

Abstract
The chemical step of protein synthesis, peptide bond formation, is catalyzed by the large subunit of the ribosome. Crystal structures have demonstrated that the active site for peptide bond formation is composed entirely of RNA1. Recent work has focused on how an RNA active site is able to catalyze this fundamental biological reaction at a suitable rate for protein synthesis. Based on the absence of important ribosomal functional groups2, lack of a dependence on pH3, and the dominant contribution of entropy to catalysis4, it has been suggested that the role of the ribosome is limited to bringing the substrates into close proximity. Alternatively, the importance of the 2′-hydroxyl of the peptidyl-tRNA5 and a Bronsted coefficient near zero6 were taken as evidence that the ribosome coordinates a proton-transfer network. Here we report the transition state of peptide bond formation based upon kinetic isotope effect analysis at five positions at the reaction center of a peptidyl-tRNA mimic. Our results indicate that in contrast to the uncatalyzed reaction, formation of the tetrahedral intermediate and proton-transfer from the nucleophilic nitrogen both occur in the rate-limiting step. Unlike previous proposals, the reaction is not fully concerted, instead breakdown of the tetrahedral intermediate occurs in a separate fast step. This suggests that in addition to substrate positioning, the ribosome is contributing to chemical catalysis by changing the rate-limiting transition state.
Several reaction mechanisms have been proposed for peptide bond formation (Figure 1). The peptidyl transferase reaction occurs through nucleophilic attack of the α-amino group of aminoacyl-tRNA on the carbonyl carbon of the peptidyl-tRNA. A peptide bond forms and the ester bond linking the peptide to the 3′-oxygen of peptidyl-tRNA breaks, leaving a deacylated tRNA and a peptide lengthened by one amino acid. If substrate positioning is the sole contribution to catalysis, the mechanism on the ribosome is expected to be equivalent to the well-studied uncatalyzed reactions where a pathway involving two tetrahedral intermediates is followed (T± and T-, Figure 1, black pathway, see figure legend for explanation of mechanisms and intermediates). At high pH, the rate-limiting transition state for the uncatalyzed reaction was predicted to occur during deprotonation of the zwitterionic T± intermediate based on the pH-rate dependence of the reaction and a Bronsted coefficient near 1 (transition state D)7. At low pH, breakdown of the negatively charged T- is rate-limiting (transition state F).
Figure 1.

Proposed reaction mechanisms. Ground states are shown in black, intermediates in grey, and transition states in blue. The ribosomal reaction may involve two intermediates like the uncatalyzed reaction (black pathway). Nucleophilic attack leads to an intermediate with a positively charged nitrogen and negatively charged carbonyl oxygen (T+/−). Deprotonation leads to a negatively charged intermediate (T-), which breaks down to products. Alternatively, one or both of these steps may be concerted, with only one intermediate (red and orange pathways), or no intermediates (green). The identities of B1 and B2 are uncertain; the 2′-hydroxyl may be one, both, or neither.
It has also been suggested that the peptidyl-tRNA 2′-hydroxyl acts as a “proton shuttle” that abstracts a proton from the nucleophilic amine and simultaneously donates another proton either directly or indirectly to the leaving group oxygen8,9. This could happen in a stepwise fashion (Figure 1, red and orange pathways) involving a tetrahedral intermediate. Alternatively, the reaction has been proposed to be fully concerted, with the amide bond to the nucleophile formed at the same time that the ester bond to the leaving group is broken (green pathway). Recently, computational methods were used to evaluate concerted proton shuttle mechanisms with and without an additional water molecule, and favored a fully concerted eight-member proton shuttle(similar to transition state A)10. Alternatively, other computational studies have indicated a stepwise proton shuttle would be favorable11,12. All of these possibilities have been proposed4,8–13.
Each mechanism predicts a different transition state and hence a different role for the ribosome in catalysis. Therefore we sought to obtain experimental constraints for modeling the transition state of ribosome-catalyzed peptide bond formation using kinetic isotope effect (KIE) analysis (reviewed in 14). Generally, if the rate effect of isotopic substitution (defined as klight/kheavy ) is greater than one (termed “normal”), it indicates bonding to that isotope is weaker in the transition state. Reaction coordinate motion of a substituted atom will also contribute to a normal isotope effect. Alternatively, if the isotope effect is less than one (termed “inverse”) it indicates stronger bonding. The magnitude of the effect is correlated with the change in bond order. To make this analysis possible, we previously synthesized a complete series of molecules that differ by a single isotopic substitution at each atom within the reaction center15,16. These molecules are substrates for the ribosomal 50S fragment reaction17,18. Previous studies indicate chemistry is rate-limiting for this assay and that unlike the 70S reaction, which has a complete commitment to catalysis, this reaction is amenable to KIE analysis18. The Bronsted coefficient of the nucleophile and the structure of the active site are within experimental error for 50S and 70S ribosomes, indicating the mechanism of catalysis is similar6,19.
To achieve the necessary precision, KIEs were measured by a competitive assay. The light and heavy substrates were incubated in the same reaction, and the change in their ratio as the reaction proceeded was used to determine their relative reaction rates. We used two remote radiolabels, 32P and 33P, attached to the 5′-ends of the P-site substrate and performed scintillation counting to define the relative abundance of the two substrates as previously described for the uncatalyzed reaction20,21.
Given the small magnitude of the expected effects (typically in the range of 0.95 to 1.05) and the complexity of the experimental system (a 1.5 MDa complex altered by 1–2 Da), we performed several controls to determine the extent of random and systematic error. First, we demonstrated that the effect from the remote label alone [1.001 ± 0.002 (standard error)] is much smaller than the effects we expect for atoms at the reaction center. This value and the effect of deuterium substitution at the amino acid α-carbon were determined thirty-three and forty times, respectively. Repeated measurements in both cases yielded normally distributed data, with a significant difference between the mean of the control and the mean of the α-deuterium substitution (Figure 2). Furthermore, two sets of measurements were made for each substitution, one pairing 32P with the light isotope at the reaction center and 33P with the heavy isotope, and a second with the opposite pairings – 32P with the heavy isotope and 33P with the light isotope. These determinations gave the same values within experimental error.
Figure 2.
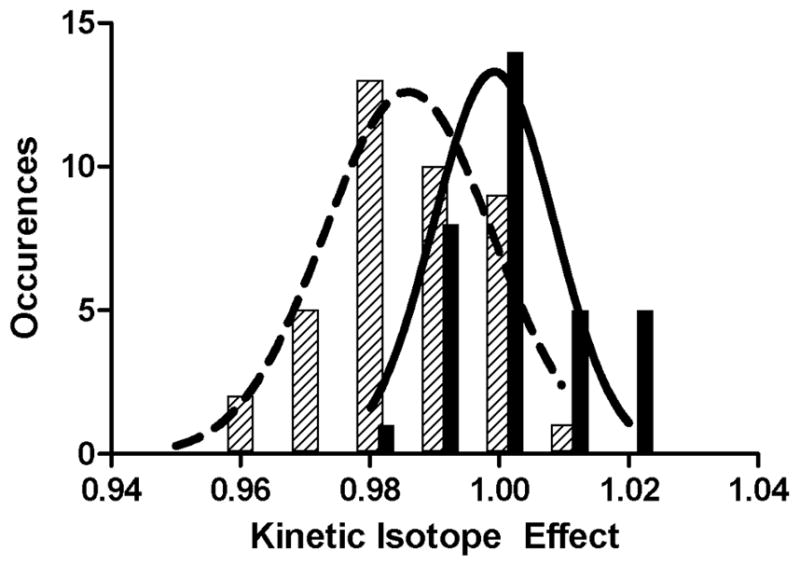
Histograms of measured isotope effects. Controls (solid bars) and α-deuterium substitution (hatched bars) both fit to normal distributions.
We measured isotope effects for five positions at the reaction center: the 3′-oxygen leaving group, the carbonyl carbon, the nucleophilic nitrogen, the hydrogen attached to the α-carbon, and the vicinal 2′-hydroxyl (Figure 3). The simplest effect to interpret is that of 18O substitution on the leaving group, which measures the extent of C-O bond dissociation in the transition state. Effects in the range of 1.02 to 1.06 have been observed when cleavage of the C-O bond is rate-limiting22,23. Conversely, if formation of the amide bond or deprotation of the T± intermediate is the rate-limiting step, then a maximum effect of only 1.01 is expected22. We observed a small effect (1.006) upon 3′-18O substitution, which indicates there is not significant C-O bond-breaking in the rate-limiting step. This is inconsistent with transition state A, the mechanism in which bond formation to the nucleophile is concerted with breaking the bond to the leaving group, and limits the possible transition states to B, C, and D (Figure 1). Notably, the leaving group oxygen effect previously predicted for fully concerted proton-shuttle mechanisms varied from 1.023 to 1.03810 (transition state A), which is inconsistent with the value of 1.006 measured here.
Figure 3.

Kinetic isotope effects. Values are reported as means ± standard errors with the number of independent trials in parentheses. The second value for the nitrogen nucleophile (asterisked) was previously determined using mass spectrometry instead of scintillation counting as the readout. Calculated values for the transition state in figure 4 are shown in italics. Full substrates are shown; cyt=cytidine, cap=caproic acid, bio=biotin, pmn=puromycin.
The primary carbonyl-13C isotope effect is derived from a combination of bond breaking to the leaving group oxygen, bond formation to the nitrogen, and the accompanying change in hybridization. Given that the bond to the leaving group is intact, the latter two factors should dominate. The observed effect for the carbonyl carbon is large and normal, 1.026. For hydrazinolysis of methyl formate, a similar effect has been interpreted as resulting from rate-limiting C-N bond formation24. However, it was also shown computationally that an effect of approximately 1.02 could be derived from rate-limiting deprotonation of the zwitterionic intermediate (transition state D)25. The remaining isotope effects made it possible to differentiate between these possibilities.
The deuterium effect on the amino acid α-carbon is primarily derived from the loss of hyperconjugation between the carbonyl π bond and the antibonding orbital of the C-H bond, which increases the strength of the C-H bond. The magnitude of this effect is largest when the ground state has a large geometric overlap between the π bond and the antibonding orbital and the transition state is very tetrahedral. Calculations and previous reactions indicate a lower limit of this isotope effect to be approximately 0.9621,26. The measured isotope effect, 0.985, indicates a significant decrease in overlap in the transition state. This is consistent with the decreased π bond character in a partially tetrahedral transition state.
The KIE of the nucleophilic nitrogen was previously measured to be 1.010, suggestive of an early transition state with partial C-N bond order18. We remeasured this value using the remote radiolabel-based assay, and again obtained a normal isotope effect, 1.014. Formation of the C-N bond is expected to produce an inverse isotope effect on the nitrogen. The normal isotope effect observed can be derived from two factors: reaction coordinate motion and deprotonation of the nitrogen. Calculations here and elsewhere indicate either of these factors alone is insufficient to result in a normal isotope effect (Supplementary Tables S1 and S2)10. This suggests that the nitrogen is being deprotonated in the rate-limiting step, simultaneous with C-N bond formation (transition state C). Such a mechanism is consistent with the near-zero Bronsted coefficient6 which indicates there is no buildup of positive charge on the nucleophile in the transition state.
The 2′-hydroxyl contributes 100- to 2000-fold to ribosome-catalyzed peptide bond formation5 and has been postulated to be involved in proton transfer8–13. The isotope effect for this position is close to unity (1.002). Changes in bond order between the 2′-oxygen and protons can be compensated by opposite changes in bond strength to the 2′-carbon; therefore this effect is relatively insensitive to the protonation state of the 2′-oxygen 27. The measured effect indicates the oxygen does not bear a large positive or negative charge, consistent with previous studies13.
To supplement the qualitative descriptions above, we calculated isotope effects for several potential transition state structures (Supplementary Tables S1–S3). Though we cannot exclude the possibility that multiple steps contribute to the isotope effects, a single transition state did yield calculated values in reasonable agreement with the experimental measurements (Figure 4). As expected for a transition state, there is a single large imaginary frequency corresponding to reaction coordinate motion (carbon-nitrogen bond formation and nitrogen deprotonation). The error inherent in these calculations and our measurements produces an uncertainty in the precise structure of the transition state; however, a single rate-limiting step is consistent with the measured isotope effects. Formation of the tetrahedral intermediate is simultaneous with deprotonation (transition state C). This step is followed by fast breakdown of the tetrahedral intermediate into products (red pathway). The data are not consistent with a fully concerted reaction mechanism.
Figure 4.

Electrostatic (top) and geometric (bottom) structures of the substrates and transition states for peptide bond formation shown in similar orientations. Bond lengths are shown in angstroms. Protons on the nucleophilic nitrogen, 2′-hydroxyl, and water are displayed; for clarity, all others are omitted.
This mechanism is markedly different from what has been observed for similar uncatalyzed reactions7,21. For uncatalyzed reactions at low pH, stepwise formation of T- is rate-limiting. At high pH, or with the strong nucleophile hydroxylamine, breakdown of the T-intermediate is rate-limiting. Our results indicate that the ribosome not only alters the relative rates of formation and breakdown of tetrahedral intermediates, but also substantially alters the energy landscape such that nucleophilic attack and deprotonation are coordinated.
Concerted attack and deprotonation has also been proposed for aminolysis of the acyl-enzyme intermediate by chymotrypsin28,29. This conclusion was supported in part by a low Bronsted coefficient, similar to that observed for peptide bond formation. This indicates that the pKa of the intermediate in the context of these active sites is lower than that of a catalytic group, so that T+/− does not have a finite lifetime and instead T- is the only stable intermediate7. Although the pKa of the amine is initially high, it decreases dramatically as the C-N bond is formed. For chymotrypsin, a catalytic histidine is responsible for deprotonating the nucleophile. For the ribosome, the lack of a pH dependence of the reaction argues against general-acid or -base catalysis3. The 2′-hydroxyl is important for the ribosome reaction, but the pKa of a hydroxyl dictates that if it abstracts the amino proton it must donate its proton elsewhere. This mechanism, termed the “proton shuttle” has been proposed in a variety of forms for the ribosome8–12. Alternatively, the 2′-hydroxyl may provide an important transition state hydrogen bond, with another group or a water molecule deprotonating the nucleophile. Either possibility would be consistent with measured solvent isotope effects, which indicate more than one proton is in motion at the transition state (S. Kuhlenkoetter and M. Rodnina, personal communication).
The destination of the proton originating from the nucleophile is uncertain. Two computational studies favor the carbonyl oxygen, which would otherwise develop a negative charge11,12. This would result in an uncharged tetrahedral intermediate, which would be consistent with the lack of pH dependence of the reaction. Alternatively, a water molecule observed in crystal structures is correctly positioned to accept a proton from the 2′-hydroxyl and later donate it to the 3′-oxygen leaving group. Finally, the 3′-oxygen could receive the proton from the 2′-hydroxyl. Our data cannot distinguish these possibilities; for simplicity, we have modeled a water molecule as the proton acceptor.
The ribosome increases the rate of peptide bond formation by an estimated ten-million-fold through an altered chemical mechanism. The rate of nucleophilic attack is likely increased through positioning effects – first, the increased likelihood of the two substrates being in proximity and second, precise orientation of the nucleophile (possibly by the 2′-hydroxyl of A24512). The ribosome also plays a significant catalytic role, beyond substrate positioning, to coordinate nucleophilic attack and deprotonation in a single rate-limiting step. Catalysis by the ancient, conserved, RNA active site of the ribosome fundamentally alters the reaction pathway for peptide bond formation relative to the uncatalyzed reaction.
Methods Summary
A general strategy for measuring isotope effects with these substrates using a competitive assay has been described21. The P-site substrate cytidylyl-(3′5′)-cytidylyl-(3′5′)-3′ (2′)-O-(N-(6-D-(+)-biotinoylaminohexanoyl)-L-phenylalanyl)adenosine (CCApcb) was synthesized as described. For each measurement, two substrates differing by a single isotopic substitution at the reaction center were mixed. Each substrate was labeled with either 32P or 33P to allow differentiation by scintillation counting.
Reactions contained 7 mM MgCl2, 140 mM NH4Cl, 25 mM HEPES pH 8.5, trace P-site substrate, 250–500 mM A-site substrate, and 6–10 μM 50S ribosomes. At these concentrations the isotope effect is on kcat/KM for the P-site substrate. Timepoints were quenched at 15–30% reacted and near the reaction endpoint. Substrate and product were separated on an acrylamide gel and the fraction reacted determined by phosphorimaging. Each band was eluted and scintillation counted to determine the ratio of 32P to 33P in each sample.
To account for substrate that hydrolyzed before the reaction was started, the amount of 32P- and 33P-labeled product at zero time was subtracted from all determinations of the product isotope ratio. Additionally, some substrate was unreactive even at long timepoints. The amounts of 32P- and 33P-labeled unreactive substrate were similarly subtracted from all determinations of the substrate isotope ratio.
Isotope effects were calculated as previously described21, using hybrid density functional methods implemented in Gaussian03. Transition state structures were optimized and frequencies computed on the optimized structures using the three-parameter Becke exchange functional, the LYP correlation functional and the standard 6-31G (d,p) basis set. Isotope effects were calculated from the computed frequencies using ISOEFF 9830. Methylamine was used to model the nucleophile. Calculated isotope effects for tetrahedral intermediate formation were repeated with a set of nucleophiles of increasing size and complexity; these effects varied by less than 0.003.
Methods
Reaction Assay
A general strategy for measuring isotope effects with these substrates using a competitive assay has been described21. The P-site substrate cytidylyl-(3′5′)-cytidylyl-(3′5′)-3′ (2′)-O-(N-(6-D-(+)-biotinoylaminohexanoyl)-L-phenylalanyl)adenosine (CCApcb) was synthesized as previously described15,16. Substrates were prepared that differed only by a single isotopic substitution at the reaction center. Each substrate was 5′-end labeled with either γ32P- or γ33P-ATP by polynucleotide kinase and purified on a pH 6 denaturing 15% polyacrylamide gel. 32P- and 33P-CCApcb were mixed at an approximately 1:4 ratio and purified on a pH 6 non-denaturing 12% polyacrylamide gel. Purified mixes were used as soon as possible to minimize hydrolysis in the starting material.
The isotopic enrichment of each sample was determined by high-resolution Fourier-transform – ion cyclotron resonance mass spectrometry. Each molecule had 95% or greater isotopic purity.
Ribosome reactions contained 7 mM MgCl2, 140 mM NH4Cl, 25 mM HEPES pH 8.5, trace P-site substrate, 250–500 mM A-site substrate, and 6–10 μM 50S ribosomes. At these concentrations the A-site substrate is saturating but the ribosome concentration is not; therefore the isotope effect is on kcat/KM for the P-site substrate. All reactions were performed at room temperature. Reaction aliquots at 15–30% reacted (approximately 1 minute) and near the reaction endpoint (greater than 30 minutes) were quenched with three volumes of formamide loading buffer. Each timepoint was run on a pH 6 denaturing 15% polyacrylamide gel to separate substrate from product.
Substrate and product bands were visualized using a Storm 840 PhosphorImager with a two-ply sheet of duck tape between the gel and screen to block 33P emission. The fraction of 32P-labeled substrate reacted could then be determined. The ratio of 32P to 33P was determined by scintillation counting. Each product band was excised from the gel and eluted into 1 mL of 50mM NaCl overnight. This elution was then added to 13 mL of Optima Gold scintillation fluid and counted for thirty minutes along with 32P and 33P standards.
Data Analysis
Counts per minute were divided into two channels, 0–400 keV and 400–2000 keV. Approximately 70% of the 32P standard was detected in the high energy channel, and greater than 99% of the 33P sample was detected in the low energy channel. The ratio of 32P to 33P in each sample could be determined using equation 1:
| (1) |
where A is the counts per minute in the low energy channel, B is the counts per minute in the high energy channel, and r is the fraction of total emission of a 32P standard detected in the low energy channel. To account for substrate that hydrolyzed before the reaction was started, the amount of 32P- and 33P-labeled product at zero time was determined. These were subtracted from all other determinations of the product isotope ratio. Additionally, some substrate was unreactive even at long timepoints. The amounts of 32P- and 33P-labeled unreactive substrate were similarly subtracted from all other determinations of the substrate isotope ratio. The observed isotope effect was then determined from the ratio in the midpoint and endpoint samples and the fraction reacted using equation 2:
| (2) |
where f is the fraction reacted, Rp is the isotope ratio in the product at that fraction reacted, and R0 is the isotope ratio in the product at the reaction endpoint. For high fraction reacted samples (greater than 50%) the isotope effect was also determined from substrate and endpoint samples using equation 3:
| (3) |
where Rs is the isotope ratio in the remaining substrate at f fraction reacted, and R0 is as above. This value was corrected for incomplete isotopic incorporation as determined by mass spectrometry with equation 4:
| (4) |
where e is the isotopic enrichment of the heavy sample. This equation assumes a negligible amount of heavy isotope in the light sample14,21.
For the control and α-deuterium isotope effects, enough trials were performed to plot a histogram of the data (Figure 2). Both datasets were fit to a normal distribution, equation 5:
| (5) |
where c is the number of counts in a given bin, A is the amplitude, μ is the mean and σ is the standard deviation. For some substitutions, the standard errors for trials performed on the same day were smaller than those from different days, indicating each trial may not be independent. Therefore, for all substitutions each set of experiments from a single day were averaged. The mean and standard error of multiple datasets are reported.
Computation of Transition States
Transition state structures that reproduced the experimental KIEs were determined in vacuo using hybrid density functional methods implemented in Gaussian03. Tetrahydro-4,5-dihydroxy-2-(methoxymethyl)furan-3-yl 2-acetamidopropanoate was used to model the P-site substrate and methylamine was used to model the A-site nucleophile. Calculated isotope effects for tetrahedral intermediate formation were repeated with a set of nucleophiles of increasing size and complexity; the calculated isotope effects varied by less than 0.003.
Structures of the transition states were optimized and the frequencies were computed on the optimized structures using the three-parameter Becke (B3) exchange functional, the LYP correlation functional and the standard 6-31G (d,p) basis set. The 5′-methoxy group and the reaction center were constrained during the optimization and many of these constraints were modified to match the experimental KIEs.
Kinetic and equilibrium isotope effects (KIEs and EIEs, respectively) were calculated from the computed frequencies using ISOEFF 9830. KIEs and EIEs were calculated at the temperature of 298 K and the frequencies were scaled using a factor of 0.964, corresponding to B3LYP/6-31G(d, p). KIEs were calculated whenever the magnitude of the imaginary frequency was greater than 50i cm−1, otherwise EIEs were computed. All vibrational modes were used to calculate isotope effects.
Geometric and the electrostatic models were generated by iteratively optimizing the transition states by modifying the applied constraints until the computed isotope effects closely match the experimental KIEs.
The natural bond orbital (NBO) calculations were performed on optimized structures by including the pop = nbo keyword in the route section of input files. The molecular electrostatic potential (MEP) surfaces were calculated by the CUBE subprogram of Gaussian03. The formatted checkpoint files used in the CUBE subprogram were generated by constrained geometry optimization at the B3LYP level of theory with the 6-31G** basis set. MEP surfaces of the substrate and the transition states were visualized using Molekel4.0 at a density of 0.008 electrons/bohr3. Geometric figures were created using PyMol.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
This work was supported by a NIH postdoctoral fellowship to DAH, a Brown-Coxe fellowship to VS and NIH grant GM54839.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions
DAH and SAS devised experiments, MZ synthesized substrates, DAH collected and analyzed isotope effect data, VS calculated theoretical isotope effects, DAH and SAS wrote the paper.
Author Information
Reprints and permissions information is available at npg.nature.com/reprintsandpermissions. The authors declare no competing financial interests.
The authors declare no competing financial interests.
References
- 1.Nissen P, Hansen J, Ban N, Moore PB, Steitz TA. The Structural Basis of Ribosome Activity in Peptide Bond Synthesis. Science. 2000;289:920–930. doi: 10.1126/science.289.5481.920. [DOI] [PubMed] [Google Scholar]
- 2.Erlacher MD, et al. Chemical engineering of the peptidyl transferase center reveals an important role of the 2′-hydroxyl group of A2451. Nucl Acids Res. 2005;33:1618–1627. doi: 10.1093/nar/gki308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bieling P, Beringer M, Adio S, Rodnina MV. Peptide bond formation does not involve acid-base catalysis by ribosomal residues. Nat Struct Mol Biol. 2006;13:423–428. doi: 10.1038/nsmb1091. [DOI] [PubMed] [Google Scholar]
- 4.Sievers A, Beringer M, Rodnina MV, Wolfenden R. The ribosome as an entropy trap. P Natl Acad Sci USA. 2004;101:7897–7901. doi: 10.1073/pnas.0402488101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zaher HS, Shaw JJ, Strobel SA, Green R. The 2′-OH group of the peptidyl-tRNA stabilizes an active conformation of the ribosomal PTC. EMBO J. 2011 doi: 10.1038/emboj.2011.142. advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kingery DA, et al. An Uncharged Amine in the Transition State of the Ribosomal Peptidyl Transfer Reaction. Chem Biol. 2008;15:493–500. doi: 10.1016/j.chembiol.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Satterthwait AC, Jencks WP. Mechanism of the aminolysis of acetate esters. J Am Chem Soc. 1974;96:7018–7031. doi: 10.1021/ja00829a034. [DOI] [PubMed] [Google Scholar]
- 8.Dorner S, Panuschka C, Schmid W, Barta A. Mononucleotide derivatives as ribosomal P-site substrates reveal an important contribution of the 2′-OH to activity. Nucl Acids Res. 2003;31:6536–6542. doi: 10.1093/nar/gkg842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schmeing TM, Huang KS, Kitchen DE, Strobel SA, Steitz TA. Structural Insights into the Roles of Water and the 2′ Hydroxyl of the P Site tRNA in the Peptidyl Transferase Reaction. Mol Cell. 2005;20:437–448. doi: 10.1016/j.molcel.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 10.Wallin G, Åqvist J. The transition state for peptide bond formation reveals the ribosome as a water trap. P Natl Acad of Sci USA. 2010;107:1888–1893. doi: 10.1073/pnas.0914192107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Q, Gao J, Liu Y, Liu C. Validating a new proton shuttle reaction pathway for formation of the peptide bond in ribosomes: A theoretical investigation. Chem Phys Lett. 2010;501:113–117. [Google Scholar]
- 12.Rangelov MA, Petrova GP, Yomtova VM, Vayssilov GN. Catalytic Role of Vicinal OH in Ester Aminolysis: Proton Shuttle versus Hydrogen Bond Stabilization. J Org Chem. 2010;75:6782–6792. doi: 10.1021/jo100886p. [DOI] [PubMed] [Google Scholar]
- 13.Huang KS, Carrasco N, Pfund E, Strobel SA. Transition State Chirality and Role of the Vicinal Hydroxyl in the Ribosomal Peptidyl Transferase Reaction†. Biochemistry. 2008;47:8822–8827. doi: 10.1021/bi800299u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cleland WW. Isotope effects: Determination of enzyme transition state structure. Enzyme Kinetics and Mechanism Part D: Developments in Enzyme Dynamics. 1995;249(13):341–373. doi: 10.1016/0076-6879(95)49041-8. [DOI] [PubMed] [Google Scholar]
- 15.Zhong M, Strobel SA. Synthesis of the Ribosomal P-Site Substrate CCA-pcb. Org Lett. 2006;8:55–58. doi: 10.1021/ol052484f. [DOI] [PubMed] [Google Scholar]
- 16.Zhong M, Strobel SA. Synthesis of Isotopically Labeled P-Site Substrates for the Ribosomal Peptidyl Transferase Reaction. J Org Chem. 2008;73:603–611. doi: 10.1021/jo702070m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Monro RE, Marcker KA. Ribosome-catalysed reaction of puromycin with a formylmethionine-containing oligonucleotide. J Mol Biol. 1967;25:347–350. doi: 10.1016/0022-2836(67)90146-5. [DOI] [PubMed] [Google Scholar]
- 18.Seila AC, Okuda K, Nunez S, Seila AF, Strobel SA. Kinetic Isotope Effect Analysis of the Ribosomal Peptidyl Transferase Reaction†. Biochemistry. 2005;44:4018–4027. doi: 10.1021/bi047742f. [DOI] [PubMed] [Google Scholar]
- 19.Selmer M, et al. Structure of the 70S ribosome complexed with mRNA and tRNA. Science. 2006;313:1935–1942. doi: 10.1126/science.1131127. [DOI] [PubMed] [Google Scholar]
- 20.Unrau PJ, Bartel DP. RNA-catalysed nucleotide synthesis. Nature. 1998;395:260–263. doi: 10.1038/26193. [DOI] [PubMed] [Google Scholar]
- 21.Hiller DA, Zhong M, Singh V, Strobel SA. Transition States of Uncatalyzed Hydrolysis and Aminolysis Reactions of a Ribosomal P-Site Substrate Determined by Kinetic Isotope Effects. Biochemistry. 2010;49:3868–3878. doi: 10.1021/bi901458x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O’Leary MH, Marlier JF. Heavy-atom isotope effects on the alkaline hydrolysis and hydrazinolysis of methyl benzoate. J Amer Chem Soc. 1979;101:3300–3306. [Google Scholar]
- 23.Sawyer CB, Kirsch JF. Kinetic isotope effects for reactions of methyl formate-methoxyl-18O. J Amer Chem Soc. 1973;95:7375–7381. [Google Scholar]
- 24.Marlier JF, Haptonstall BA, Johnson AJ, Sacksteder KA. Heavy-Atom Isotope Effects on the Hydrazinolysis of Methyl Formate. J Amer Chem Soc. 1997;119:8838–8842. [Google Scholar]
- 25.Singleton DA, Merrigan SR. Resolution of Conflicting Mechanistic Observations in Ester Aminolysis. A Warning on the Qualitative Prediction of Isotope Effects for Reactive Intermediates. J Amer Chem Soc. 2000;122:11035–11036. [Google Scholar]
- 26.Hogg JL, Rodgers J, Kovach I, Schowen RL. Kinetic isotope-effect probes of transition-state structure. Vibrational analysis of model transition states for carbonyl addition. J Amer Chem Soc. 1980;102:79–85. [Google Scholar]
- 27.Gawlita E, et al. H-Bonding in Alcohols Is Reflected in the Cα H Bond Strength: Variation of C–D Vibrational Frequency and Fractionation Factor. J Amer Chem Soc. 2000;122:11660–11669. [Google Scholar]
- 28.Inward PW, Jencks WP. The reactivity of nucleophilic reagents with furoyl-chymotrypsin. J Biol Chem. 1965;240:1986. [PubMed] [Google Scholar]
- 29.Zeeberg B, Caplow M. Transition State Charge Distribution in Reactions of an Acetyltyrosylchymotrypsin Intermediate. J Biol Chem. 1973;248:5887–5891. [PubMed] [Google Scholar]
- 30.Anisimov V, Paneth P. ISOEFF98. A program for studies of isotope effects using Hessian modifications. J Math Chem. 1999;26:75–86. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.