

Abstract
A concise, diastereoselective total synthesis of (±)-cortistatin J has been completed in 20 steps from furan. Key steps include an intramolecular [4 + 3] cyclization of a disubstituted furan with a (Z)-2-(trialkylsilyloxy)-2-enal to construct the tetracyclic core and a (Z)-vinylsilane/iminium ion cyclization to form the A ring.
The cortistatins are a family of novel steroidal alkaloids recently isolated by Kobayashi and co-workers from the marine sponge Corticium simplex collected off Flores Island, Indonesia.1 The unprecedented reorganization of the steroidal framework in addition to the unusual amino and isoquinoline substituents contribute to the appeal of these natural products as targets for total synthesis. However, their biological properties are even more significant. Cortistatins A-D1a and J-L1c inhibited the proliferation of human umbilical vein endothelial cells (HUVECs) with high selectivity. Cortistatins J (1) and A (2) (Figure 1) are the most active and selective of the natural products, having shown cytostatic and anti-proliferative activity (IC50 = 8 and 1.8 nM for 1 and 2 respectively) against HUVECs with a selective index of 300–1100 for 1 and greater than 3000 for 2 in comparison with that of normal human dermal fibroblast or several tumor cell lines. The anti-angiogenic properties of 2 were also evaluated and it was found that the migration and tubular formation of HUVECs induced by VEGF or bFGF could be inhibited at 2 nM concentration. Thus, the cortistatins represent exciting new leads for anticancer drug discovery efforts based upon the inhibition of angiogenesis.2 Accordingly, these natural products have attracted significant attention from the synthetic community, resulting in numerous approaches3 and several total4 and formal5 syntheses.
Figure 1.

Structures of cortistatin J (1) and A (2).
We recognized that the cortistatins represented the ideal targets to apply our methodology for the [4 + 3] cyclization of (Z)-2-(trialkylsilyloxy)-2-enals,6 which are readily prepared via retrocycloaddition of 5-(trialkylsilyloxy)-1,3-dioxins.7 Two representative and relevant examples are shown in Scheme 1. Thus, thermally promoted retrocycloaddition of dioxins 3 provided (Z)-enals 4, which underwent Lewis acid catalyzed [4 + 3] cyclizations8 with a variety of dienes, in this case furan to yield cycloadducts 5.9 During the course of this study we made several key findings: (1) endo adducts are uniformly preferred over exo adducts, (2) the stereoselectivity is better with smaller silyl substituents (e.g. TES vs. TBS) and (3) the endo/exo ratio can be significantly improved by the choice of Lewis acid. These results encouraged us to advance a retro-synthetic analysis of cortistatin J, outlined in Scheme 2. Thus, the dimethylaminocyclohexene A ring could be stereoselectively constructed by intramolecular addition of (Z)-vinylsilane 6 onto the equatorially oriented exo-iminium ion following the Overman protocol.10 The (Z)-vinylsilane moiety of 6 could be installed by a Sonagashira coupling of triflate 7 with (trime-thylsilyl)acetylene followed by semi-reduction with DIBAL-H. Oxidation of the alcohol functionality would provide the aldehyde precursor to the iminium ion. Dienol triflate 7 could be obtained via triflation of the dienolate derived from enone 8. In turn, enone 8 would be provided by oxidation of α-hydroxy ketone 9 to an α-hydroxy enone and reductive removal of the superfluous hydroxyl functionality. Stille coupling with of the enol triflate moiety of 9 with 7-(trimethylstannyl)isoquinoline would install the isoquinoline functionality. In the key step, it was hoped the tetracyclic core 9 could be constructed by a diastereoselective, endo [4 + 3] cyclization between the disubstituted furan and (Z)-2-(triethylsilyloxy)-2-enal moiety in 10, which could be obtained by stereoselective retrocycloaddition of 5-(triethylsilyloxy)-1,3-dioxin 11.
Scheme 1.

[4 + 3] Cyclizations of (Z)-2-(Silyloxy)-2-enals
Scheme 2.

Retrosynthetic Analysis of Cortistatin J (1)
The preparation of the key dioxin 11 began with utilization of Kim’s protocol for the conjugate addition of organoaluminates to α,β unsaturated ketones.11 Thus, TIPS-protected 2-furylethanol12 12 was metalated with n-butyllithium and then converted to the lithium trimethylaluminate, which underwent trimethysilyl triflate-promoted conjugate addition to 2-methylcyclopenten-1-one to afford the TMS enol ether 13 (Scheme 3). The enolate derivative of 13, generated upon treatment of with methyllithium, was diastereoselectivly alkylated with methyl iodoacetate to provide ketone 14. A simple three step sequence (preparation of the enol triflate 15, ester reduction and iodination) yielded iodide 16, which was used to alkylate the azaenolate of the dimethyl hydrazone13 of 2,2-dimethyl-1,3-dioxan-5-one. The alkylated hydrazone was hydrolyzed during workup to yield dioxanone 17. Silylation of the kinetic enolate of dioxanone 16 followed by thermally-promoted retrocycloaddition of the resultant silyloxydioxin gave the requisite (Z)-2-(triethylsilyloxy)-2-enal 10. After much experimentation,14 it was found that subjection of silyloxyenal 10 to catalytic triflic acid in dichloromethane at −78 °C for 1 hour effected the desired [4 + 3] cyclization with TES cleavage in the same pot to provide the tetracyclic core 9 as a single diastereomer, whose structural assignment was based on extensive nOe experiments.
Scheme 3.

Tetracyclic Core Synthesis by [4 + 3] Cyclization
With gram quantities of the tetracyclic core 9 in hand, we turned our attention to installation of the isoquinoline functionality at this point in the synthesis since its presence was not expected to complicate subsequent steps. (Scheme 4). Thus, Stille coupling of the enol triflate moiety of 9 with 7-trimethylstannylisoquinoline 1815 proceeded uneventfully to give isoquinoline 19. Stereoselective and global alkene reduction was achieved with diimide16 and provided tetrahydrofuran 20 in excellent yield. A three step oxidation/deoxygenation sequence (Swern oxidation of α hydroxy ketone 20, α-hydroxy enone triflation and Pd-catalyzed reduction of triflate 21) gave enone 8. The 9(11),10(19)-diene unit was introduced as a dienol triflate via generation of the dienolate of enone 8 with LiHMDS and sulfonylation with PhNTf2. Removal of the TIPS protecting group with aqueous HCl yielded alcohol 7. The (Z)-vinylsilane was incorporated in one step17 utilizing the known potassium trifluoroborate reagent 2218 and Molander’s conditions for Suzuki-Miyaura coupling.19 Parikh-Doering oxidation of the alcohol smoothly provided key aldehyde 23. To our delight, we found that heating aldehyde 23 in the presence of excess dimethylamine hydrochloride in aceto-nitrile at 60 °C overnight provided cortistatin J as a single diastereomer in excellent yield. The cyclization presumably proceeds through iminium ion conformer 6 (Scheme 2), with the dimethyliminium ion adopting a pseudo-equatorial position to avoid an incipient 1,3-diaxial interaction with the ethylene bridge of the tetrahydrofuran.
Scheme 4.

Completion of the Total Synthesis of Cortistatin J
In summary, we have completed a concise total synthesis of (±)-cortistatin J in 20 steps from furan. The tetracyclic core was assembled by an intramolecular, diastereoselective [4 + 3] cyclization of a disubstituted furan and a (Z)-2-(triethylsilyloxy)-2-enal, obtained by retrocycloaddition of a 5-(triethylsilyloxy)-1,3-dioxin, and represents the first application of our methodology. The total synthesis was completed with the Overman-type (Z)-vinylsilane/iminium ion cyclization to construct the A ring. The preparation of other cortistatin congeners, in particular, cortistatins A and K via the pivotal enone 8 are now under investigation.
Supplementary Material
Acknowledgments
We appreciate the financial support provided by the National Institutes of Health (GM28553). We thank David J. Roach for model system studies.
Footnotes
ASSOCIATED CONTENT
Supporting Information. Spectroscopic data and experimental details for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Cortistatins A-D: Aoki S, Watanabe Y, Sanagawa M, Setiawan A, Kotoku N, Kobayashi M. J Am Chem Soc. 2006;128:3148–3149. doi: 10.1021/ja057404h.E-H: Watanabe Y, Aoki S, Tanabe D, Setiawan A, Kobayashi M. Tetrahedron. 2007;63:4074–4079.J-L: Aoki S, Watanabe Y, Tanabe D, Setiawan A, Arai M, Kobayashi M. Tetrahedron Lett. 2007;48:4485–4448.
- 2.(a) Folkman J, Shing Y. J Biol Chem. 1992;267:10931–10934. [PubMed] [Google Scholar]; (b) Carmeliet P. Nature. 2005;438:932–936. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- 3.(a) Craft DT, Gung BW. Tetrahedron Lett. 2008;49:5931–5934. doi: 10.1016/j.tetlet.2008.07.155. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dai M, Danishefsky SJ. Tetrahedron Lett. 2008;49:6610–6612. doi: 10.1016/j.tetlet.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Dai M, Wang Z, Danishefsky SJ. Tetrahedron Lett. 2008;49:6613–6616. doi: 10.1016/j.tetlet.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kotoku N, Sumii Y, Hayashi T, Kobyashi M. Tetrahedron Lett. 2008;49:7078–7081. [Google Scholar]; (e) Kürti L, Czakó B, Corey EJ. Org Lett. 2008;10:5247–5250. doi: 10.1021/ol802328n. [DOI] [PubMed] [Google Scholar]; (f) Yamashita S, Iso K, Hirama M. Org Lett. 2008;10:3413–3415. doi: 10.1021/ol8012099. [DOI] [PubMed] [Google Scholar]; (g) Dai M, Danishefsky SJ. Heterocycles. 2009;77:157–161. doi: 10.3987/com-08-s(f)6. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Liu L, Gao Y, Che C, Wu N, Wang DZ, Li CC, Yang Z. Chem Commum. 2009:662–664. doi: 10.1039/b817376a. [DOI] [PubMed] [Google Scholar]; (i) Magnus P, Littich R. Org Lett. 2009;11:3938–3941. doi: 10.1021/ol901537n. [DOI] [PubMed] [Google Scholar]; (j) Frie JL, Jeffrey CS, Sorensen EJ. Org Lett. 2009;11:5394–5397. doi: 10.1021/ol902168g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Baumgartner C, Ma S, Liu Q, Stoltz BM. Org Biomol Chem. 2010;8:2915–2917. doi: 10.1039/c004275g. [DOI] [PubMed] [Google Scholar]; (l) Yu F, Li G, Gao P, Gong H, Liu Y, Wu Y, Cheng B, Zhai H. Org Lett. 2010;12:5135–5137. doi: 10.1021/ol102058f. [DOI] [PubMed] [Google Scholar]; (m) Liu LL, Chiu P. Chem Commun. 2011;47:3416–3417. doi: 10.1039/c1cc00087j. [DOI] [PubMed] [Google Scholar]; (n) Kotoku N, Sumii Y, Kobayashi M. Org Lett. 2011;13:3514–3517. doi: 10.1021/ol201327u. [DOI] [PubMed] [Google Scholar]
- 4.(a) Shenvi RA, Guerrero CA, Shi J, Li CC, Baran PS. J Am Chem Soc. 2008;130:7241–7243. doi: 10.1021/ja8023466. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nicolaou KC, Sun YP, Peng XS, Polet D, Chen DYK. Angew Chem, Int Ed. 2008;47:7310–7313. doi: 10.1002/anie.200803550. [DOI] [PubMed] [Google Scholar]; (c) Lee HM, Nieto-Oberhuber C, Shair MD. J Am Chem Soc. 2008;130:16864–16866. doi: 10.1021/ja8071918. [DOI] [PubMed] [Google Scholar]; (d) Nicolaou KC, Peng XS, Sun YP, Polet D, Zou B, Lim CS, Chen DYK. J Am Chem Soc. 2009;131:10587–10597. doi: 10.1021/ja902939t. [DOI] [PubMed] [Google Scholar]; (e) Flyer AN, Si C, Myers AG. Nature Chem. 2010;2:886–892. doi: 10.1038/nchem.794. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Yamashita S, Iso K, Kitajima K, Himuro M, Hirama M. J Org Chem. 2011;76:2408–2425. doi: 10.1021/jo2002616. [DOI] [PubMed] [Google Scholar]; (g) Shi J, Manolikakes G, Yeh CH, Guerrero CA, Shenvi RA, Shigesha H, Baran PS. J Am Chem Soc. 2011;133:8014–8027. doi: 10.1021/ja202103e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Simmons EM, Hardin AR, Guo X, Sarpong R. Angew Chem, Int Ed. 2008;47:6650–6653. doi: 10.1002/anie.200802203. [DOI] [PubMed] [Google Scholar]; (b) Yamashita S, Kitajama K, Iso K, Hirama M. Tetrahedron Lett. 2009;50:3277–3279. [Google Scholar]; (c) Simmons EM, Hardin-Narayan AR, Guo X, Sarpong R. Tetrahedron. 2010;66:4696–4700. doi: 10.1016/j.tet.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Fang L, Chen Y, Huang J, Liu L, Quan J, Chuang-chuang L, Yang Z. J Org Chem. 2011;76:2479–2487. doi: 10.1021/jo102202t. [DOI] [PubMed] [Google Scholar]
- 6.The preparation and [4 + 3] cyclization of the parent compound, 2-(trimethylsilyloxy)acrolein was originally disclosed by Sasaki, see: Sasaki T, Ishibashi Y, Ohno M. Tetrahedron Lett. 1982;23:1693.Harmata and co-workers have also examined the TIPS congener of this compound, see: Harmata M, Sharma U. Org Lett. 2000;2:2703–2705. doi: 10.1021/ol006281x.
- 7.Aungst RA, Jr, Funk RL. Org Lett. 2001;3:3553–3555. doi: 10.1021/ol016668f. [DOI] [PubMed] [Google Scholar]
- 8.For reviews on [4 + 3] cycloadditions, see: Harmata M. Chem Commun. 2010;46:8904–8922. doi: 10.1039/c0cc03621h.Harmata M. Chem Commun. 2010;46:8886–8903. doi: 10.1039/c0cc03620j.Harmata M. Adv Synth Catal. 2006;348:2297–2306.Rigby JH, Pigge FC. Org React. 1997;51:351–478.
- 9.A possible explanation for the preferred endo adduct and improved diastereoselectivity with smaller silyl substituents is provided by DFT analysis (B3LYP/6-31G* level). The reaction of furan with 2-(trimethylsilyloxy)acrolein was calculated to proceed via a stepwise mechanism involving conjugate addition of the complexed aldehyde, closure of the silyl enol ether onto the resulting furonium ion and rate limiting silyl transfer, see: Sáez JA, Arnó M, Domingo LR. Tetrahedron. 2005;61:7538–7545.(b) A more direct, concerted parthway involves a [4 + 3] cycloaddition of a complexed aldehyde to afford the same silyl oxocarbenium ion intermediate followed by silyl transfer and decomplexation, see reference 8. An alternative pathway involving a Diels-Alder cycloaddition followed by an α-ketol rearrangement has been advanced, see: Davies HML, Dai X. J Am Chem Soc. 2004;126:2692–2693. doi: 10.1021/ja039908q.
- 10.For the only examples of exocyclic iminium ion/vinylsilane cyclizations, see: Overman LE, Burk RM. Tetrahedron Lett. 1984;25:5739–5742.For a review of the electrophilic substitution of allyl- and vinylsilanes, see: Fleming I, Dunogués J, Smithers R. Org React. 1989;37:57–575.
- 11.Kim S, Park JH. Synlett. 1995:163–164. [Google Scholar]
- 12.Prepared via alkylation of 2-lithiofuran with (2-iodoethoxy)triisopropylsilane, see Supporting Information.
- 13.Prepared via condensation of 2,2-dimethyl-1,3-dioxan-5-one with dimethylhydrazine, see Supporting Information.
- 14.Lewis acids such as Sc(OTf)3, TESOTf, and BF3·OEt2 effected the desired [4 + 3] cyclization, but cleaved the TIPS group. Interestingly, cyclizations with a derivative of enal 10 with the isoquinoline in place of the triflate substituent provided a mixture of endo/exo products.
-
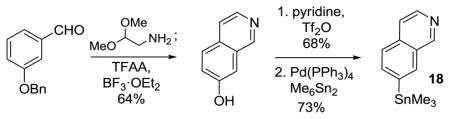
15.Prepared on a 20 gram scale in three steps from 3-benzyloxybenzaldehyde.
- 16.Conventional heterogeneous and homogeneous hydrogenations resulted in reduction of the isoquinoline and/or decomposition.
- 17.Attempts to reduce the TMS-alkyne product of Sonagashira coupling of triflate 7 with (trimethylsilyl)acetylene resulted in reduction of the isoquinoline.
- 18.Molander GA, Ellis NM. J Org Chem. 2008;73:6841–6844. doi: 10.1021/jo801191v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Molander GA, Felix LA. J Org Chem. 2005;70:3950–3956. doi: 10.1021/jo050286w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.