

Abstract
Drosophila melanogaster Heterochromatin Protein 1a (HP1a) is essential for compacted heterochromatin structure and associated gene silencing. Its chromo shadow domain (CSD) is well-known for binding to peptides that contain a PXVXL motif. Heterochromatin protein 2 (HP2) is a nonhistone chromosomal protein that associates with HP1a in the pericentric heterochromatin, telomeres and the fourth chromosome. Using NMR spectroscopy, fluorescence polarization and site-directed mutagenesis, we identified an LCVKI motif in HP2 that binds to the HP1a CSD. The binding affinity of the HP2 fragment is approximately two orders of magnitude higher than that of peptides from PIWI (with a PRVKV motif), AF10 (with a PLVVL motif), or CG15356 (with LYPLL and LSIVA motifs). To delineate differential interactions of the HP1a CSD, we characterized its structure, backbone dynamics and dimerization constant. We find that the dimerization constant is bracketed by the affinities of HP2 and PIWI, which dock to the same HP1a homodimer surface. This suggests that HP2, but not PIWI, interaction can drive homodimerization of HP1a. Interestingly, the integrity of the disordered C-terminal extension (CTE) of HP1a is essential for discriminatory binding, whereas swapping the PXVXL motifs does not confer specificity. Serine phosphorylation at the peptide binding surface of the CSD is thought to regulate heterochromatin assembly. Glutamic acid substitution at these sites destabilizes HP1a dimers, but improves the interaction with both binding partners. Our studies underscore the importance of CSD dimerization and cooperation with the CTE in forming distinct complexes of HP1a.
Keywords: Drosophila, Heterochromatin, HP1, HP2, PIWI
INTRODUCTION
Many critical components required for the initiation and spread of heterochromatin packaging in Drosophila melanogaster have been identified through genetic screens and cytological assays. These proteins include structural components of heterochromatin, histone modifying enzymes, and proteins involved in the RNAi pathway [1]. While a role for these classes of proteins has been established both by demonstrating localization to heterochromatin with immunofluorescence, and by showing dominant loss of heterochromatin-mediated silencing in mutant lines, a clear picture of how these components function together at the molecular level is still lacking.
One major structural component of heterochromatin is Heterochromatin Protein 1a (HP1a) [2], a protein conserved in many of the fungi and all of the animals [3]. HP1a is an essential protein that consistently localizes to heterochromatic regions, including pericentromeric domains, telomeres, and (in the case of Drosophila) the small fourth chromosome [4]. It is thought to act as an adapter protein, as many of the other critical components of heterochromatin bind to HP1a. The heterochromatin-specific mark of di- and tri-methylated histone H3 lysine 9 (H3K9 me2/me3) recruits the N-terminal HP1a chromodomain (CD) [5], while multiple proteins associate with heterochromatin by binding to the dimeric chromo shadow domain (CSD) [for review see [6]]. Some protein partners (e.g. Ku70) bind to the hinge region (demonstrated for mammalian HP1γ), and do not require HP1 dimerization [7]. The CD and CSD, which are connected by the disordered hinge, have been shown to fold independently in HP1a [5b] (see Figure 1a).
Figure 1. The disordered HP2 fragment binds to the HP1a CSD using a critical valine.

A. HP1a and the HP2 recombinant fragment from D. melanogaster used here are diagrammed. HP1a is made up of a chromodomain (CD) linked to a chromo shadow domain (CSD) by a flexible linker; the C-terminal extension (CTE) is also indicated. In HP2, blocks G and H represent regions conserved among D. melanogaster; D. pseudoobscura; D. willistoni; and D. virilis. B and C. 2D [15N-1H] HSQC spectra demonstrate the specific interaction of the CSD with the HP2 recombinant fragment from A. Resonances that disappear after titration are boxed (blue). In panel B, the 15N-labeled full length HP1a protein is shown in the absence (black) and presence (red) of the HP2 recombinant fragment; the resulting chemical shift perturbations all map to the CSD and CTE, not to the CD or hinge. In C, the 15N labeled HP2 recombinant fragment is shown in the absence (black) and presence (red) of the CSD [residues 131–203 as defined by [23]]. The HP2 fragment appears disordered. D and E. The CSD complex with the HP2 fragment is disrupted by mutations W200A and I191E in HP1a or by mutation V2468E in HP2. Antibody WA192 (specific for HP1a) was used to immunoprecipitate S-35 labeled HP1a and the bound fragment; the immunoprecipitated proteins were resolved by SDS-PAGE and detected by autoradiography. TP corresponds to the test peptide, the HP2 recombinant fragment (seen in A).
Reported HP1a CSD partners include chromatin modifying proteins (e.g. Su(var)3–9, CKII), proteins involved in DNA replication and repair (e.g. HOAP, ORC1–6), heterochromatin-associated proteins (e.g. Su(var)3–7, Arp4, Hip, HP2), the RNA binding protein PIWI, and the transcription regulators AF10 and the EMSY-like CG15356 (for appropriate references see [8]). For many HP1a partners, a short peptide consisting of a PXVXL motif is sufficient for CSD binding; this motif was originally discovered by screening a phage display library for binding with the CSD module [9]. Interestingly, HP2 binding to HP1a has been suggested to utilize a different mechanism [10]. The Drosophila HP1a CSD is presumed to form a conserved homodimer related to the yeast Swi6 [11], mouse HP1β [12], and human HP1β [8b] CSDs. Structures of HP1β have revealed that the PXVXL sequences bind across the interface of the CSD homodimer. Two consecutive PXVXL motifs were captured in the HP1β complex with EMSY. These tandem PXVXL motifs are conserved in the Drosophila ortholog of EMSY, CG15356, suggesting that they may bind to HP1a [8b]. The affinity of these putative partners for HP1a CSD has not been reported previously. Whether these peptide interactions can differentially alter the properties of HP1a dimers is also not known.
The large number of HP1a binding partners raises questions as to how HP1a discriminates among them. In vivo, the problem of target discrimination may be simplified by variation in the spatiotemporal availability of the partner proteins. However, in the presence of different concentrations of competitively interacting partners, the affinity of these partners for HP1a must come into play. Variables that potentially influence protein partner affinity in this case include: different hydrophobic residues in the PXVXL motif, differences in the amino acid sequence outside of the PXVXL motif, binding through an alternative binding surface that does not coincide with the PXVXL platform, and post-translational modifications in HP1a or the partner protein. In this study we focus on the biophysical characterization of the HP1a interaction with HP2, and compare this interaction with that of the previously-identified HP1a CSD-binding motif, PXVXV, found in PIWI [13]. HP2 has been characterized as a structural component of heterochromatin that tracks with HP1a as shown by immunofluorescent staining of polytene chromosomes; genetic analysis has shown a dosage-dependent requirement for this protein, which plays an essential role in heterochromatin formation and mitotic chromosome integrity [14].
Using NMR spectroscopy, we identified a short peptide of HP2 which is sufficient for specific binding to HP1a. The HP2 peptide bound to the common PXVXL binding surface using an LCVKI motif, forming a distinct complex with the HP1a CSD dimer. Surprisingly, we found that the HP2 peptide binds approximately two orders of magnitude better than the PIWI, AF10 and CG15356 peptides, despite its deviation from the pentapeptide consensus sequence. Interestingly, the binding coefficients of these two groups of peptides bracket the dimerization constant of HP1a. HP1a selectivity is derived from cooperation between the CSD module (amino acids 141–201) and the CTE (C-terminal extension, amino acids 202–206), residues which we find are disordered. This mechanism results in preferential interactions with different peptides containing a PXVXL motif. Further studies showed that HP1a modulates partner affinity through phosphorylation at the PXVXL platform. We suggest that a tight binding partner such as HP2 could drive HP1a dimerization and thus promote the chromatin condensation typical of heterochromatin.
RESULTS
A recombinant HP2 fragment binds specifically to the CSD dimer
To determine the binding surface used by HP2 to bind HP1a CSD we relied on NMR spectroscopy. We performed 2D [15N-1H] HSQC experiments using the 15N labeled full length HP1a to determine chemical shift perturbations in the absence and presence of excess amounts of the recombinant HP2 fragment (Figure 1B). Upon interaction with HP2, a subset of resonances is perturbed and exhibits line broadening consistent with conformational exchange. Perturbed resonances map to the CSD, indicating that the HP2 fragment interacts only with residues in the CSD, in agreement with the earlier yeast two-hybrid study [14b].
To investigate the hypothesis that tertiary structure in HP2 [10] is required for binding, we performed the reciprocal experiment, examining the 2D [15N-1H] HSQC spectrum of the 15N labeled HP2 fragment in the presence of unlabeled CSD (Figure 1C). We find that in the absence of the CSD the HP2 fragment is disordered. Both 1D 1H and 2D [15N-1H] HSQC spectra lack chemical shift dispersion. In the presence of the CSD, a limited set of resonances become perturbed (Figure 1C). Although we did not perform a sequential assignment of the HP2 spectrum, we observed that the single Trp side chain signal (Trp 2478) exhibits line broadening and an Asn side chain shifts its signals. This result indicates that a short portion of the HP2 fragment containing the Trp with a nearby Asn residue binds the CSD.
The atomic structure of the Drosophila HP1a CSD has not been characterized previously. However, the structure of mouse HP1β has been reported [12]. The key residues of HP1a that form the PXVXL binding platform and the dimerization interface have been predicted based on sequence conservation with HP1β to be Trp 200 and Ile 191 respectively [12b]. As an initial test of the dependence of the complex of HP1a and HP2 on the PXVXL platform or on HP1a dimerization, we looked for co-immunoprecipitation of the HP2 fragment with wild-type and the W200A mutant or the I191E mutant in full length HP1a. We found that indeed both mutations abrogate the interaction of HP1a with HP2 (Figure 1D; compare lane 5 with lane 8 and lane 11), suggesting that dimerization and the PXVXL platform are required for binding.
Given that the PXVXL motif is variable and that the interaction must take place in Block H, RKLCVKINRRPYNKWL, (Figure 1A), we decided to test whether the sequence, LCVKI surrounding Val 2468 was mediating the interaction of HP2 with HP1a. In a CoIP experiment, we found that binding of the HP2 fragment to the CSD was abrogated by the point mutation V2468E in HP2 (Figure 1E, compare lane 4 with lane 5). Together, our results from NMR and co-immunoprecipitation suggest that a group of neighboring residues in HP2 block H may be sufficient for CSD binding.
Characterization of the HP1a CSD reveals dimerization affinity
Studies on mouse HP1β have indicated that the CSD dimerizes prior to peptide binding. By gel filtration we examined whether this mechanism is valid for D. melanogaster HP1a CSD as well. Due to the independence of the CD and CSD, subsequent experiments were performed with the CSD alone or CSD and CTE. Gel filtration data (when compared to molecular weight standards) shows that the wild-type Drosophila CSD elutes with an approximately two-fold higher molecular weight compared to the I191E construct, which is unable to dimerize (Supp. Figure 1). Thus, the I191E mutant of the CSD forms homogenous monomers. The W200A interaction mutant eluted as a homogenous dimer, suggesting that this mutation has no impact on dimerization.
To measure the self-association of the HP1a CSD we performed analytical ultracentrifugation (AUC) experiments, and found that the wild type dimer has a dissociation constant of 3.0 ± 0.2 μM (Figure 2A) while the I191E mutant is a fixed monomer (Figure 2B). We measured similar dimerization constants for the full length and the CSD fragment of HP1a (dimerization constants of 1.4 ± 0.2 μM and 1.6 ± 0.4 μM, respectively, under similar buffer conditions containing 150 mM NaCl). Therefore, neither the chromodomain nor the hinge region affects the dimerization properties of HP1a (Supp. Figure. 2). Interestingly, we found a two-fold weakening of the HP1a dimerization constant due to the W200A mutation (Figure 2C). In contrast, the Δ204 construct (lacking the CTE) showed improved dimerization (2.0 ± 0.2 μM instead of 3.0 ± 0.2 μM), suggesting that the CTE residues influence the stability of the dimers. Together our findings suggest that the CSD and CTE motifs of HP1a cooperate to organize weakly associated dimers with a free energy of −7.3 kcal/mol. We note that the weaker dimerization constant for HP1a (compared to HP1β) suggests that binding partners may be important in stabilizing the former.
Figure 2. Characterization of D. melanogaster CSD dimerization by AUC and Far-UV CD.

Equilibrium AUC absorbance curves (bottom plot) were globally fit to a monomer-dimer model to obtain the converged dissociation constant Kd with the residual plot shown (top plot) for wild-type CSD (A), and its mutants W200A (C) and Δ204 (D). Data for the mutant I191E polypeptide converged with a single-species fit, exhibiting a molecular weight of 9.62 kD, as expected for a single monomer species; residuals are shown in the upper panel (B). E. A superposition of the far-UV CD spectra of wild-type and I191E CSD polypeptides indicates no difference in the secondary structure, even though the I191E CSD construct does not dimerize.
The dimerization constant of the full length mammalian HP1β protein has not been previously reported, whereas that of the HP1β CSD fragment lacking the CTE was estimated at 150 nM [8b]. Therefore, we measured the dimerization constant of the full length HP1β, and found it to be 3 times tighter than that of HP1a under the same buffer conditions (0.50 ± 0.24 μM) (Supp. Figure 3). We suggest that the cooperation of the CSD and CTE in HP1β is responsible for the weaker dimerization constant as compared to the CSD alone.
Our results for HP1a show that only the CSD I191E mutant abrogates dimerization and that the other constructs associate with low micromolar affinity – a critical result for our subsequent measurement of protein interactions with the CSD. In order to compare the structures of the CSD monomer with dimer, we analyzed the far-UV CD spectrum of the wild-type and I191E constructs. The spectrum of the CSD shows negative bands corresponding to the presence of both helical (at 208 and 222 nm) and β-sheet (at 218 nm) structure (Figure 2E). These features are intact in the monomeric construct, suggesting that the CSD dimerization does not lead to the formation of additional secondary structure, and supporting the conclusion that the loss of HP2 binding to the I191E mutant form reflects a requirement for the dimer.
The high resolution structure of the CSD defines its ordered and disordered regions
We used NMR spectroscopy (15N backbone dynamics studies) and X-ray crystallography to obtain a full picture of both the ordered and disordered regions of the wild-type CSD construct. By measuring the heteronuclear NOE (Nuclear Overhauser Effect) data for backbone amides, we identified disordered regions, which exist at the N-terminus (residues 131 to 140 of intact HP1a), and the CTE (residues 202 to 206), as well as segments within the CSD module (residues 158–159, 162, 170, and 172–173) (Figure 3A). To delineate the features of the ordered structure, we generated crystals of the CSD construct and determined the atomic structure at 2.3 Å resolution (Table I). The symmetric dimer resembles that of the mammalian HP1β [8b]. We found contiguous electron density corresponding to the N-terminal residue Thr 141 through the C-terminal residues Ser 199 in chain A and Tyr 201 in chain B (Figure 3B and 3C). For residues 200 and 201 in chain B we detected only the backbone electron density. The missing density for the indole ring of Trp 200 suggests that it does not participate in intramolecular or cross-dimer packing in the CSD; this is consistent with our prior observation that the W200A mutant form is maintained as a dimer. As we showed, the HP1a W200A mutant is unable to bind to HP2, suggesting that this side chain is necessary for binding to a partner protein.
Figure 3. High resolution structure and NOE data identify flexible regions of the CSD backbone.

A. Heteronuclear NOE data obtained from 15 N-relaxation analysis of the CSD fragment by NMR spectroscopy identified backbone amides that lack ordered structure ([1H]-15N NOE < 0.55). Secondary structure elements above the plot are derived from the crystal structure (rectangles = beta sheet, ovals = alpha helix). Terminal residues 131–140 and 202–206 are disordered. B, C. Ribbon diagram shows the crystal structure of HP1a CSD, which forms a homodimeric assembly of 2 CSD modules. Chain A consists of Thr 141 to Ser 199 and while chain B is resolved to Tyr 201. Flexible regions within the CSD module are colored pink; these have [1H]-15N NOE values of ≤ 0.55 (dashed line). Diagrams in B and C differ by rotating 90 degrees about the central axis.
Table I.
Data collection and refinement statistics for the crystal structure of the HP1a CSD. The Ramachandran plot was calculated using MOLPROBITY.
| Data collection | |
|---|---|
| Beamline | 19BM (APS) |
| Wavelength (Å) | 0.9792 |
| Unit cell (Å) | a= 44.8, b= 56.6, c=67.3 |
| Space group | P212121 |
| Solvent content (%) | 55 |
| Number of protein chains in asymmetric unit | 2 |
| Resolution range (Å) | 50.0–2.3 |
| Highest resolution shell (Å) | 2.30–2.34 |
| Unique reflections | 8023(397) |
| Redundancy | 7.1(7.3) |
| Completeness(%) | 99.4(98.8) |
| Raverage.(%) | 4.7(47.5) |
| Average I/σ (I) | 37.2(4.3) |
| Refinement | |
| R(%) | 19.9 |
| Rfree (%) | 24.6 |
| Mean B value (Å2) | 62.5 |
| B from Wilson plot (Å2) | 45.7 |
| RMS deviation bond lengths (Å) | 0.019 |
| RMS deviation bond angles (°) | 1.4 |
| Number of amino acid residues | 116 |
| Number of non-H atoms | 984 |
| Number of water molecules | 37 |
| Ramachandran plot | |
| Most favored regions (%) | 99.1 |
| Additional allowed regions (%) | 0.9 |
Using a 3D alignment of the HP1a CSD with the human HP1β CSD [analyzed in a complex with the EMSY fragment, pdb 2FMM [8b]], we found that the domains are similarly assembled. The two 111 residue chains of the dimer were superimposed with those of HP1β using COOT [15] which uses secondary structure matching [16] and resulted in a 1.4 Å RMSD (Root Mean Squared Deviation) for the backbone Cα atoms. Individual monomers were also compared in this manner. Superposition of only chain A or chain B resulted in 1.2 Å and 1.4 Å RMSD respectively. In contrast to this structure of the HP1a CSD in the absence of peptide, the complex of HP1β CSD with EMSY shows the indole ring of the C-terminal Trp engaged in cross-dimer interactions, but not peptide interaction. This suggests that peptide binding enhances cross-dimer contacts of the CSD module.
We investigated whether HP1a CSD exhibits backbone flexibility, which could permit changes that could increase cross-dimer contacts upon peptide binding. We mapped the regions that exhibited heteronuclear NOE values ≤ 0.55 onto the crystal structure (Fig. 3B and C). We found that the turn between the β1 and β2 strands (residues 158–161) and residue 196 in the last α-helix form a cluster near Trp 200 (Figures 3B and 3C). This region is adjacent to the interface of peptide binding in the mammalian HP1β structure, and is shifted significantly toward the other monomer in the complex with the peptide. Therefore, the flexibility we detected for these sites may have general significance for stabilizing cross-dimer contacts. By combining results of NMR and crystallography, we conclude that the CSD module is made up of residues 141 through 201 in the HP1a. The module is flanked by the disordered N-terminal residues and CTE residues that point away from opposite faces of the dimer (Figure 3B and 3C). (Note that the N-terminus is an artificial one; however, in the intact protein, this region is flanked by the disordered linker.) Inherently flexible residues near the CTE region agree with the evidence for alterations in the backbone due to peptide binding.
An HP2 peptide binds to the HP1a dimer with high affinity
Next, we investigated the complex formed by HP1a and HP2. For this experiment the CSD construct Δ204 was used, which lacks three amino acids at the C-terminus. Gel filtration showed that the addition of the HP2 fragment to the CSD forms a complex consistent with a 1:2 stoichiometry (Supp. Figure 4). To determine the affinity of HP2 for the HP1a CSD we performed fluorescence polarization assays. We designed a short HP2 peptide (22 aa) with the critical valine residue near the center. For comparison to a known PXVXL partner of Drosophila HP1a, we used PIWI [13] and AF10 (i.e. Alhambra) [17] peptides. We also designed a CG15356 peptide by homology to the mammalian EMSY interaction site for HP1β[8b]. To our surprise we found that the HP2 peptide, with the pentamer sequence LCVKI, binds to the CSD far better than the PIWI, AF10 or CG15356 peptides (Figure 4A). Since the affinities of PIWI, AF10 and CG15356 are quite similar we decided to follow the effects of PIWI due to its potential role in heterochromatin initiation – a complementary role to HP2’s maintenance function. A two-order of magnitude difference in dissociation constant (sub-micromolar to micromolar range) was measured (Table II and Figure 4A). To ensure that the tight binding is not due to oxidation of the Cys residue in the HP2 peptide, we performed the binding assay using an HP2 peptide with the pentamer sequence LSVKI, and found little change in binding affinity. Using this assay, we observed that both HP2 and PIWI binding are shifted to the millimolar range by I191E and W200A mutations in HP1a. Thus, both peptides bind using a similar mechanism; they bind to the CSD dimer and require the PXVXL platform (Figure 4A).
Figure 4. The HP2 peptide binds CSD dimers with high affinity.

A. Fluorescence polarization assays were used to determine the Kd for the binding of the HP2 peptide to wild-type HP1a-CSD and mutants W200A and I191E. The HP1a-CSD binding of the HP2 peptide is compared to PIWI, AF10, and CG15356 peptides, which have varying conservation of the PXVXL sequence motif. Data were fit as described in the Methods. Peptide sequences are listed above the plot and the critical pentapeptide motifs are underlined. The binding curves corresponding to HP2 compared to PIWI, CG15356, and AF10 exhibit a significant difference in Kd which brackets the modeled homodimerization binding curve of the CSD in the absence of peptide. The HP2 and PIWI peptides show no evidence of binding with the W200A mutant (binding curves shown); similar results were obtained with the I191E mutant. B&C. Determination of the molecular weight of the HP2 peptide alone (B) and of its complex (C) with the HP1a CSD by equilibration AUC demonstrates that 2 CSD molecules and 1 HP2 peptide form the complex. D. A model for HP1a dimerization and peptide association. The dissociation constants, KDimer and KPeptide are independent of each other. The high affinity of HP2 could drive dimerization of HP1a.
Table 2.
Elucidation of the binding surface of the HP1a-CSD. This table summarizes an exploration of some of the factors that contribute to HP1a-CSD ligand binding. Line 1. HP1a-CSD binds to the HP2 peptide (either wild-type or with a C2467S substitution) with tenths of micromolar affinity, tenfold less than the dimerization constant of HP1a-CSD. HP1a-CSD binds to the PIWI peptide with tens of micromolar affinity, tenfold more than the dimerization constant of HP1a-CSD. Lines 2–3. Consistent with the behavior of known PXVXL platform binding peptides, the HP1a W200A and I191E substitutions abrogate binding of the HP2 and PIWI peptides. Line 4. Although PXVXL is the published consensus polypeptide sequence for binding to the HP1a-CSD dimer, LXVXI can bind equally well in the context of PIWI (compare lines 1 and 4 in the PIWI Kd column). Line 5. Removing the last three residues of HP1a-CSD weakens peptide binding 3- to 16-fold, while not shifting the dimerization constant appreciably. Line 6. HP2 is impacted twice as much as PIWI is by mutating HP1a E195. Line 7. An equivalent fold change occurs when HP1a A174 is mutated. Line 8. The phosphorylation mimic mutations in HP1a-CSD decrease the dimerization constant of HP1a-CSD while improving the binding of the HP2 22mer and the PIWI 21mer.
| HP1a CSD Construct | Peptide | HP1a Dimerization Constant | Kd for HP2 (μM) | Kd for PIWI (μM) |
|---|---|---|---|---|
| WT | HP2 (HP2 LCVKI) PIWI | 3.0±0.2 | 0.30±0.02 (0.14±0.07) | 20.0±0.5 |
| I191E | HP2 PIWI | N/A | >400 | >2000 |
| W200A | HP2 PIWI | 5.8±0.6 | >500 | >2000 |
|
| ||||
| WT | Pentamer Swap | 3.0±0.2 | 0.99±0.08 | 21.3±1.3 |
|
| ||||
| Δ204 | HP2 PIWI | 2.0±0.2 | 4.9±0.6 | 60±2 |
|
| ||||
| E195Q | As above | 4.3±0.4 | 3.7±0.4 | 97±12 |
|
| ||||
| A174E | As above | (3.0±0.2) | 1.26±0.12 | 101±6 |
|
| ||||
| S199ES202E | As above | 19±1 | 0.044±0.007 | 1.7±0.1 |
Interestingly, the dimerization constant for the CSD construct in the absence of peptide is weaker than the binding constant for the HP2 peptide. Using AUC, we analyzed the short peptide of HP2 and its complex with the CSD construct, measuring the molecular weight (Figure 4B and C). This showed that the peptide is a monomeric species of 2.9 kDa alone and binds as monomer to a dimeric CSD. Together, the binding assays and AUC data indicate that the HP2 peptide promotes CSD dimerization by shifting the equilibrium from monomers to dimers. Since dimers are required for the binding of the HP2 or PIWI peptides, stable/ordered peptide complexes form in a subsequent reaction (Figure 4D). The contrast in binding coefficients of the HP2 and PIWI peptides led us to investigate the source of the differences in these interactions.
Residues within the CSD module accommodate HP2 and PIWI peptides similarly
To address whether the source of the enhanced affinity observed for HP2 compared to PIWI is attributable to the binding surface of the CSD module, we relied on NMR spectroscopy. We delineated chemical shift perturbations of the 2D [15N-1H] HSQC spectrum of 15N-labeled CSD in the absence (black) (Supp. Figure 5 and 6) and presence (red) of the HP2 and PIWI peptides (Supp. Figure 6A and 6B). This study allowed us to identify residues affected by peptide binding. Using a cutoff for chemical shift perturbations ≥ 0.1 ppm, we identified a common surface of interaction consisting of 9 residues on one face of the dimer (highlighted, Supp. Table I); these residues are not in a single continuous primary sequence. For the HP2 complex the total surface is composed of 14 residues shifted (13 mapped to the crystal structure) (Figure 5A), while for the PIWI complex a smaller surface of 13 residues is observed (10 mapped to the crystal structure) (Figure 5B). Both peptides induce changes in the CSD structure spanning β1, the loop connecting β1 and β2, β2, α2 and the CTE. The majority of the perturbations involved splitting of a resonance into two new resonances, suggesting asymmetry in the interaction of the monomer with peptide residues for both HP2 and PIWI. Specific sites of interaction with the peptides were pinpointed by utilizing available data from the CAF-1 peptide binding to HP1β (Figure 5C and 5D) [12b]. We found that Tyr 194 and Leu 198, which form the pocket for the peptide’s central valine, are shifted 0.034/0.101 ppm and 0.055/0.122 ppm in the HP2 and PIWI complexes, respectively (Supp. Figure 6, residues highlighted in yellow Figure 5C). Drosophila HP1a residues that make contact in the +2/−2 positions from the central valine residue, Ala 155, Phe 193, Arg 197, Leu 198, and Trp 200 are absolutely conserved between HP1β and HP1a (Figure 5D). These residues all show chemical shift perturbations, supporting a conserved mode of peptide binding (Supp. Figure 6, residues highlighted in pink in Figure 5C).
Figure 5. HP2 utilizes an extensive peptide binding surface in the CSD.

A and B. Residues are highlighted that are perturbed more than 0.1 ppm in the complex of the CSD with HP2 (A. Red) and PIWI (B. Blue). The upper panels show a side view; rotating the structures 90° about the horizontal axis shows the top view of the peptide binding surface in the lower panels. C. Residues in the CSD structure are highlighted according to the color scheme in D. for yellow, blue, and pink. D. Multiple sequence alignment of the Drosophila HP1a and HP1b with mouse HP1β. Highlighted in yellow are the residues that interact with valine at position 0 of the interacting peptide. Residues that interact with the +2/−2 position are pink, those that interact in the −6/−7 and +5/+6 positions are blue, and those important for the fold of the CSD are grey. Blue triangles denote residues that are important for dimerization. The green dot denotes a residue that is important for both the fold of the CSD and for peptide binding. The pink dot is a residue important for the fold of the CSD which can also be phosphorylated in HP1a; the green star is a second phosphorylation site. D. Alignment of the HP2, PIWI, and mouse Caf-1 peptides that interact with the CSD. Residues are highlighted in colors that correspond to their interaction sites with the CSD (shown in C). Yellow denotes the central valine, pink the +2/−2 positions and blue the −7/−6 and +5/+6 positions previously determined to be important for binding.
The mouse HP1β CSD residues which interact with PXVXL containing peptides at the −6/−7 and +5/+6 positions are Thr 126, Leu 135 and Leu 146, corresponding to D. melanogaster HP1a CSD residues Ser 156, Leu 165 and Met 176 (Figure 5D). The CAF-1 peptide that binds to HP1β has Phe/Ile in the −6/−7 positions while HP2 has Ile/Ser and PIWI has Arg/Gly. The chemical shift perturbations of the HP1a CSD residues in the HP2 and PIWI complexes corresponding to this region show those resonances of Ser 156 and Leu 165 shift, but not Met 176. Instead, we found that in the HP1a complex with HP2, the resonance of Ala 174 shifts. The crystal structure supports the notion that Ala 174 in cooperation with Ser 156 and Leu 165 forms the surface that accommodates amino acids at positions −6/−7 and +5/+6 of the HP2 peptide (Supp. Fig 6, highlighted residues in blue Fig 5C).
Since we observed no shift for Ala 174 in the PIWI complex, the data suggested that this site might contribute to discriminatory peptide binding (Supp. Figure 6B). To test this hypothesis we prepared an A174E mutant and measured the binding affinity. We found that this mutation weakened the binding of HP2 and PIWI peptides by equal amounts (Table II). Therefore, although the backbone amide only shifts for the HP2 complex, the integrity of the Ala 174 side chain is critical for complementary interactions with both peptides. Together these observations indicate that a conserved and broadly utilized surface for peptide binding is presented by the CSD module.
Residues Glu 195 and Glu 196 are conserved between HP1a, HP1b, and mouse HP1β (Figure 5D). Comparing the structures of free and complexed HP1β, we noticed the side chains of Glu 196 and Arg 197 form hydrogen bonds in one of the CSD monomers after the peptide binds. Examining our data, we found that the Glu 196 and Arg 197 backbone resonances are dramatically shifted and split into two new peaks in both HP2 and PIWI complexes (Supp. Figure 6A and 6B). The large shifts indicate that peptide binding causes a major change in their chemical environment, and their splitting indicates that this residue is no longer involved in a symmetric environment in the dimer. In the crystal structure of the HP1a CSD construct, we found that the side chains of Glu 196 and Arg 197 are distinctly apart. Interestingly, NMR backbone dynamics identified Glu 196 as a flexible spot in the last α-helix, suggesting a critical function for peptide recognition (Figure 3A and 3B).
To quantitate the impact of conserved Glu side chains on dimerization and peptide binding, we generated an E195Q mutation. This mutation has a subtle effect on the dimerization constant (Supp. Figure 7A; Table II). However, it results in a 10-fold drop in affinity for HP2, and a 5-fold drop in affinity for PIWI (Table II). We suggest that the presence of the Glu 195 and 196 side chains is fundamental for efficient and selective peptide binding at the PXVXL platform.
The PXVXL pentamer sequence does not determine binding selectivity
To test whether the sequence LSVKI in HP2 is the dominant factor influencing peptide affinity, we swapped that sequence with PIWI’s pentamer PRVKV in the HP2 peptide and vice versa. We found that this synthetic HP2 sequence causes a 3-fold weakening in affinity, whereas the synthetic mutant PIWI sequence causes no change (Figure 6; Table II). This subtle effect indicates that the specific and discriminatory binding of these peptides requires the residues that flank the pentapeptide motif. This idea is also supported by our NMR analysis of the 15N labeled recombinant HP2 fragment which shows that 26 resonances were perturbed. These residues include the Trp side chain located at the +10 position with respect to the Val in the LSVKI motif, suggesting that this and other peripheral hydrophobic residues in the peptide chains contact the CSD (see Figure 5E for peptide alignment). Previous studies on the mammalian HP1β have found that such distant positions in the peptide become ordered in a complex with the CSD [12b]. In contrast, PIWI has no hydrophobic side chains at peripheral locations, and this is in agreement with a weaker binding constant and smaller interaction surface. Thus although the PXVXL motif is crucial for binding, the peripheral residues determine the binding surface and strength of interaction.
Figure 6. The CTE and phosphorylation of HP1a contribute to affinity; variation in the binding pentapeptide is not a major factor.
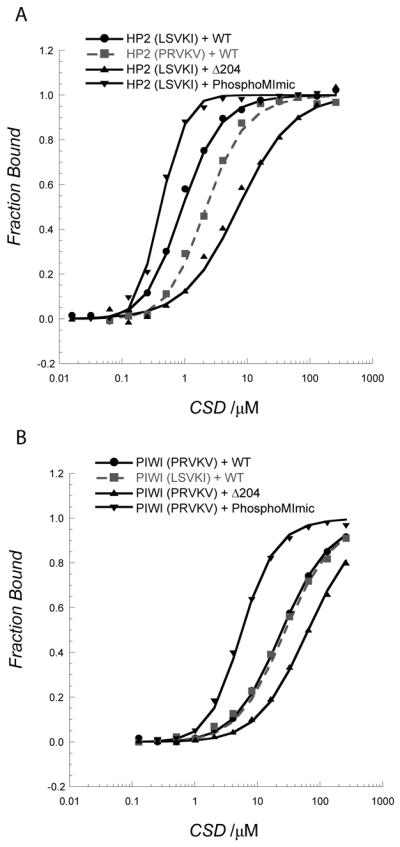
A. Swapping the pentamer sequence of PIWI into the HP2 peptide results in a 3-fold weakening of binding; in contrast, removing the last three residues in HP1a-CSD (the CTE) reduces binding 16-fold. Phosphomimic charges on the CSD at positions S199 and S202 result in a 7-fold enhancement in binding the HP2 peptide, with increased cooperativity. B. A similar situation is found in PIWI, although more subtle. Adding the HP2 pentamer sequence to the PIWI peptide does not change its affinity for HP1a-CSD. Clipping off the last three residues in the CSD results in a 3 fold weakening of binding. The phosphorylation mimic enhances binding 12-fold.
The CTE impacts CSD selectivity and affinity
It is clear that the ordered HP1a CSD plays a role in the peptide interaction surface however, in addition some of the observed perturbed resonances were found in the disordered HP1a CTE, residues 203–206 (Figure 3A). For example we found significant chemical shift perturbations in residues 202 and 204 for both complexes, suggesting that an ordered structure is formed in the presence of the peptide. To test what role the CTE is playing in protein partner selection, we studied the interaction of the Δ204 mutant with HP2 and PIWI peptides. We found that the CSDΔ204 mutant binds the HP2 fragment (Supp. Figure 4) with a major difference in the affinity for the HP2 peptide. The Δ204 construct showed a 16-fold weaker binding to the HP2 peptide, but only a 3-fold weaker binding to the PIWI peptide (Figure 6A and 6B, Table II). This result indicates that CTE interaction with each peptide modulates strength of binding.
C-terminal phosphorylation of HP1a enhances peptide affinity
During DNA damage response, mouse HP1β is phosphorylated by casein kinase 2 (CK2) at the chromodomain, which eliminates binding to the H3K9 methyl mark and induces HP1β mobilization [18]. In contrast, the function of phosphorylation of the HP1a-CSD at serines 199 and 202 by the same enzyme is suggested to be critical for heterochromatin assembly (Figure 5D) [19]. To investigate whether HP1a-CSD phosphorylation affects peptide binding, a phosphorylation mimic was generated (S199E and S202E double mutant). We found the affinity for both HP2 and PIWI enhanced 10-fold in response to this change (Figure 6; Table II), even though the dissociation constant for the CSD dimer was weakened 6-fold (Supp. Figure 7B). The HP2 peptide binding also became cooperative with a Hill coefficient of 1.7 ± 0.3. We propose that the phosphorylated state increases the flexibility near the C-terminus and supports productive interactions between the hydrophobic residues of the CSD and the peptide.
Conclusion
HP1a is a central component of heterochromatin in some fungi and all animals examined to date. A conserved feature is the multiplicity of partners, with about a dozen different chromosomal proteins identified as binding to HP1a, most often through the chromo shadow domain. To identify how HP1a CSD discriminates among its binding partners we performed a physical study of HP1a dimerization and CSD’s interaction with HP2 and PIWI.
We found the dimerization of HP1a to be weaker than that observed for the mammalian HP1β, and this may be attributed to sequence differences in their CSDs. Of the residues important in the dimerization interface [12b], there are two that differ between the species; Gln 158 and Thr 169 in HP1β are replaced with Arg 188 and Ser 199 in Drosophila melanogaster, respectively. These residues are located at the two ends of the dimerization helix α2 (Figure 3). In HP1β, Gln 158 in chain A interacts with Asn 153 of chain B in a van der Waals interaction. In HP1a, Gln is replaced with Arg 188 and forms an electrostatic interaction with Glu 184. These differences likely contribute to the different dimerization constants measured for HP1a and HP1β. Our findings underscore the importance of delineating the dimerization properties of other HP1 variants to understand the basis for their respective complexes.
To date the only binding surface in the CSD shown to be functional is the PXVXL platform. At the outset of our work we thought that HP2 was using an alternative surface to bind to the CSD. However, as we show here, HP2 binds to the CSD using its peptide sequence LXVXI, which shares the properties of PXVXL sequences in terms of binding surface and stoichiometry. Given the variability of the PXVXL motif, the failure of a protein to interact with the HP1a mutants W200A and I191E and evidence of interaction at the CSD, would seem to be the best initial indicators of use of the PXVXL platform. Several additional cases can be noted where recognition of a PXVXL domain has been difficult. Positively charged residues were emphasized in the discussion of the binding sequence of HP1a-interacting protein HIP/HP4; although these residues may be important for binding or peptide flexibility, PXVXL motifs as defined by a central valine surrounded by hydrophobic residues in the +2/–2 positions are present (8 MIVKI, 37 CRVNL, 64 CIVRV) [20]. In the case of Su(var)3–9, the residues interacting with HP1a [21] have not yet been identified. The region of binding that overlaps between the two studies cited contains a single valine residue that is not surrounded by hydrophobic amino acids in the +2/−2 positions. The interaction has, however, been shown to be sensitive to the W200A mutation in HP1a [21a], arguing that the PXVXL surface is being employed to bind Su(var)3–9.
Although a central valine residue is necessary for binding, it is a poor predictor of affinity for HP1a binding. Our pentamer swap between HP2 and PIWI showed no improvement in PIWI binding when the higher affinity peptide HP2’s pentamer was swapped into the lower affinity peptide. That both pentamer sequences contained a valine residue and both interactions were abrogated by the W200A mutation in HP1a, argues that this residue is important for binding. However the presence of a central valine surrounded by hydrophobic residues in the +2/−2 positions does not demand binding. Multiple examples of PXVXL motifs have been examined (including a PSVSL sequence in HP2 at residue 1665) which fail to show binding to HP1 [for HP1α see [22], for HP1a see [10]]. We conclude that a central valine residue is necessary for binding but that residues outside the pentamer are critical for modulating the affinity of a peptide.
To search for potential contacts to pinpoint a particular surface of the CSD that might have differential interactions with HP2 and PIWI peptides, we utilized NMR and crystallography for a structural analysis. The structural results demonstrated that the PXVXL binding surface is created through dimerization of an ordered CSD ‘module’ and a disordered CTE (Figure 3). Mapping the perturbed residues in the HP2 and PIWI complexes with the CSD indicates that there is a larger surface for the HP2 peptide that is significantly perturbed (Supp. Table I and Fig 6A and 6B). There is agreement between the shifted residues from mouse HP1β and D. melanogaster HP1a. So in the CSD module the differential affinity must be derived from side chain interactions. In addition, when we tested the function of the last three residues of the disordered CTE; we found that their removal has a dramatic impact on the affinity for the HP2 peptide (16-fold decrease), while having only a 3-fold impact on affinity for the PIWI peptide (Figure 6, Table II). This indicates that there is cooperation between these two regions of the HP1a-CSD that is utilized by some binding partners but not others, providing discrimination. Thus, critical side chain interactions in the CSD and CTE account for the observed difference in affinity between the HP2 and PIWI peptides.
HP1 paralogs (HP1a, HP1b, HP1c, Rhino/HP1d, HP1e) in Drosophila are quite varied, but all contain a chromodomain and a chromo shadow domain. While the chromo shadow domain does not change significantly among these proteins, the C-terminal residues (CTEs) vary considerably in length and in sequence (Supp. Figure 8) [2c, 23]. Here we have shown that the CTE is important for the tight binding observed in the CSD:HP2 interaction, and we speculate that in other HP1 paralogs, the CTE plays a critical role in partner selectivity at the chromo shadow domain. For example, yeast two-hybrid studies found that HP1a, but not HP1b, interacts with PIWI [13] despite the absolute conservation of critical residues within the CSD modules of HP1a and HP1b (Figure 5D). The data suggest that the 88-residue CTE region of HP1b (Supp. Fig. 8) blocks binding to partners of HP1a. This observation suggests a basis for the differential interactions of HP1 paralogs observed in Drosophila, through the paralog specific CTE motifs.
We also considered whether post-translational modifications of HP1a impacted its binding and ability to discriminate among its many partners. Not all binding partners have appropriately placed basic residues to complement phosphorylation, so there is likely to be some partner selectivity based on this modification (Figure 5D) [12b]. Dynamic regulation of this modification is necessary for full silencing effects [19b]. Here we observe that mutation of HP1a residues Ser 199 and Ser 202 to glutamic acid (as a phosphorylation mimic) enhances binding for both peptides (Figure 6; Table II).
HP2 is a structural component of heterochromatin [10, 14b] while PIWI is a critical nuclear component of the RNAi system which potentially mimicks in D. melanogaster the RNAi functions observed in fungi and plants [24]. The high affinity of HP2 for the HP1a dimer and lack of binding to the HP1a monomer (Figure 4A, Table II) suggests that HP2 could play a major role in stabilizing the dimer, potentially promoting chromatin condensation in vivo, while PIWI, with a lower binding affinity would not have a similar effect. The weaker interaction with PIWI may be essential, in that one might anticipate a more transient interaction during assembly, while the tighter affinity observed for HP2 is consistent with the observation that HP2 and HP1a colocalize consistently. The shift observed on HP1a “phosphorylation,” while weakening dimerization, indicates that both PIWI and HP2 could populate the dimer under these conditions (Table II). PIWI is abundant in the female germline and 1–2.5 hr embryo, while HP2 is absent from the female germ line, but abundant throughout embryogenesis [25]. This suggests a transition in HP1a partners during the critical assembly of heterochromatin in the early embryo. Such dynamic changes in peptide binding by the CSD might play a critical role in heterochromatin assembly [19b].
EXPERIMENTAL METHODS
Cloning and preparation of constructs
Full length HP1a expressed from the pET16b vector, as previously described [5b], was used for NMR studies. Full length HP1a expressed from the pET28a vector, as previously described [10], was used for immuno-precipitation studies. Mutations I191E and W200A in the full length protein were made in the pET28a vector using the QuikChange kit (Agilent Technologies). The full length HP1a and mammalian HP1β (sequences of the human and mouse HP1β are identical) as well as the recombinant fragments below, were cloned in the pET11a vector using NdeI and BamHI sites. Each contains a permanent N-terminal his-tag (MKKHHHHHH). They also have two lysines in the C-terminus for improved solubility with the exception of HP1β. The HP2 recombinant fragment corresponds to residues 2415–2512, the HP1a-CSD corresponds to residues 131-carboxy terminus (residue 206), and HP1a-CSDΔ204 corresponds to residues 131–203. Point mutations were prepared in the HP1a-CSD construct using the QuikChange kit (Agilent Technologies).
Protein expression of wild-type and mutant versions of HP1a
Vectors were transformed into BL21-CodonPlus (DE3)-RIL cells (Strategene) and expressed following induction with IPTG (1 mM) for 4 hours at 17–20°C. Typically 4 L of culture were grown in LB media, and the harvested cells were sonicated for 30 minutes in lysis/wash buffer (180 mL, 50 mM NaH2PO4, 500 mM NaCl, 20 mM imidazole, pH 8.0). All constructs yielded protein in the soluble fraction. The supernatant of the lysate was loaded onto 2–4 mL of Ni-NTA agarose (Qiagen, Valencia, CA), and washed with 1 L of lysis/wash buffer as reported above. Highly pure protein was eluted in each case using the elution buffer containing NaH2PO4 (50 mM, pH 8.0), NaCl (100 mM), imidazole (250 mM). The protein concentrations were determined by absorption using computed extinction coefficients HP1a, ε280=26930 M−1 cm−1, HP1a-CSD, ε280=8480 M−1 cm−1; HP1a-CSD-W200A, ε280=2980 M−1 cm−1; HP2, ε280=6990 M−1 cm−1, HP1β, ε280=28085 M−1 cm−1.
NMR Spectroscopy
NMR data were collected using a Bruker AVANCE (600 MHz or 800 MHz) spectrometer equipped with a cryoprobe. For 2D 1H-15N HSQC experiments, proteins were 15N-labeled using minimal media containing 15NH4Cl. HP1a spectra were collected and analyzed as described [5b]. Chemical shift perturbations were assigned by comparing them to the previously assigned HP1a spectrum [5a]. To determine the sequential assignments of the CSD in the absence and presence of the bound peptide, we used 15N and 13C-labeled protein produced by growth on minimal media containing 15NH4Cl and 13C-glucose. Protein concentrations used for HSQC spectra and titration studies ranged from 100 to 600 μM. For example, a 100 μM 15N-labeled HP2 was measured at 600 MHz in the absence and presence of 600 μM unlabeled full-length HP1a or CSD protein in buffer containing phosphate (25 mM, pH 7.0), NaCl (150 mM), DTT (1 mM), D2O (5%) at 20 °C (details available upon request).
Sequential assignment for the backbone of the CSD in the presence of each peptide was completed by collecting data from triple resonance experiments HNCA, HNCOCA, HNCO, and 15N-edited NOEY at 800 MHz [26]. A 200 μM 15N, 13C-labeled CSD protein in the presence of unlabeled 400 μM HP2 or PIWI peptide was used. The weighted average chemical shift difference due to peptide binding at each amino acid was calculated by using the relationship 0.5[(ΔHN)2 + (ΔN/5)2]1/2, where Δi is the chemical shift difference for resonance i in the free and complexed states.
The {1H}-15N steady-state heteronuclear NOEs for the backbone 15N nuclei of the free CSD (600 μM) were measured by collecting 2D spectra at 800 MHz with a water flip-back method [27]. Two different spectra were recorded in an interleaved manner with and without proton saturation during 3 seconds. The heteronuclear NOE value for each residue was calculated as the ratio of the crosspeak intensity NOE/reference, and error was estimated from the baseline noise in spectra. All spectra were processed using NMRPipe [28] and analyzed with NMRView [29].
Gel Filtration
A 12 inch Superdex 75 gel filtration column was used and loaded with 100 μL of each sample in buffer containing phosphate (50 mM, pH 7.2), NaCl (150 mM), at 4°C. The flow rate was 0.5 mL/min. To study the stoichiometry of the HP1 complex with HP2, 167 μM HP1-CSDΔ204 (considered as one unit) and various ratios of the HP2 fragment were loaded.
Analytical Ultracentrifugation
We performed equilibrium sedimentation using a Beckman Optima XLA instrument, analyzing HP1-CSD in buffer containing phosphate (50 mM, pH 8.0) and NaCl (25 mM). For the mutant HP1a-CSD-W200A, full length HP1a, mouse/human HP1β, and one run of HP1a-CSD-CTE the buffer contained NaCl (150 mM). The speeds used were 30, 35, and 45 krpm at 15 °C for the chromoshadow domain units and 10, 15, and 20 krpm at 15 °C for the full length protein of HP1. The program Ultrascan was used to process and analyze the data [30]. A single species model was used first to fit the data. The density of the buffer was 1.0037 g/cm3, the partial specific volume was calculated based on the amino acid composition (0.7232 cm3/g for HP1a-CSD, HP1a-CSDΔ204, and the phosphomimic; 0.7159 cm3/g for HP1a; 0.7204 cm3/g for HP1a-CSD-I191E; 0.7251 cm3/g for mammalian HP1β). If this approach was insufficient to give a random distribution of residuals, the monomer-dimer model was tested with the molecular weight fixed to the calculated size, and the data fitted to determine the dimerization constant. The molecular weights are 23.2 kDa for HP1a, 10.02 kDa for HP1a-CSD and HP1a-CSD-E195Q, 9.66 kDa for HP1a-CSDΔ204, 10.04kDa for HP1a-CSD-I191E (fitted 9.62 kDa), 9.91 kDa for HP1a-CSD-W200A, 3.09 kDa for the HP2 peptide, and 21.4 kDa for mammalian HP1β. To estimate the variation in the equilibrium constant we assumed 8% error in the data to insure that we sampled the possible equivalent fits and did not sample noise, and 95% confidence intervals were obtained.
Circular Dichroism
A Jasco J-815 instrument with a 1 mm pathlength cuvette was used to measure the far-UV spectrum of 1 mg/mL (100 μM) HP1a-CSD wild-type or I191E mutant in a buffer containing phosphate (50 mM, pH 8.0), NaCl (25 mM) at 15° C. Scan speed was set to 50 nm/min with 4 replicates, data pitch of 0.2 nm, response of 1 sec and the band width of 1 nm. The molar ellipticity was calculated and plotted [31].
Coimmunoprecipitation experiments
Immunoprecipitation experiments were carried out as described previously [10]. In brief, Promega’s Quick coupled rabbit reticulocyte lysate system was used to express full length HP1a and the HP2 recombinant fragment (Figure 1A) from the pET28a vector, incubating at 30°C for 60–90 minutes. Protein interactions were identified by pull-down with the antibody WA192 generated against the chromodomain of HP1a. This antibody does not cross-react with HP2 (see lane 3 in Figure 1D).
Synthetic Peptides
Peptides were synthesized at the Yale University Keck facility. Non-native residues are underlined including a Tyr added to the PIWI peptide for monitoring concentration. For fluorescence assays the peptides were synthesized with an N-terminal fluorescein group.
HP2 peptide wild type: NH2-QNISPRKLCVKINRRPYNKWLR-COOH,
HP2 peptide Cys to Ser mutant: NH2-QNISPRKLSVKINRRPYNKWLR-COOH,
HP2 peptide with PIWI pentamer: NH2-QNISPRKPRVKVNRRPYNKWLR-COOH,
PIWI peptide: PIWI NH2-TSRGSGDPRVKVFRGSSSGDY-COOH
PIWI peptide with HP2 pentamer: NH2-TSRGSGDLSVKIFRGSSSGDY-COOH.
The AF10 peptide: NH2-PATTSPLVVLSNQLPLNSVAASY-COOH, and the CG15356 peptide: NH2-SEGRRLYPLLPRISPQTALSIVANDLAESD-COOH were labeled as previously described [32].
Fluorescence Polarization Binding Assay
This assay was carried out as previously described [32] using N-terminally fluoresceinated peptides. Assays were performed at 15 °C in buffer containing phosphate (50 mM, pH 8.0) and NaCl (25 mM). The binding constants were determined using Scientist (Micromath, St Louis, MO) assuming a binding model that accounts for HP1a dimers binding to a peptide. Plots were prepared using Kaleidagraph (Synergy Software). Since we are following changes in peptide polarization, the fraction of the total peptide bound was plotted:
| (1) |
where KPeptide is the affinity of the peptide for the dimer and HP1aDimer is the concentration of the free HP1a dimers. The concentration of free HP1a dimers is given in the conservation of mass equation:
| (2) |
and is a function of the total concentration of HP1a and HP1a’s self association constant. Thus
| (3) |
Rearranging (3) to solve for the number of monomers as a function of total HP1a concentration via the quadratic formula we have:
| (4) |
The conservation of mass allows for the concentration of HP1a monomers to be calculated which when subtracted from the total HP1a pool gives a concentration for the amount of HP1a dimers:
| (5) |
In the case where the Hill coefficient, nH, was included, equation (1) had [HP1adimers] raised to the nH power.
Equations (1), (4), and (5) were used in the Scientist to find the least squares solution to KPeptide. In this model we exclude binding to monomers of HP1a. Given that the data show a 100-fold weaker binding to the monomer as compared to the dimer, inclusion of the binding to the monomer does not change the KPeptide value (Table II).
X-ray Crystallography
Crystallization was carried out using the vapor diffusion, hanging drop method. 10 mg/mL of protein solution was prepared in imidazole (10 mM, pH 7) and NaCl (40 mM). Crystals grew from PEG400 (12%), HEPES (0.1 M, pH 7.5) at 4 °C and were cryoprotected in PEG400 (12%), HEPES (0.1 M, pH 7.5) and glycerol (33%). The crystals diffracted to 2.3 Å at Argonne National Laboratory beam-line 19 BM and were indexed with HKL2000 [33]. The structure was solved by molecular replacement using the human HP1β CSD (PDB 2FMM) as the search model (Table I). The structure was determined by molecular replacement using PHASER [34]. The program PHENIX [35], COOT [15] and REFMAC [36] were used for the refinement. MOLPROBITY was used for structure validation [37]. Diagrams were prepared with PyMOL [38].
Supplementary Material
Acknowledgments
We thank members of the Elgin and Khorasanizadeh labs for critical review of the manuscript; Thomas Ream and the C. Pikaard lab for use of their FPLC; the R. Blankenship lab for use of their FPLC and CD instruments. We thank Jeff Iwig, Roberto Galleto, and the Biochemistry Department at Washington University for their assistance and use of the AUC. Katrina Clines and Kimberly Wiggins for the HP1β construct.
Deanna Mendez expressed protein and made the wild type and mutant constructs, expressed and purified protein, and performed the AUC, fluorescence polarization binding assays, gel filtration, and CoIP (Figure 1E) experiments and wrote the paper. Daesung Kim performed NMR spectroscopy and analysis and made crystals. Maksymilian Chruszcz acquired X-ray diffraction data under W. Minor’s supervision. Gena Stephens performed CoIP experiments for Figure 1D. SCR Elgin and S Khorasanizadeh assisted with experimental design and interpretation, as well as with preparing the manuscript. This work was supported by NIH grants R01 GM 68388 (SCRE) and GM 070558 (SK). Crystallography results are derived in part from work performed at the Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by U Chicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357.
Footnotes
Accession Numbers
Coordinates and structure factors have been deposited in the Protein Data Bank with accession number 3P7J.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
Contributor Information
Prof. Sepideh Khorasanizadeh, Email: sepideh@sanfordburnham.org.
Prof. Sarah C.R. Elgin, Email: selgin@biology.wustl.edu.
References
- 1.Huisinga KL, Brower-Toland B, Elgin SC. Chromosoma. 2006;115:110. doi: 10.1007/s00412-006-0052-x. [DOI] [PubMed] [Google Scholar]
- 2.a) Eissenberg JC, James TC, Foster-Hartnett DM, Hartnett T, Ngan V, Elgin SC. Proc Natl Acad Sci U S A. 1990;87:9923. doi: 10.1073/pnas.87.24.9923. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) James TC, Elgin SC. Mol Cell Biol. 1986;6:3862. doi: 10.1128/mcb.6.11.3862. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Vermaak D, Malik HS. Annu Rev Genet. 2009;43:467. doi: 10.1146/annurev-genet-102108-134802. [DOI] [PubMed] [Google Scholar]
- 3.Hiragami K, Festenstein R. Cell Mol Life Sci. 2005;62:2711. doi: 10.1007/s00018-005-5287-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.James TC, Eissenberg JC, Craig C, Dietrich V, Hobson A, Elgin SC. Eur J Cell Biol. 1989;50:170. [PubMed] [Google Scholar]
- 5.a) Jacobs SA, Khorasanizadeh S. Science. 2002;295:2080. doi: 10.1126/science.1069473. [DOI] [PubMed] [Google Scholar]; b) Jacobs SA, Taverna SD, Zhang Y, Briggs SD, Li J, Eissenberg JC, Allis CD, Khorasanizadeh S. EMBO J. 2001;20:5232. doi: 10.1093/emboj/20.18.5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grewal SI, Jia S. Nat Rev Genet. 2007;8:35. doi: 10.1038/nrg2008. [DOI] [PubMed] [Google Scholar]
- 7.Lomberk G, Bensi D, Fernandez-Zapico ME, Urrutia R. Nat Cell Biol. 2006;8:407. doi: 10.1038/ncb1383. [DOI] [PubMed] [Google Scholar]
- 8.a) Kwon SH, Workman JL. Mol Cells. 2008;26:217. [PubMed] [Google Scholar]; b) Huang Y, Myers MP, Xu RM. Structure. 2006;14:703. doi: 10.1016/j.str.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 9.Smothers JF, Henikoff S. Curr Biol. 2000;10:27. doi: 10.1016/s0960-9822(99)00260-2. [DOI] [PubMed] [Google Scholar]
- 10.Stephens GE, Slawson EE, Craig CA, Elgin SC. Biochemistry. 2005;44:13394. doi: 10.1021/bi051006+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cowieson NP, Partridge JF, Allshire RC, McLaughlin PJ. Curr Biol. 2000;10:517. doi: 10.1016/s0960-9822(00)00467-x. [DOI] [PubMed] [Google Scholar]
- 12.a) Brasher SV, Smith BO, Fogh RH, Nietlispach D, Thiru A, Nielsen PR, Broadhurst RW, Ball LJ, Murzina NV, Laue ED. EMBO J. 2000;19:1587. doi: 10.1093/emboj/19.7.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Thiru A, Nietlispach D, Mott HR, Okuwaki M, Lyon D, Nielsen PR, Hirshberg M, Verreault A, Murzina NV, Laue ED. EMBO J. 2004;23:489. doi: 10.1038/sj.emboj.7600088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brower-Toland B, Findley SD, Jiang L, Liu L, Yin H, Dus M, Zhou P, Elgin SC, Lin H. Genes Dev. 2007;21:2300. doi: 10.1101/gad.1564307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a) Shaffer CD, Cenci G, Thompson B, Stephens GE, Slawson EE, Adu-Wusu K, Gatti M, Elgin SC. Genetics. 2006;174:1189. doi: 10.1534/genetics.106.057604. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Shaffer CD, Stephens GE, Thompson BA, Funches L, Bernat JA, Craig CA, Elgin SC. Proc Natl Acad Sci U S A. 2002;99:14332. doi: 10.1073/pnas.212458899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Emsley P, Cowtan K. Acta Crystallogr D Biol Crystallogr. 2004;60:2126. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 16.Krissinel E, Henrick K. Acta Crystallogr D Biol Crystallogr. 2004;60:2256. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 17.Linder B, Gerlach N, Jackle H. EMBO Rep. 2001;2:211. doi: 10.1093/embo-reports/kve039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ayoub N, Jeyasekharan AD, Bernal JA, Venkitaraman AR. Nature. 2008;453:682. doi: 10.1038/nature06875. [DOI] [PubMed] [Google Scholar]
- 19.a) Zhao T, Eissenberg JC. J Biol Chem. 1999;274:15095. doi: 10.1074/jbc.274.21.15095. [DOI] [PubMed] [Google Scholar]; b) Zhao T, Heyduk T, Eissenberg JC. J Biol Chem. 2001;276:9512. doi: 10.1074/jbc.M010098200. [DOI] [PubMed] [Google Scholar]
- 20.Schwendemann A, Matkovic T, Linke C, Klebes A, Hofmann A, Korge G. Proc Natl Acad Sci U S A. 2008;105:204. doi: 10.1073/pnas.0705595105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.a) Eskeland R, Eberharter A, Imhof A. Mol Cell Biol. 2007;27:453. doi: 10.1128/MCB.01576-06. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Schotta G, Ebert A, Krauss V, Fischer A, Hoffmann J, Rea S, Jenuwein T, Dorn R, Reuter G. EMBO J. 2002;21:1121. doi: 10.1093/emboj/21.5.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lechner MS, Schultz DC, Negorev D, Maul GG, Rauscher FJ., 3rd Biochem Biophys Res Commun. 2005;331:929. doi: 10.1016/j.bbrc.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 23.Li Y, Kirschmann DA, Wallrath LL. Proc Natl Acad Sci U S A. 2002;99(Suppl 4):16462. doi: 10.1073/pnas.162371699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.a) Grewal SI, Elgin SC. Nature. 2007;447:399. doi: 10.1038/nature05914. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Brennecke J, Aravin AA, Stark A, Dus M, Kellis M, Sachidanandam R, Hannon GJ. Cell. 2007;128:1089. doi: 10.1016/j.cell.2007.01.043. [DOI] [PubMed] [Google Scholar]
- 25.Gauhar Z, Ghanim M, Herreman T, Lambert JD, Li TR, Mason C, Rifkin S, Sun L, White KO, Costello JC, Andrews JR. Flybase. 2008. FBrf0205914. [Google Scholar]
- 26.a) Kay LE, Xu GY, Yamazaki T. Journal of magnetic resonance. 1994;A109:129. [Google Scholar]; b) Clore GM, Gronenborn AM. NMR of Proteins. CRC Press; Ann Arbor: 1993. Determination of structures of larger proteins in solution by three-and four-dimensional heteronuclear magnetic resonance spectroscopy. [Google Scholar]
- 27.a) Piotto M, Saudek V, Sklenar V. J Biomol NMR. 1992;2:661. doi: 10.1007/BF02192855. [DOI] [PubMed] [Google Scholar]; b) Grzesiek S, Bax A. Journal of the American Chemical Society. 1993;115:12593. [Google Scholar]
- 28.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. J Biomol NMR. 1995;6:277. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 29.Johnson BA, Blevins RA. Journal of Biomolecular Nmr. 1994;4:603. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 30.Demeler B. UltraScan A Comprehensive Data Analysis Software Package for Analytical Ultracentrifugation Experiments. Royal Society of Chemistry; UK: 2005. [Google Scholar]
- 31.Kelly SM, Jess TJ, Price NC. Biochim Biophys Acta. 2005;1751:119. doi: 10.1016/j.bbapap.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 32.Jacobs SA, Fischle W, Khorasanizadeh S. Methods Enzymol. 2004;376:131. doi: 10.1016/S0076-6879(03)76009-1. [DOI] [PubMed] [Google Scholar]
- 33.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 34.McCoy AJ, Grosse-Kunstleve, Adams RW, Winn MD, Storoni LC, Read RJ. Journal of Applied Crystallograpy. 2007;40:658. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCoy AJ. Acta Crystallogr D Biol Crystallogr. 2007;63:32. doi: 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murshudov GN, Vagin AA, Dodson EJ. Acta Crystallogr D Biol Crystallogr. 1997;53:240. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 37.Lovell SC, Davis IW, Arendall WB, 3rd, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Proteins. 2003;50:437. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 38.DeLano WL. DeLano Scientific. San Carlos, CA: 2004. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.