastic transformation, albeit aneuploidy per se acts to facilitate the development of tumors in a predisposed background. How aneuploidy does this remains unclear. One possibility is that aneuploidy may result in the duplication of a chromosome containing an oncogenic allele or the loss of a chromosome possessing the remaining wild type copy of a tumor suppressor gene (known as loss of heterozgozity (LOH)). Consistent with this hypothesis, aneuploidy caused by haploinsufficiency of Mad2 or Mad1;Mad2 has been shown to increase both the frequency and number of tumors in a p53+/- backgound47. By contrast, however, Bub3 haploinsufficiency did not alter the rate or frequency of tumorigenesis in p53 or Rb1 heterozygous mice39. While these studies appear contradictory, it is notable that the incidence of aneuploidy is considerably higher in Mad2+/- compared with Bub3+/- MEFs (Table 1). This suggests the difference in tumor susceptibility may be a result of a higher level of LOH in Mad2 haploinsufficient mice.
An alternative explanation for aneuploidy's tumor promoting activity is that additional chromosomes help protect aneuploid cells against the effect of deleterious mutations in essential and haplo-insufficient genes. Aneuploidy may, therefore, allow cells to survive longer in the presence of ongoing DNA damage, allowing more time for cells to accumulate critical growth promoting and transforming mutations. Identifying the lesions that cooperate with aneuploidy to promote cellular transformation will be an important area for future research.
Doubling up: tetraploidy and cancer
While some aneuploid human cancers have minor imbalances in chromosome numbers, a substantial number also exhibit large-scale aneuploidy, often containing a near tetraploid number of chromosomes4. Tetraploidy can arise through a number of mechanisms, including cell fusion, mitotic slippage and cytokinesis failure (Figure 2A). In addition to a doubling of the chromosome content, tetraploid cells typically contain twice the normal complement of centrosomes. Supernumerary centrosomes promote aberrant mitotic divisions and chromosome missegregation at a high frequency and thus, tetraploidy is an inherently unstable state that acts as a catalyst to promote further aneuploidy and genomic instability ((Box 2)48. Indeed, tetraploidy has been shown to initiate CIN and has been found to precede the development of CIN and aneuploidy in several cancers49-51.
There is now compelling evidence to suggest that the uncontrolled proliferation of tetraploid cells can trigger cellular transformation and tumor formation. The most direct evidence came from the observation that tetraploid p53-/- mouse cells initiate tumor formation when transplanted into immunocompromised mice, while isogenic diploid cells did not48. Importantly, the tetraploid-derived tumors displayed large-scale numerical and structural chromosomal aberrations, demonstrating that tetraploidy can initiate massive genomic instability. Interestingly, cells derived from mice overexpressing MAD2 have a substantial increase in the number of tetraploid cells, which may explain the increase in structural chromosome aberrations and high tumor susceptibility of these animals46.
Consistent with a causative role for tetraploidy in cancer, several established oncogenes and tumor suppressor genes have also been shown to induce tetraploidization. For instance, AURORA A is frequently overexpressed in human cancers and increased levels have been shown to cause cytokinesis failure52. Overexpression of AURORA A in the mammary gland of mice leads to an increase in tetraploidization, CIN and the formation of mammary tumors53. In addition, mutations in the APC tumor suppressor are commonly found in human colon cancers and have been shown to cause cytokinesis failure and tetraploidization in mice54.
Aneuploidy can act as a tumor suppressor
Although aneuploidy has long been implicated in driving cancer, in certain cases aneuploidy can suppress tumorigenesis (Table 1). CENP-E haplo-insufficiency reduces the incidence of carcinogen-induced tumors and dramatically extends the survival of mice lacking the p19ARF tumor suppressor38. Moreover, mice heterozygous for BubR1 develop ~50% fewer tumors in the sensitized APCMin/+ background37, while deletion of Securin reduces the incidence of pituitary tumors by ~50% in Rb heterozygous animals55 (admittedly, in this last case it remains unclear if tumor suppression results from increased levels of aneuploidy).
Tumor repression has also been observed in stably aneuploid mice that are trisomic for ~50% of the orthologue genes on human chromosome 2156. One explanation for these observations is that exposure to carcinogens or loss of tumor suppressor function results in low levels of genetic damage and/or chromosome missegregation, which when combined with aneuploidy, drive rates of genetic instability above a threshold compatible with cell viability. Consistently, p19ARF-/- and carcinogen treated MEFs exhibit a level of aneuploidy that is exacerbated by CENP-E haploinsufficiency5. Moreover, aneuploidy and apoptosis is also increased in the intestines of BubR1+/-; APCMin/+ mice, providing evidence that too much aneuploidy may promote cell death and inhibit tumor growth37.
The Yin and Yang of aneuploidy in tumorigenesis
Unlike point mutations that only affect a small number of genes, the gain or loss of a single chromosome alters the transcription of hundreds of genes and has the capacity to disturb a large array of cellular processes. This imbalance imparts a stress that can hamper the growth of aneuploid cells. Indeed, yeast strains containing one or more additional chromosomes grow more slowly than their haploid counterparts. Moreover, mouse cells engineered to be trisomic for specific chromosomes exhibit a proliferation delay, as do human fibroblasts derived from individuals with Down's syndrome57-59. Consistently, when aneuploidy is introduced into a normally diploid cancer cell line, the aneuploid cells are outcompeted by diploid cells25. Thus, under normal circumstances, aneuploidy may act as a barrier to suppress tumorigenesis by reducing the growth of preneoplastic cells.
If the majority of the karyotpes generated by random chromosome missegregation will confer a growth disadvantage to cells or cause lethality, how then can aneuploidy promote tumorigenesis in some contexts? One interesting possibility proposed by Amon and coworkers is that aneuploidy provides a selective pressure for the accumulation of additional mutations that allow cells to tolerate the adverse effects of chromosomal imbalances60. The unbalanced gene expression caused by aneuploidy may increase the rate at which cells acquire the mutations necessary for their survival and proliferation. Once gained, these adaptations would unlock the oncogenic potential of aneuploidy, allowing cells to survive and continue to proliferate in the face of increased genomic instability.
Conclusions: context matters
A century after Boveri initially proposed aneuploidy to drive tumorigenesis an overriding message is now clear: aneuploidy is able to alter the course of tumor development. However, whether it does so in a positive or negative manner depends upon the cell type and the genetic context. For example, while mice heterozygous for CENP-E exhibit an increase in the rate of spontaneous lung and spleen tumors, these animals demonstrate a decreased incidence of liver tumors38. Moreover, Down's syndrome patients carrying an extra copy of chromosome 21 have a significant increase in hematological cancers, but a reduced incidence of solid tumors61-63. Therefore, the effect of aneuploidy may not be driven by a particular combination of chromosomes per se, but rather by the specific interaction of the karyotype with the various genetic contexts and microenvironments found in different tissues. This explains why some tissues, such as lung epithelial cells, seem to have a higher propensity for malignant progression in aneuploid mice (Table 1). A clear goal for the future is to establish under which genetic contexts and cell types aneuploidy promotes or suppresses tumorigenesis.
Moreover, while current mouse modeling has predominantly focused on deregulation of mitotic checkpoint genes as a course for driving aneuploidy in vivo, checkpoint dysfunction does not appear to be a primary cause of CIN in human cancers. Therefore, new models of chromosomal instability that faithfully mimic the lesions and pathways frequently deregulated in aneuploid cancer cells are needed, especially models that can drive inducible or transient chromosomal instability, whose use may reveal novel therapeutic avenues to exploit the tumor suppressive effect of aneuploidy.
References
- 1.Boveri T. Ueber mehrpolige Mitosen als Mittel zur Analyse des Zellkerns. Verh Phys-med Ges Würzburg NF. 1902;35:67–90. [Google Scholar]
- 2.Hansemann D. Ueber asymmetrische Zelltheilung in Epithelkrebsen und deren biologische Bedeutung. Arch Pathol Anat Physiol Klin Medicin. 1980;119:299–326. [Google Scholar]
- 3.Boveri T. Zur Frage der Entstehung maligner Tumoren. Fischer, Jena. 1914 [Google Scholar]
- 4.Weaver BA, Cleveland DW. Does aneuploidy cause cancer? Curr Opin Cell Biol. 2006;18:658–67. doi: 10.1016/j.ceb.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 5.Weaver BA, Cleveland DW. Aneuploidy: instigator and inhibitor of tumorigenesis. Cancer Res. 2007;67:10103–5. doi: 10.1158/0008-5472.CAN-07-2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kops GJ, Weaver BA, Cleveland DW. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat Rev Cancer. 2005;5:773–85. doi: 10.1038/nrc1714. [DOI] [PubMed] [Google Scholar]
- 7.Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol. 2007;8:379–93. doi: 10.1038/nrm2163. [DOI] [PubMed] [Google Scholar]
- 8.Rieder CL, Cole RW, Khodjakov A, Sluder G. The checkpoint delaying anaphase in response to chromosome monoorientation is mediated by an inhibitory signal produced by unattached kinetochores. J Cell Biol. 1995;130:941–8. doi: 10.1083/jcb.130.4.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanks S, et al. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat Genet. 2004;36:1159–61. doi: 10.1038/ng1449. [DOI] [PubMed] [Google Scholar]
- 10.Matsuura S, et al. Monoallelic BUB1B mutations and defective mitotic-spindle checkpoint in seven families with premature chromatid separation (PCS) syndrome. Am J Med Genet A. 2006;140:358–67. doi: 10.1002/ajmg.a.31069. [DOI] [PubMed] [Google Scholar]
- 11.Barber TD, et al. Chromatid cohesion defects may underlie chromosome instability in human colorectal cancers. Proc Natl Acad Sci U S A. 2008;105:3443–8. doi: 10.1073/pnas.0712384105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang N, et al. Overexpression of Separase induces aneuploidy and mammary tumorigenesis. Proc Natl Acad Sci U S A. 2008;105:13033–8. doi: 10.1073/pnas.0801610105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pei L, Melmed S. Isolation and characterization of a pituitary tumor-transforming gene (PTTG). Mol Endocrinol. 1997;11:433–41. doi: 10.1210/mend.11.4.9911. [DOI] [PubMed] [Google Scholar]
- 14.Yu R, Lu W, Chen J, McCabe CJ, Melmed S. Overexpressed pituitary tumor-transforming gene causes aneuploidy in live human cells. Endocrinology. 2003;144:4991–8. doi: 10.1210/en.2003-0305. [DOI] [PubMed] [Google Scholar]
- 15.Zhang X, et al. Structure, expression, and function of human pituitary tumor-transforming gene (PTTG). Mol Endocrinol. 1999;13:156–66. doi: 10.1210/mend.13.1.0225. [DOI] [PubMed] [Google Scholar]
- 16.Cimini D. Merotelic kinetochore orientation, aneuploidy, and cancer. Biochim Biophys Acta. 2008;1786:32–40. doi: 10.1016/j.bbcan.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 17.Cimini D, Moree B, Canman JC, Salmon ED. Merotelic kinetochore orientation occurs frequently during early mitosis in mammalian tissue cells and error correction is achieved by two different mechanisms. J Cell Sci. 2003;116:4213–25. doi: 10.1242/jcs.00716. [DOI] [PubMed] [Google Scholar]
- 18.Cimini D, Fioravanti D, Salmon ED, Degrassi F. Merotelic kinetochore orientation versus chromosome mono-orientation in the origin of lagging chromosomes in human primary cells. J Cell Sci. 2002;115:507–15. doi: 10.1242/jcs.115.3.507. [DOI] [PubMed] [Google Scholar]
- 19.Cimini D, et al. Merotelic kinetochore orientation is a major mechanism of aneuploidy in mitotic mammalian tissue cells. J Cell Biol. 2001;153:517–27. doi: 10.1083/jcb.153.3.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brinkley BR. Managing the centrosome numbers game: from chaos to stability in cancer cell division. Trends Cell Biol. 2001;11:18–21. doi: 10.1016/s0962-8924(00)01872-9. [DOI] [PubMed] [Google Scholar]
- 21.Nigg EA. Origins and consequences of centrosome aberrations in human cancers. Int J Cancer. 2006;119:2717–23. doi: 10.1002/ijc.22245. [DOI] [PubMed] [Google Scholar]
- 22.Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997;386:623–7. doi: 10.1038/386623a0. [DOI] [PubMed] [Google Scholar]
- 23.Cahill DP, et al. Mutations of mitotic checkpoint genes in human cancers. Nature. 1998;392:300–3. doi: 10.1038/32688. [DOI] [PubMed] [Google Scholar]
- 24.Gascoigne KE, Taylor SS. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell. 2008;14:111–22. doi: 10.1016/j.ccr.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 25.Thompson SL, Compton DA. Examining the link between chromosomal instability and aneuploidy in human cells. J Cell Biol. 2008;180:665–72. doi: 10.1083/jcb.200712029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bakhoum SF, Thompson SL, Manning AL, Compton DA. Genome stability is ensured by temporal control of kinetochore-microtubule dynamics. Nat Cell Biol. 2008 doi: 10.1038/ncb1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iwanaga Y, et al. Heterozygous deletion of mitotic arrest-deficient protein 1 (MAD1) increases the incidence of tumors in mice. Cancer Res. 2007;67:160–6. doi: 10.1158/0008-5472.CAN-06-3326. [DOI] [PubMed] [Google Scholar]
- 28.Michel LS, et al. MAD2 haplo-insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature. 2001;409:355–9. doi: 10.1038/35053094. [DOI] [PubMed] [Google Scholar]
- 29.Babu JR, et al. Rae1 is an essential mitotic checkpoint regulator that cooperates with Bub3 to prevent chromosome missegregation. J Cell Biol. 2003;160:341–53. doi: 10.1083/jcb.200211048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kalitsis P, Earle E, Fowler KJ, Choo KH. Bub3 gene disruption in mice reveals essential mitotic spindle checkpoint function during early embryogenesis. Genes Dev. 2000;14:2277–82. doi: 10.1101/gad.827500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perera D, et al. Bub1 maintains centromeric cohesion by activation of the spindle checkpoint. Dev Cell. 2007;13:566–79. doi: 10.1016/j.devcel.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 32.Putkey FR, et al. Unstable kinetochore-microtubule capture and chromosomal instability following deletion of CENP-E. Dev Cell. 2002;3:351–65. doi: 10.1016/s1534-5807(02)00255-1. [DOI] [PubMed] [Google Scholar]
- 33.Wang Q, et al. BUBR1 deficiency results in abnormal megakaryopoiesis. Blood. 2004;103:1278–85. doi: 10.1182/blood-2003-06-2158. [DOI] [PubMed] [Google Scholar]
- 34.Baker DJ, et al. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet. 2004;36:744–9. doi: 10.1038/ng1382. [DOI] [PubMed] [Google Scholar]
- 35.Jeganathan K, Malureanu L, Baker DJ, Abraham SC, van Deursen JM. Bub1 mediates cell death in response to chromosome missegregation and acts to suppress spontaneous tumorigenesis. J Cell Biol. 2007;179:255–67. doi: 10.1083/jcb.200706015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weaver BA, et al. Centromere-associated protein-E is essential for the mammalian mitotic checkpoint to prevent aneuploidy due to single chromosome loss. J Cell Biol. 2003;162:551–63. doi: 10.1083/jcb.200303167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rao CV, et al. Colonic tumorigenesis in BubR1+/-ApcMin/+ compound mutant mice is linked to premature separation of sister chromatids and enhanced genomic instability. Proc Natl Acad Sci U S A. 2005;102:4365–70. doi: 10.1073/pnas.0407822102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weaver BA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell. 2007;11:25–36. doi: 10.1016/j.ccr.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 39.Kalitsis P, et al. Increased chromosome instability but not cancer predisposition in haploinsufficient Bub3 mice. Genes Chromosomes Cancer. 2005;44:29–36. doi: 10.1002/gcc.20215. [DOI] [PubMed] [Google Scholar]
- 40.Baker DJ, et al. Early aging-associated phenotypes in Bub3/Rae1 haploinsufficient mice. J Cell Biol. 2006;172:529–40. doi: 10.1083/jcb.200507081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dai W, et al. Slippage of mitotic arrest and enhanced tumor development in mice with BubR1 haploinsufficiency. Cancer Res. 2004;64:440–5. doi: 10.1158/0008-5472.can-03-3119. [DOI] [PubMed] [Google Scholar]
- 42.Jeganathan KB, Malureanu L, van Deursen JM. The Rae1-Nup98 complex prevents aneuploidy by inhibiting securin degradation. Nature. 2005;438:1036–9. doi: 10.1038/nature04221. [DOI] [PubMed] [Google Scholar]
- 43.Jeganathan KB, Baker DJ, van Deursen JM. Securin associates with APCCdh1 in prometaphase but its destruction is delayed by Rae1 and Nup98 until the metaphase/anaphase transition. Cell Cycle. 2006;5:366–70. doi: 10.4161/cc.5.4.2483. [DOI] [PubMed] [Google Scholar]
- 44.Baker DJ, et al. Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nat Cell Biol. 2008;10:825–36. doi: 10.1038/ncb1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Diaz-Rodriguez E, Sotillo R, Schvartzman JM, Benezra R. Hec1 overexpression hyperactivates the mitotic checkpoint and induces tumor formation in vivo. Proc Natl Acad Sci U S A. 2008;105:16719–24. doi: 10.1073/pnas.0803504105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sotillo R, et al. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell. 2007;11:9–23. doi: 10.1016/j.ccr.2006.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chi YH, Ward JM, Cheng LI, Yasunaga J, Jeang KT. Spindle assembly checkpoint and p53 deficiencies cooperate for tumorigenesis in mice. Int J Cancer. 2008 doi: 10.1002/ijc.24094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fujiwara T, et al. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature. 2005;437:1043–7. doi: 10.1038/nature04217. [DOI] [PubMed] [Google Scholar]
- 49.Galipeau PC, et al. 17p (p53) allelic losses, 4N (G2/tetraploid) populations, and progression to aneuploidy in Barrett's esophagus. Proc Natl Acad Sci U S A. 1996;93:7081–4. doi: 10.1073/pnas.93.14.7081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Olaharski AJ, et al. Tetraploidy and chromosomal instability are early events during cervical carcinogenesis. Carcinogenesis. 2006;27:337–43. doi: 10.1093/carcin/bgi218. [DOI] [PubMed] [Google Scholar]
- 51.Ornitz DM, Hammer RE, Messing A, Palmiter RD, Brinster RL. Pancreatic neoplasia induced by SV40 T-antigen expression in acinar cells of transgenic mice. Science. 1987;238:188–93. doi: 10.1126/science.2821617. [DOI] [PubMed] [Google Scholar]
- 52.Meraldi P, Honda R, Nigg EA. Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53-/-cells. EMBO J. 2002;21:483–92. doi: 10.1093/emboj/21.4.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang X, et al. Overexpression of aurora kinase A in mouse mammary epithelium induces genetic instability preceding mammary tumor formation. Oncogene. 2006;25:7148–58. doi: 10.1038/sj.onc.1209707. [DOI] [PubMed] [Google Scholar]
- 54.Caldwell CM, Green RA, Kaplan KB. APC mutations lead to cytokinetic failures in vitro and tetraploid genotypes in Min mice. J Cell Biol. 2007;178:1109–20. doi: 10.1083/jcb.200703186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chesnokova V, Kovacs K, Castro AV, Zonis S, Melmed S. Pituitary hypoplasia in Pttg+/-mice is protective for Rb+/-pituitary tumorigenesis. Mol Endocrinol. 2005;19:2371–9. doi: 10.1210/me.2005-0137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sussan TE, Yang A, Li F, Ostrowski MC, Reeves RH. Trisomy represses Apc(Min)-mediated tumours in mouse models of Down's syndrome. Nature. 2008;451:73–5. doi: 10.1038/nature06446. [DOI] [PubMed] [Google Scholar]
- 57.Williams BR, et al. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science. 2008;322:703–9. doi: 10.1126/science.1160058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Segal DJ, McCoy EE. Studies on Down's syndrome in tissue culture. I. Growth rates and protein contents of fibroblast cultures. J Cell Physiol. 1974;83:85–90. doi: 10.1002/jcp.1040830112. [DOI] [PubMed] [Google Scholar]
- 59.Torres EM, Williams BR, Amon A. Aneuploidy: cells losing their balance. Genetics. 2008;179:737–46. doi: 10.1534/genetics.108.090878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Torres EM, et al. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science. 2007;317:916–24. doi: 10.1126/science.1142210. [DOI] [PubMed] [Google Scholar]
- 61.Yang Q, Rasmussen SA, Friedman JM. Mortality associated with Down's syndrome in the USA from 1983 to 1997: a population-based study. Lancet. 2002;359:1019–25. doi: 10.1016/s0140-6736(02)08092-3. [DOI] [PubMed] [Google Scholar]
- 62.Hasle H, Clemmensen IH, Mikkelsen M. Risks of leukaemia and solid tumours in individuals with Down's syndrome. Lancet. 2000;355:165–9. doi: 10.1016/S0140-6736(99)05264-2. [DOI] [PubMed] [Google Scholar]
- 63.Satge D, et al. A tumor profile in Down syndrome. Am J Med Genet. 1998;78:207–16. [PubMed] [Google Scholar]
- 64.Buffin E, Emre D, Karess RE. Flies without a spindle checkpoint. Nat Cell Biol. 2007;9:565–72. doi: 10.1038/ncb1570. [DOI] [PubMed] [Google Scholar]
- 65.Hoyt MA, Totis L, Roberts BT. S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function. Cell. 1991;66:507–17. doi: 10.1016/0092-8674(81)90014-3. [DOI] [PubMed] [Google Scholar]
- 66.Li R, Murray AW. Feedback control of mitosis in budding yeast. Cell. 1991;66:519–31. doi: 10.1016/0092-8674(81)90015-5. [DOI] [PubMed] [Google Scholar]
- 67.Dobles M, Liberal V, Scott ML, Benezra R, Sorger PK. Chromosome missegregation and apoptosis in mice lacking the mitotic checkpoint protein Mad2. Cell. 2000;101:635–45. doi: 10.1016/s0092-8674(00)80875-2. [DOI] [PubMed] [Google Scholar]
- 68.Kitagawa R, Rose AM. Components of the spindle-assembly checkpoint are essential in Caenorhabditis elegans. Nat Cell Biol. 1999;1:514–21. doi: 10.1038/70309. [DOI] [PubMed] [Google Scholar]
- 69.Kops GJ, Foltz DR, Cleveland DW. Lethality to human cancer cells through massive chromosome loss by inhibition of the mitotic checkpoint. Proc Natl Acad Sci U S A. 2004;101:8699–704. doi: 10.1073/pnas.0401142101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kulukian A, Han JS, Cleveland DW. Unattached kinetochores catalyze production of an anaphase inhibitor that requires a Mad2 template to prime Cdc20 for BubR1 binding. Dev Cell. 2009;16:105–17. doi: 10.1016/j.devcel.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Holland AJ, Taylor SS. Many faces of separase regulation. SEB Exp Biol Ser. 2008;59:99–112. [PubMed] [Google Scholar]
- 72.Basto R, et al. Centrosome amplification can initiate tumorigenesis in flies. Cell. 2008;133:1032–42. doi: 10.1016/j.cell.2008.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang Z, Loncarek J, Khodjakov A, Rieder CL. Extra centrosomes and/or chromosomes prolong mitosis in human cells. Nat Cell Biol. 2008;10:748–51. doi: 10.1038/ncb1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Quintyne NJ, Reing JE, Hoffelder DR, Gollin SM, Saunders WS. Spindle multipolarity is prevented by centrosomal clustering. Science. 2005;307:127–9. doi: 10.1126/science.1104905. [DOI] [PubMed] [Google Scholar]
- 75.Basto R, et al. Flies without centrioles. Cell. 2006;125:1375–86. doi: 10.1016/j.cell.2006.05.025. [DOI] [PubMed] [Google Scholar]
- 76.Kwon M, et al. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev. 2008;22:2189–203. doi: 10.1101/gad.1700908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Habedanck R, Stierhof YD, Wilkinson CJ, Nigg EA. The Polo kinase Plk4 functions in centriole duplication. Nat Cell Biol. 2005;7:1140–6. doi: 10.1038/ncb1320. [DOI] [PubMed] [Google Scholar]
- 78.Peloponese JM, Jr., Haller K, Miyazato A, Jeang KT. Abnormal centrosome amplification in cells through the targeting of Ran-binding protein-1 by the human T cell leukemia virus type-1 Tax oncoprotein. Proc Natl Acad Sci U S A. 2005;102:18974–9. doi: 10.1073/pnas.0506659103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang X, et al. sSgo1, a major splice variant of Sgo1, functions in centriole cohesion where it is regulated by Plk1. Dev Cell. 2008;14:331–41. doi: 10.1016/j.devcel.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang XX, Liu R, Jin SQ, Fan FY, Zhan QM. Overexpression of Aurora-A kinase promotes tumor cell proliferation and inhibits apoptosis in esophageal squamous cell carcinoma cell line. Cell Res. 2006;16:356–66. doi: 10.1038/sj.cr.7310046. [DOI] [PubMed] [Google Scholar]