

Abstract
Objectives
Few effective options are available for the treatment of unresectable hepatocellular carcinoma (HCC). Several phase I trials suggest promising activity of a combination of gemcitabine and docetaxel.
Methods
Patients with unresectable or metastatic HCC were treated with docetaxel 40 mg/m2 (later reduced to 30 mg/m2) and gemcitabine 800 mg/m2 on days 1, 8 every 3 weeks. Twenty-five patients were enrolled in 26 months. Median age was 64 (range 27-78), 17 were male, 14 had liver-only disease and, 11 had extrahepatic disease.
Results
Of 25 patients evaluable for the primary endpoint (response), 2 (8%) have a confirmed partial response. The median TTP is 2.76 months (95% CI 1.84-6.64 months). Median survival was 12.8 months (95% CI: 5.26-28.00). Two patients died on-study due to adverse events (1 hepatic and 1 renal failure), neither of which were attributed to the study medications. Twenty patients (81%) have experienced grade 3+ adverse events, including 11 with grade 4+ adverse events, primarily neutropenia, thrombocytopenia, diarrhea, and fatigue.
Conclusions
While this combination appears to have potential benefit, as measured by overall survival, its toxicity and the recent introduction of sorafenib has further limited the use of chemotherapy. Approaches other than chemotherapy are likely to be of the greatest potential benefit.
Introduction
Prior trials of chemotherapy for locally advanced or metastatic hepatocellular carcinoma (HCC) have shown limited benefit. For many years doxorubicin served as a standard of therapy with a response rates of 10% or less. Until the recent introduction of sorafenib, there had been on going efforts to identify newer chemotherapy agents with potential activity in HCC. Gemcitabine has broad activity in a variety of solid tumors including upper gastrointestinal tumors.1 In preclinical studies gemcitabine has shown strong activity against liver cancer cell lines.2 However, phase II clinical trials with single agent gemcitabine have shown limited activity with response rate ranging from 0-20%.3-8
Docetaxel has also shown a wide spectrum of antitumor activity.9 A large number of phase I and II trials of gemcitabine and docetaxel have been performed, a portion of these trials have been summarized.9 Several different schedules of administration have been used including four-week, three-week, and every other week regimens. With the three week regimen Bhargava et al found a maximum tolerated dose of gemcitabine 800 mg/m2 and docetaxel 40 mg/m2 with both drugs given on days 1 and 8.10 The major limitation to its use in HCC is based on its liver metabolism. Docetaxel undergoes extensive hepatic metabolism. Patients with significant hepatic dysfunction will have a reduced clearance of docetaxel.11-13 In one trial a 30% reduction in docetaxel clearance was observed.12
No prior studies have formally assessed the activity of gemcitabine and docetaxel in HCC. However, in a previously reported phase I trial of this combination a patient with HCC had a reported response.14 The patient had a 33% reduction in tumor mass and a 90% reduction in the alpha-fetoprotein. The response was maintained for 8 cycles. Based on broad preclinical activity of gemcitabine and docetaxel, as well as evidence of activity from a phase I trial, a phase II trial of this combination was conducted in patients with HCC. This trial also included an assessment of docetaxel pharmacokinetics given uncertainty of its clearance in patients receiving gemcitabine.
Methods
Eligibility
Patients with histologic or cytologic confirmation of HCC deemed unresectable and not candidates for liver transplant were eligible for enrollment in this phase II trial. Patients were required to be at least 18 years old and to have an Eastern Cooperative Oncology Group (ECOG) performance score of 0-2. Hematologic and chemistry parameters were to be in the following ranges: absolute neutrophil count ≥ 1.5 × 109/L, platelets ≥ 100 × 109/L, total bilirubin ≤ 1.5 times the institutional upper normal limit (UNL), AST ≤ 2.5 times the UNL, and creatinine ≤ 1.5 times the UNL. Patients also needed to have some evidence of tumor progression (radiologic changes, biochemical changes, increase in tumor marker) to be eligible.
Prior use of gemcitabine and prior radiation to >25% of bone marrow were not allowed. Patients were not allowed to be pregnant or lactating and required to use adequate contraception methods to prevent pregnancy during treatment. Other contraindications included a history of brain or other CNS metastases not amenable to local therapy.
Patients with locally treatable disease were eligible if treatment was completed at least 4 weeks prior and there no evidence of CNS progression. Prior biologic, hormonal, or immunologic therapy was allowed if greater than 4 weeks of study entry. Any history of prior malignancy diagnosed within 5 years was not allowed, with the exception of basal or squamous cell carcinoma or the skin and cervical carcinoma in situ.
This study was approved by the Mayo Institutional Review Board (IRB) and by the IRBs of the individual memberships of the NCCTG that elected to participate in this study. A signed written informed consent was obtained from all patients prior to initiating therapy. This study was funded through the NCCTG grant from the National Cancer Institute. There was no direct industrial funding of this study.
Treatment
One 3-week course of treatment involved the administration of dexamethasone, docetaxel (provided by sanofi-aventis Pharmaceuticals), and gemcitabine (provided by Eli Lilly and Company) given on days 1 and 8. Dexamethasone 10 mg was given intravenously (IV) over 30 minutes prior to docetaxel. An initial dose of docetaxel 40 mg/m2 was used in this trial. However, after excessive toxicity was observed in the first seven patients the dose was reduced. For the remainder of the trial docetaxel 30 mg/m2 was administered over 60-minutes in 250 mL D5W or normal saline followed by gemcitabine 800 mg/m2 over 30-minutes in 250 ml solution of normal saline. Cycles were repeated every 3 weeks if patients met criteria for further therapy. Dose adjustments were made as per a study defined dose modification table depending on the type and severity of treatment related toxicities. Treatment was given until progression, intolerance, or patient's request to discontinue.
Patient Evaluation
Within 14 days prior to enrollment, patients were required to undergo a complete history, physical exam, serum pregnancy test, and tumor assessment via computer tomography (CT) or magnetic resonance imaging (MRI). A chest x-ray was required within 28 days prior to enrollment. Blood chemistries collected included: creatinine, AST, alkaline phosphatase, and total and direct bilirubin. Hemoglobin, platelets, differential, and white blood counts were also recorded.
During the course of treatment, hematologic parameters were collected weekly. Prior to the next course of treatment, a history of adverse events experienced was collected and blood chemistries were repeated. Adverse events were collected using the National Cancer Institute's Common Toxicity Criteria Version 2.0 (NCI CTC V2.0, http://ctep.cancer.gov/reporting/ctc.html). Unless noted otherwise, all adverse events are reported, regardless of attribution to study treatment. Tumor measurements were repeated at 6 and 12 weeks, then every 6-12 weeks unless more frequent assessments were needed to document response. A chest x-ray was to be performed as clinically indicated during treatment. Following the discontinuation of study treatment, patients were observed for disease progression every 3 months for 1 year, then every 6 months for up to 4 years past their registration date. At the time of disease progression, patients were monitored for their status for a maximum of 4 years post-registration.
Disease Assessment
A measurable indicator lesion was defined as a tumor mass with clearly defined bidimensional measurements whose minimum size is at least 1.0 cm on chest radiograph or at least 2.0 cm on computed tomography scan, magnetic resonance imaging, or ultrasound. An assessable indicator lesion was defined as a tumor mass on physical examination or imaging study that can be assessed as to its changes in size but cannot be clearly measured in two dimensions. Response was defined as any one or more of the following: total disappearance of tumor (complete response [CR]), a reduction of at least 50% in the sum of the products of the perpendicular diameters of a measurable lesion (partial response [PR]), or definite clinical evidence of a decrease in the size of an assessable lesion (regression [REG]). Confirmed response was defined as two consecutive evaluations at least 6 weeks apart showing one of these reponses (CR, PR, REG). Progression was defined as the appearance of new lesions; a 25% or greater increase (definite increase) in the size of the indicator lesion from its smallest size for those patients who achieved a PR (REG); or a 25% or greater increase (definite increase) in the size of the indicator lesion from its pretreatment size for those patients who did not achieve a PR (REG). Stable disease is the failure to meet the criteria for response or progression.
Patients were also considered to have progressed in cases of significant clinical deterioration that could not be attributed to study treatment or other medical conditions. These conditions included worsening of tumor-related symptoms, ≥ 10% weight loss, or a decline in PS of > 1 level. Subsequent treatment was at the treating physician's discretion.
Duration of response was calculated from the first date of a patient's objective status of either a complete response (CR) or partial response (PR) to the date of disease progression (or last tumor assessment). Time to disease progression is calculated from the date of registration to the date of disease progression (or last tumor assessment). Time to death is calculated from the date of registration to the date of death (or last contact). Patients having been lost to follow-up were counted as having no progression (alive) on their date of last tumor assessment (contact).
Statistical Considerations
The primary endpoint of this trial was confirmed response within the first 6 cycles. A confirmed tumor response was defined to be either a CR or PR noted as the objective status on 2 consecutive evaluations at least 6 weeks apart. The two-stage Simon design was sized to give 90% power, and a significance of 0.04, when testing for a 20% response rate versus a 5% response rate for standard care. The first stage enrolled 22 patients and if 2 or more responses were seen the second stage would have enrolled an additional 18 patients.
Docetaxel Analysis
Non-linear mixed effects modeling of the plasma concentration data was performed using NONMEM (version VI).15 The data were fit to a three-compartment population pharmacokinetic model for docetaxel in 280 cancer patients developed by Bruno et al.16 Modeling of the plasma concentration data was performed essentially as previously described by Adjei et al.17 The structural model from this study was fitted to the data from the eleven patients who received docetaxel and gemcitabine using the ADVAN 5 (TRANS=1) subroutine in NONMEM. AUC and half-life estimates were calculated as secondary parameters for each individual. The data were first analyzed using a naïve approach in which the observed plasma concentrations from the eleven patients in this study were fitted to the model assuming no prior knowledge of the pharmacokinetic parameter estimates. The influence of dose level and body surface area (BSA) on individual parameter estimates were tested using generalized additive models (GAM) implemented in Xpose (version 3.1).18 To determine the influence of co-administration of gemcitabine on the pharmacokinetics of docetaxel, twenty data sets were simulated in NONMEM using the model parameters developed by Bruno et al,16 and the sampling times and doses from the patients in this study. This data which simulated the effect of administration of docetaxel alone on 220 patients was combined with the observed data from the eleven patients in this study and then analyzed by NONMEM using the structural model mentioned above. Gemcitabine was included as a categorical covariate and tested against all of the pharmacokinetic parameters using forward inclusion and backward elimination.19 During forward inclusion the influence of gemcitabine was evaluated using the likelihood ratio test in which the change in the objective function value (-2 log-likelihood difference [-2 LLD]) approximates the Chi-squared distribution (χ2[1, 0.05]>3.8). Gemcitabine effect was then eliminated as a covariate from each parameter one at a time, and evaluated again using the likelihood ratio test, but using more stringent statistical conditions (χ2[1, 0.01]>6.63).
Results
Patient Characteristics and Outcomes
Between October 23, 2001, and November 26, 2003, 25 patients were enrolled to this trial at the time it was stopped for slow accrual and an interim analysis. The median age of enrolled patients was 64, with approximately two-thirds of the patients being male and two-thirds being Caucasian (Table 1). Limited information was available on pre-existing liver disease. In patients where information was available 9 patients had a clinical history of cirrhosis. Six patients had chronic viral hepatitis, 3 had alcoholic cirrhosis, and 1 had hemochromatosis. The HCC was restricted to the liver in 14 patients.
At the time of this analysis in March 2008, of the 25 patients enrolled in the trial 23 patients have died. Two patients remain alive and both these patients have completed protocol defined follow-up. Eighteen patients discontinued treatment due to disease progression, 4 due to an adverse event, 1 had alternate treatment, 1 died on study, and 1 completed treatment per protocol. Twenty-four patients progressed while 1 patient died seven days after going on study without evidence of progression.
All 25 patients are considered evaluable for response. The overall confirmed response rate, consisting of two partial responses, was 8% (95% CI: 1.0, 26.0). The duration of responses for these two patients were 4.4 and 14.7 months, respectively. An additional 11 patients had stable disease.
Ninety-two percent (23/25) of patients have died and 96% (24/25) have progressed. One patient died on study without clinical evidence of progression. The follow-up for the 2 surviving patients was 54.8 and 59.3 months, respectively, at the time of the last analysis. Median overall survival and time-to-progression was 12.8 months (95% CI: 5.26-28.00) and 2.76 months (95% CI: 1.84-6.64), respectively (Figure 1).
Figure 1.
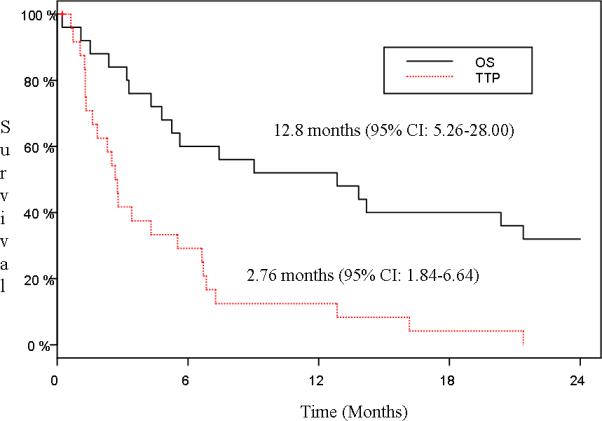
Overall Survival (OS) and Time to Progression (TTP) (N=25)
Drug Administration
The trial was temporarily closed on January 31, 2002, for an evaluation of toxicity due to a death on study. The study was re-opened on June 28, 2002, with a revised dose of 30mg/m2 for docetaxel. The original dose of docetaxel of 40mg/m2 was administered to seven patients. Patients received a median of 3 cycles (range 1-16) of treatment. One patient received approximately 20% more than the intended dose of docetaxel for cycles 1 and 2 because of an error in the calculation of the dose. Only 6 patients received at least 6 cycles of treatment, while 8 patients received only one cycle of treatment. A summary of the first 6 cycles of docetaxel and gemcitabine for percent of target dose is in Table 2. The percentage of dose delivered did not differ between patients at the two different dose levels of docetaxel.
Toxicity
Toxicities are summarized in Table 3. Twenty patients experienced at least one grade 3+ adverse event. Eleven patients experienced a grade 4+ adverse event. The most common severe adverse events (grade 3+) were neutropenia (52%), thrombocytopenia (28%), fatigue (16%), and diarrhea (16%). Six patients experienced a grade 4 neutropenia. Two grade 5 events were observed for patients on treatment. The first death was due to hepatic failure and was unlikely to be related to study treatment. The second death was due to renal failure and was not felt to be related to study treatment by the treating physician.
Docetaxel Pharmacokinetics
Naïve fitting of the plasma concentration data from the eleven patients that received docetaxel and gemcitabine to the three compartment model reported by Bruno et al16 showed no significant relationship between either BSA or dose on any of the population pharmacokinetic parameters. Combining simulated data from 220 patients receiving docetaxel alone with the observed data from eleven patients who received docetaxel and gemcitabine allows direct testing of the influence of gemcitabine on the pharmacokinetics of docetaxel. Population pharmacokinetic parameter estimates from this analysis are shown in Table 4, alongside the estimates from Bruno et al.16 Clearance of docetaxel from the central compartment was estimated to be 36.7 L/hr (95%CI, 34.3 to 39.2 L/hr). This is very similar to the estimated for clearance of docetaxel of 38.5 L/hr (95% CI, 36.2 to 38.7 L/hr) reported by Bruno et al in 280 patients (Table 4).16 The influence of gemcitabine as a categorical covariate is not significant on clearance or volume of distribution for the central compartment. However, co-administration of gemcitabine does appear to cause a 211% increase (p<0.01) in the intercompartmental rate constant k21.
Discussion
Until the recent introduction of sorafenib as a potential therapeutic option for patients with advanced HCC, chemotherapy had remained as the primary form of treatment for patients with disease not amenable to local modalities of therapy. While doxorubicin had been viewed as a standard of care, it has limited efficacy. Objective responses to doxorubicin have generally been around 10% with an associated short median survival.20-22 A variety of phase II studies using an anthracycline combined with another agent have not shown consistent improvement in outcome compared to single agent trials.23 Based on the limited activity of doxorubicin a variety of other chemotherapy agents have been evaluated.
Gemcitabine used alone is of limited benefit.3-8 However, gemcitabine combinations have shown more promising results. The combination of gemcitabine and doxorubicin was evaluated in a phase I-II trial.24 Of the 34 evaluable patients in this trial, a partial response rate of 12% was observed. Overall, 56% of patients had stable or responsive disease. The median survival was 4.6 months. A trial using the combination of gemcitabine and oxaliplatin has also been reported. Using a two-day schedule every two weeks of this combination resulted in an 18% response rate together with a disease stabilization rate of 58%.25. The median survival was 11.5 months.
In our trial the combination of gemcitabine and docetaxel had moderately severe toxicity. The overall response rate was 8% and disease stabilization rate was 12% with a median survival of 12.8 months. While the response rate was lower than that reported for gemcitabine and oxaliplatin, the overall survival appears similar. While the use of response rates has served as a surrogate marker for clinical trials in oncology, its use in trials for HCC is of less certain significance given the increased difficulty of accurately measuring a mass within a cirrhotic liver. Overall survival remains as the most important endpoint.26 While the survival data in our trial appear promising, it is important to consider the characteristics of the patients enrolled relative to other trials. The proportion of patients in our trial with liver-only disease (44%) was lowered compared to patients enrolled in the trial with gemcitabine and oxaliplatin (50%). While the indolent nature of some HCC tumors may have also influenced the outcome, our trial did require evidence of progressive disease prior to enrollment.
The combination of gemcitabine and docetaxel did not appear to cause a meaningful alteration in the clearance or volume of distribution of docetaxel based on the pharmacokinetic studies performed. The dose of docetaxel was reduced part way through this trial, in part related to events were of uncertain relationship to the use of the chemotherapy regimen used in this trial. Pending further validation of the doses for this combination, when used in patients with HCC, it is recommended that the lower dose of gemcitabine 800 mg/m2 and docetaxel 30 mg/m2 be used. While this combination appears to have some potential benefit, the recent introduction of sorafenib has further limited the use of chemotherapy in HCC.
Footnotes
This study was conducted as a trial of the North Central Cancer Treatment Group and Mayo Clinic and was supported in part by Public Health Service grants CA-25224, CA-37404, CA-35103, CA-35272, CA-35431, CA-35195, CA-45450, CA-35269, and CA-63849 from the National Cancer Institute Department of Health and Human Services.
Additional participating institutions include: Duluth CCOP, Duluth, MN 55805 (Daniel Nikcevich, M.D.); Missiouri Valley Cancer Consortium, Omaha, NE 68131 (Gamini Soori, M.D.)
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Guchelaar HJ, Richel DJ, van Knapen A. Clinical, toxicological and pharmacological aspects of gemcitabine. Cancer Treat Rev. 1996;22(1):15–31. doi: 10.1016/s0305-7372(96)90014-6. [DOI] [PubMed] [Google Scholar]
- 2.Graziadei I, Kelly T, Schirmer M, Geisen FH, Vogel W, Konwalinka G. Antitumor effect of the nucleoside analogs 2-chlorodeoxyadenosine and 2',2'-difluorodeoxycytidine on human hepatoma HepG2 cells. J Hepatol. 1998;28(3):504–9. doi: 10.1016/s0168-8278(98)80326-7. [DOI] [PubMed] [Google Scholar]
- 3.Parikh PM, Fuloria J, Babu G, et al. A phase II study of gemcitabine and cisplatin in patients with advanced hepatocellular carcinoma. Trop Gastroenterol. 2005;26(3):115–8. [PubMed] [Google Scholar]
- 4.Guan Z, Wang Y, Maoleekoonpairoj S, Chen Z, et al. Prospective randomised phase II study of gemcitabine at standard or fixed dose rate schedule in unresectable hepatocellular carcinoma. Brit J Cancer. 2003;89(10):1865–9. doi: 10.1038/sj.bjc.6601369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fuchs CS, Clark JW, Ryan DP, et al. A phase II trial of gemcitabine in patients with advanced hepatocellular carcinoma. Cancer. 2002;94(12):3186–91. doi: 10.1002/cncr.10607. [DOI] [PubMed] [Google Scholar]
- 6.Kubicka S, Rudolph KL, Tietze MK, Lorenz M, Manns M. Phase II study of systemic gemcitabine chemotherapy for advanced unresectable hepatobiliary carcinomas. Hepato-Gastroenterol. 2001;48(39):783–9. [PubMed] [Google Scholar]
- 7.Ulrich-Pur H, Kornek GV, Fiebiger W, Schull B, Raderer M, Scheithauer W. Treatment of advanced hepatocellular carcinoma with biweekly high-dose gemcitabine. Oncology. 2001;60(4):313–5. doi: 10.1159/000058526. [DOI] [PubMed] [Google Scholar]
- 8.Yang TS, Lin YC, Chen JS, Wang HM, Wang CH. Phase II study of gemcitabine in patients with advanced hepatocellular carcinoma. Cancer. 2000;89(4):750–6. doi: 10.1002/1097-0142(20000815)89:4<750::aid-cncr5>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 9.van Oosterom AT, Schrijvers D, Schriivers D. Docetaxel (Taxotere), a review of preclinical and clinical experience. Part II: Clinical experience. Anti-Cancer Drugs. 1995;6(3):356–68. doi: 10.1097/00001813-199506000-00002. erratum appears in Anticancer Drugs 1995 Aug;6(4):618. [DOI] [PubMed] [Google Scholar]
- 10.Bhargava P, Marshall JL, Fried K, et al. Phase I and pharmacokinetic study of two sequences of gemcitabine and docetaxel administered weekly to patients with advanced cancer. Cancer Chemother Pharmacol. 2001;48(2):95–103. doi: 10.1007/s002800100317. [DOI] [PubMed] [Google Scholar]
- 11.Clarke SJ, Rivory LP. Clinical pharmacokinetics of docetaxel. Clin Pharmacokinetics. 1999;36(2):99–114. doi: 10.2165/00003088-199936020-00002. [DOI] [PubMed] [Google Scholar]
- 12.Bruno R, Vivier N, Veyrat-Follet C, Montay G, Rhodes GR. Population pharmacokinetics and pharmacokinetic-pharmacodynamic relationships for docetaxel. Invest New Drugs. 2001;19(2):163–9. doi: 10.1023/a:1010687017717. [DOI] [PubMed] [Google Scholar]
- 13.Donelli MG, Zucchetti M, Munzone E, D'Incalci M, Crosignani A. Pharmacokinetics of anticancer agents in patients with impaired liver function. Eur J Cancer. 1998;34(1):33–46. doi: 10.1016/s0959-8049(97)00340-7. [DOI] [PubMed] [Google Scholar]
- 14.Rizvi NA. Docetaxel (Taxotere) and gemcitabine in combination therapy. Semin Oncol. 1999;26(3 Suppl 11):19–22. [PubMed] [Google Scholar]
- 15.Beal SL, Sheiner LB. NONMEN users guide. ed 3 NONMEN Project Group, University of California at San Francisco; San Francisco: 1989. [Google Scholar]
- 16.Bruno R, Vivier N, Vergniol JC, De Phillips SL, Montay G, Sheiner LB. A population pharmacokinetic model for docetaxel (Taxotere): model building and validation. J Pharmacokinetics & Biopharmaceutics. 1996;24(2):153–72. doi: 10.1007/BF02353487. [DOI] [PubMed] [Google Scholar]
- 17.Adjei AA, Klein CE, Kastrissios H, et al. Phase I and pharmacokinetic study of irinotecan and docetaxel in patients with advanced solid tumors: preliminary evidence of clinical activity. J Clin Oncol. 2000;18(5):1116–23. doi: 10.1200/JCO.2000.18.5.1116. [DOI] [PubMed] [Google Scholar]
- 18.Jonsson EN, Karlsson MO. Xpose--an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Computer Methods & Programs in Biomedicine. 1999;58(1):51–64. doi: 10.1016/s0169-2607(98)00067-4. [DOI] [PubMed] [Google Scholar]
- 19.Jonsson EN, Karlsson MO. Automated covariate model building within NONMEM. Pharmaceutical Research. 1998;15(9):1463–8. doi: 10.1023/a:1011970125687. [DOI] [PubMed] [Google Scholar]
- 20.Chlebowski RT, Brzechwa-Adjukiewicz A, Cowden A, Block JB, Tong M, Chan KK. Doxorubicin (75 mg/m2) for hepatocellular carcinoma: clinical and pharmacokinetic results. Cancer Treat Report. 1984;68(3):487–91. [PubMed] [Google Scholar]
- 21.Lai CL, Wu PC, Chan GC, Lok AS, Lin HJ. Doxorubicin versus no antitumor therapy in inoperable hepatocellular carcinoma. A prospective randomized trial. Cancer. 1988;62(3):479–83. doi: 10.1002/1097-0142(19880801)62:3<479::aid-cncr2820620306>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 22.Sciarrino E, Simonetti RG, Le Moli S, Pagliaro L. Adriamycin treatment for hepatocellular carcinoma. Experience with 109 patients. Cancer. 1985;56(12):2751–55. doi: 10.1002/1097-0142(19851215)56:12<2751::aid-cncr2820561205>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 23.Leung TWT, Johnson PJ. Systemic therapy for hepatocellular carcinoma. Semin Oncol. 2001;28:514–20. doi: 10.1016/s0093-7754(01)90144-7. [DOI] [PubMed] [Google Scholar]
- 24.Yang TS, Wang CH, Hsieh RK, Chen JS, Fung MC. Gemcitabine and doxorubicin for the treatment of patients with advanced hepatocellular carcinoma: a phase I-II trial. Ann Oncol. 2002;13(11):1771–8. doi: 10.1093/annonc/mdf303. [DOI] [PubMed] [Google Scholar]
- 25.Louafi S, Boige V, Ducreux M, et al. Gemcitabine plus oxaliplatin (GEMOX) in patients with advanced hepatocellular carcinoma (HCC): results of a phase II study. Cancer. 2007;109(7):1384–90. doi: 10.1002/cncr.22532. [DOI] [PubMed] [Google Scholar]
- 26.Llovet JM, Di Bisceglie AM, Bruix J, et al. Design and endpoints of clinical trials in hepatocellular carcinoma. J Natl Cancer Inst. 2008;100(10):698–711. doi: 10.1093/jnci/djn134. [DOI] [PubMed] [Google Scholar]