

Abstract
Peptidomimetics 1 – 3 were prepared from aminoacid-derived tetramic acids 7 as the key starting materials. Calculations show that preferred conformations of 1 can align their side-chain vectors with amino acids in common secondary structures more effectively than 3. A good fit was found for a preferred conformation of 2 (an extended derivative of 1) with a sheet-turn-sheet motif.
Minimalist peptidomimetics, that present amino acid side-chains without any structure to directly resemble peptide backbones, are the focal point of many recent studies inspired by Smith/Hirschmann1–5 and Hamilton.6–10 We recently designed sets of scaffolds, of which A is typical, that analog local pairs of amino acids (including non-contiguous ones) in any secondary structure, ie universal peptidomimetics.11–12 That work featured five peptidomimetic designs that together could be used to mimic almost all local pairs of amino acid side-chains in six of the most common secondary structure motifs. This paper describes an alternative scaffold design 1 that has preferred conformations which overlay well with pairs of amino acid residues in three different types of helix (310, α-, and π-), in β-strands, and in both parallel- and antiparallel-β-sheets. Further, an extended form of that same scaffold design 2 has a preferred conformation which overlays well with three amino acid side chains in a sheet/β-turn/sheet-motif (an anti-parallel β-sheet). These two scaffolds are contrasted with A and a compound in series 3 where side chains are expressed on contiguous rings.
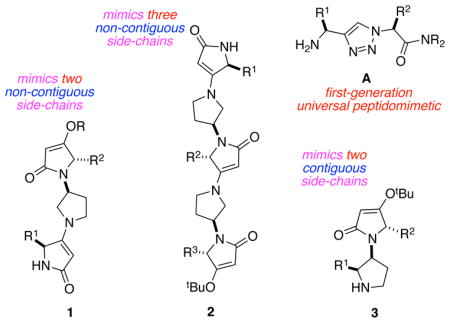
For the preparation of scaffold 1, an efficient procedure from Merck was used to decarboxylate trans-4-hydroxyproline to give more than 50 g of crystalline (R)-3-hydroxypyrrolidine.13 That pyrrolidine was N-protected to give the starting material indicated in Scheme 1. Nucleophilic displacement on a triflate-derivative of this (under conditions optimized to avoid elimination)14 gave the amino esters 4. X-ray analysis of 4d·HCl indicated its formation occurred via a single inversion. The crystalline hydrochloride salts of 4 were reacted with Bestmann’s ylide15–17 to give the pyrrolinone 5. Hydrogenolysis, then condensation of the free pyrrolidine-NH with 5-substituted 2,4-pyrrolidinediones (tetramic acids) 7 gave the featured trimers 1. The tetramic acid derivatives 7 are useful starting materials because they can be prepared from N-Boc-protected amino acids via a one-pot procedure that affords tens of gram amounts without chromatography.18–21 NMR and X-ray analysis of compound 6d indicated its formation was not complicated by epimerization. Condensation of the dimers 6 with C-deprotected derivatives of trimers 1 gave the pentamer 2, hence the overall synthesis can be divergent-convergent as in Scheme 1.
Scheme 1.

Syntheses of trimers 1 and pentamers 2
NMR studies to detect preferred conformations in these types of molecules are inappropriate due to conformational averaging. Consequently, two complementary molecular modeling methods were used. Quenched molecular dynamics (QMD)22–25 probes thermodynamic accessibilities of conformational states, as described previously.11 Briefly, this technique generates 600 minimized structures; ones that are energetically below a user-defined cut-off from the minimum energy conformer located (here 3.0 kcal/mol) are clustered into families based on RMS (root mean squared) deviations from user-defined atoms (0.5 Å). We have postulated11 that matching Cα - Cβ bond vectors forms a good basis for measuring fit to secondary structures, thus preferred conformations of scaffolds are defined by frameworks with only Me-side chains, ie Ala-analogs, like 1aa and 2aaa. For this reason, preferred conformers of 1aa and 2aaa were clustered based on Cα - Cβ coordinates, and representative members of each cluster were tested for fit on Cα - Cβ atom positions of ideal secondary structure motifs.
In our experience, a good fit of structures based on two amino acid side chains corresponds to an RMSD of 0.3 Å or less. Using this standard, Table 1 reveals preferred conformers of compound 1 fit two different residue pairs on the most elongated helical form (310), they fit three on an α-helix, and three on the most compressed helical form, the π-helix (with another at RMSD 0.31 Å). Other conformers overlay well on various side-chain pairs of β-strand and parallel-β-sheet residues, while the overall best match was for a parallel β-sheet. Interestingly, several of the favored overlays involved non-contiguous five-membered rings in structures 1 and 2 on contiguous amino acids in the secondary structures; this reflects the way the featured molecules can coil. All the preferred conformations highlighted in Table 1 were within 3.0 kcal/mol of the minimum conformation identified, hence these structures are thermodynamically accessible.
Table 1.
Fit of most appropriate conformers of mimics A, 1, and 3 on secondary structures (based on QMD analyses).a
| structure | seq. | A | 1 | 3 | |||
|---|---|---|---|---|---|---|---|
| ΔG | rmsd | ΔG | rmsd | ΔG | rmsd | ||
| 310-helix | i-i+1 | 2.55 | 0.22 | 2.09 | 0.55 | ||
| i-i+2 | 1.66 | 0.28 | 2.43 | 0.25 | |||
| i-i+3 | 1.27 | 0.49 | |||||
|
| |||||||
| α-helix | i-i+1 | 2.00 | 0.47 | 2.09 | 0.50 | ||
| i-i+2 | 1.62 | 0.25 | 2.55 | 0.30 | |||
| i-i+3 | 0.73 | 0.14 | |||||
| i-i+4 | 1.27 | 0.18 | |||||
|
| |||||||
| π-helix | i-i+1 | 2.55 | 0.14 | 2.09 | 0.54 | ||
| i-i+2 | 1.63 | 0.33 | 2.99 | 0.30 | |||
| i-i+3 | 2.43 | 0.31 | |||||
| i-i+4 | 0.85 | 0.35 | |||||
| i-i+5 | 1.27 | 0.27 | |||||
|
| |||||||
| β-strand | i-i+1 | 2.24 | 0.46 | ||||
| i-i+2 | 1.19 | 0.19 | |||||
|
| |||||||
| β-sheet (parallel) | i-i+1 | 1.01 | 0.10 | ||||
| i-i+2 | 2.55 | 0.20 | |||||
|
| |||||||
| sheet/β-turn/sheet | i-i+1 | 0.19 | 0.23 | 2.25 | 0.49 | ||
| i-i+2 | 1.19 | 0.12 | |||||
| i-i′ | 2.74 | 0.27 | |||||
| i-i′ +1 | 0.59 | 0.08 | |||||
| i-i′ +2 | 0.81 | 0.18 | |||||
| it-i+1 | 1.38 | 0.09 | |||||
| i-it′ | 0.14 | 0.06 | 0.75 | 0.25 | |||
| it-it′ +1 | 1.00 | 0.42 | |||||
All RMSD values (Å) are for the conformers (within 3.0 kcal/mol of the minimum energy) that overlay on the secondary structures shown (ΔG values are in kcal/mol). See supporting for secondary structure templates.
Table 1 also compares the overlay of scaffold A on the same elements of secondary structure. It shows scaffold A covers a more limited range of “secondary structure space”, as expected since the Cα atoms in this structure are held rigidly at one separation. A brief comparison was also made for the same elements of secondary structure with core building block of the Hirschmann/Smith pyrrolinones5 and Hamilton’s terphenyl systems (see supporting for structures). Hirschmann/Smith’s system overlaid well with parallel and antiparallel β-sheets, a β-strand, and a 310-helix (RMSD = 0.10 – 0.29 Å). A biphenyl representing part of Hamilton’s terphenyls overlaid better with 310- and π- than α-helices.26 An outline of these studies is given in the supporting, and a more extensive comparison will be reported elsewhere.
It is harder to fit six coordinates than four, so matches of the mimics involving three side chains must have higher RMSD’s. In the event, the best match for pentamer 2 was with three residues of a sheet-turn-sheet motif (1.93 kcal/mol above the minimum energy one; RMSD: 0.46 Å; Figure 1). Moreover, we found one example of a protein-protein interaction (between monomers in the RAD52 undecamer) where 2aaa matched three side-chains with an RMSD of only 0.14 Å (see supporting).
Figure 1.

Overlay of 2aaa on sheet-turn-sheet motif.
The next milestone in this study was to check that the different conformers are kinetically accessible. To do this, a density functional theory (DFT) method was used to investigate interconversion between the preferred states of 6a (Figure 2a, and supporting). A maximum energy barrier of 5.10 kcal/mol was calculated using this method (Figure 2b). This indicates conformers of 6a should rapidly interconvert on the 1H NMR time scale, and experimentally this was shown to be the case.
Figure 2.


a Structures and parameters used for DFT and QMD analyses; b low energy conformers and energy barriers for interconversion from DFT calculations; c preferred conformers from QMD calculations. All energies shown are the free energies (ΔGº) in kcal/mol.
Consideration of Newman projections through the bond indicated in Figure 2a indicates this molecule would tend to rest in predominantly three conformational states. This assertion is supported by the fact that three low energy conformational states emerged from both the DFT and QMD calculations (Figures 2b and c). The free energy differences from the DFT calculations predict that relative populations of the three conformers are on the ratio 1.00: 0.34: 0.12.
A synthesis of one compound in series 3 was developed to demonstrate that both heterocyclic rings in the “main-chain” of these peptidomimetics could be functionalized with amino acids. That route (Scheme 2) employs a known diastereoselective hydrogenation,27 and is similar to Scheme 1 except that a thioamide was introduced (9 to 10) then reduced to the amine (12 to 3).
Scheme 2.


Synthesis of 3 with side-chains on contiguous rings.
Extensive conformational analyses were performed for compound 3. Full data is shown in the supporting material, but the key point emerges from Table 1. Specifically, preferred conformers of 3 do not fit pairs of amino acid side-chains in secondary structures as well as conformers of trimers 1 do. The side chains in 3 on contiguous residues are constrained in ways that preclude good overlap on common secondary structure motifs. This is supported by the modeling studies shown here and X-ray crystallographic analyses of compound 11 (supporting). Conversely, the trimers 1 have at least one extra significant degree of freedom, and this allows them to flex into conformations that match secondary structures well.
The accessibility of tetramic acid derivatives 7 from different amino acids gives the syntheses here some considerable scope. We recently argued11–12 there are four structural criteria common to effective minimalist mimics: (i) facile syntheses with most amino acid side chains; (ii) kinetically and thermodynamically accessible conformations for induced fit; (iii) only moderate loss of entropy on docking; and, (iv) appropriate Cα - Cβ coordinates of an accessible conformation of the mimic matching those of the secondary structures. An advantage of defining a parameter such as (iv), ie matching side-chain Cα - Cβ coordinates, is that the fit can be quantitated in terms of parameters like RMSD. This provides a firm basis for comparing abilities of peptidomimetics like A and 1 – 3 to mimic secondary structures.
Supplementary Material
Acknowledgments
We thank The National Institutes of Health (GM087981) and The Robert A. Welch Foundation (A-1121) for financial support, Joseph H. Reibenspies for X-Ray crystallographic analysis, Dr. Marc Parisien for modification of the β-sheet builder program, and Dr. Dmytro Fedoseyenko for helpful discussions.
Footnotes
Supporting Information Available: Preparations and spectra of compounds 1 – 12, modeling procedures, overlays on secondary structures (preferred conformers of 1, 1aa and 2aaa on secondary structure motifs in proteins {RMSD <0.14 Å}; Hirschmann/Smith pyrrolinones and Hamilton’s terphenyl systems) and complete ref 2. CCDC 822217 – 822219 contains the supplementary crystallographic data, which can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
References
- 1.Hirschmann R, Nicolaou KC, Pietranico S, Salvino J, Leahy EM, Sprengeler PA, Furst G, Smith AB., III J Am Chem Soc. 1992;114:9217–9218. [Google Scholar]
- 2.Hirschmann R, et al. J Am Chem Soc. 1993;115:12550–12568. [Google Scholar]
- 3.Hirschmann R, Sprengeler PA, Kawasaki T, Leahy JW, Shakespeare WC, Amos B, Smith I. J Am Chem Soc. 1992;114:9699–9701. [Google Scholar]
- 4.Mowery BP, Prasad V, Kenesky CS, Angeles AR, Taylor LL, Feng J-J, Chen W-L, Lin A, Cheng F-C, Smith AB, III, Hirschmann R. Org Lett. 2006;8:4397–4400. doi: 10.1021/ol061488x. [DOI] [PubMed] [Google Scholar]
- 5.Smith AB, III, Charnley AK, Hirschmann R. Acc Chem Res. 2011;44:180–193. doi: 10.1021/ar1001186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yin H, Lee G, Sedey KA, Kutzki O, Park HS, Orner BP, Ernst JT, Wang HG, Sebti SM, Hamilton AD. J Am Chem Soc. 2005;127:10191–10196. doi: 10.1021/ja050122x. [DOI] [PubMed] [Google Scholar]
- 7.Davis JM, Truong A, Hamilton AD. Org Lett. 2005;7:5405–5408. doi: 10.1021/ol0521228. [DOI] [PubMed] [Google Scholar]
- 8.Ernst JT, Becerril J, Park H, Yin H, Hamilton AD. Angew Chem Int Ed. 2003;42:535–539. doi: 10.1002/anie.200390154. [DOI] [PubMed] [Google Scholar]
- 9.Davis JM, Tsou LK, Hamilton AD. Chem Soc Rev. 2007;36:326–334. doi: 10.1039/b608043j. [DOI] [PubMed] [Google Scholar]
- 10.Rodriguez JM, Hamilton AD. Angew Chem Int Ed. 2007;46:8614–8617. doi: 10.1002/anie.200701869. [DOI] [PubMed] [Google Scholar]
- 11.Ko E, Ling J, Perez LM, Lu G, Schaefer A, Burgess K. J Am Chem Soc. 2011;133:462–477. doi: 10.1021/ja1071916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ko E, Liu J, Burgess K. Chem Soc Rev. 2011 ASAP. [Google Scholar]
- 13.Houghton PG, Humphrey GR, Kennedy DJ, Roberts DC, Wright SHB. J Chem Soc Perking Trans I. 1993:1421–1424. [Google Scholar]
- 14.Corruble A, Davoust D, Desjardins S, Fressigne C, Giessner-Prettre C, Harrison-Marchand A, Houte H, Lasne MC, Maddaluno J, Oulyadi H, Valnot JY. J Am Chem Soc. 2002;124:15267–15279. doi: 10.1021/ja016945d. [DOI] [PubMed] [Google Scholar]
- 15.Bestmann HJ. Angew Chem Int Ed Engl. 1977;16:349–364. [Google Scholar]
- 16.Schobert R, Dietrich M, Mullen G, Urbina-Gonzalez J-M. Synthesis. 2006:3902–3914. [Google Scholar]
- 17.Schobert R, Boeckman RK, Jr, Pero JE. Org Synth. 2005;82:140–145. [Google Scholar]
- 18.Jouin P, Castro B. J Chem Soc, Perkin Trans. 1987;1:1177–1182. [Google Scholar]
- 19.Courcambeck J, Bihel F, De Michelis C, Quelever G, Kraus JL. J Chem Soc, Perkin Trans. 2001;1:1421–1430. [Google Scholar]
- 20.Ma D, Ma J, Ding W, Dai L. Tetrahedron Asymmetry. 1996;7:2365–2370. [Google Scholar]
- 21.Hosseini M, Kringelum H, Murray A, Tonder JE. Org Lett. 2006;8:2103–2106. doi: 10.1021/ol060500i. [DOI] [PubMed] [Google Scholar]
- 22.Pettitt BM, Matsunaga T, Al-Obeidi F, Gehrig C, Hruby VJ, Karplus M. Biophys J. 1991;60:1540–1544. doi: 10.1016/S0006-3495(91)82188-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Connor SD, Smith PE, Al-Obeidi F, Pettitt BM. J Med Chem. 1992;35:2870–2881. doi: 10.1021/jm00093a021. [DOI] [PubMed] [Google Scholar]
- 24.Schulten K. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Humphrey W, Dalke A, Schulten K. J Mol Graph. 199614:33–38. 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 26.Saraogi I, Hamilton AD. Chem Soc Rev. 2009;38:1726–1743. doi: 10.1039/b819597h. [DOI] [PubMed] [Google Scholar]
- 27.Hosseini M, Grau Jakob S, Sorensen Kasper K, Sotofte I, Tanner D, Murray A, Tonder Janne E. Org Biomol Chem. 2007;5:2207–2210. doi: 10.1039/b705093c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.