

Abstract
The SAMP1/YitFc mouse strain represents a model of Crohn’s disease (CD)-like ileitis that is ideal for investigating the pathogenesis of chronic intestinal inflammation. Differently from the vast majority of animal models of colitis, the ileal-specific phenotype characteristic of SAMP1/YitFc mice occurs spontaneously, without genetic, chemical or immunological manipulation. In addition, SAMP1/YitFc mice possess remarkable similarities to the human condition in regard to disease location, histologic features, incidence of extra-intestinal manifestations, and response to conventional therapies. SAMP1/YitFc mice also display a well-defined time course of a pre-disease state, and phases of acute and chronic ileitis. As such, the SAMP1/YitFc model is particularly suitable for elucidating pathways that precede the clinical phenotype that may lead to preventive, and therefore more efficacious, intervention with the natural course of disease, or alternatively, for the development of therapeutic strategies directed against chronic, established ileitis. In the following review, we summarize important contributions made by our group and others that uncover potential mechanisms in the pathogenesis of CD using this unique murine model of chronic intestinal inflammation.
Keywords: spontaneous animal model of IBD, Crohn’s disease, genetics, epithelial barrier function, cytokines, intestinal microflora, adhesion molecules, leukocyte trafficking
Introduction
Crohn’s disease (CD) is a chronic, relapsing inflammatory bowel disease (IBD) of unknown origin that can affect any portion of the gastrointestinal tract, from mouth to anus. In approximately two-thirds of cases, the primary disease location is the small intestine, specifically the terminal ileum; however, in one-third of Crohn’s cases, only colonic involvement exists (1). Despite significant advances in uncovering the cause of CD, including the discovery of several susceptibility genes (2–9), to date, the precise etiology has not yet been fully elucidated. It is generally accepted, however, that CD is the result of an abnormal immune response to environmental factors in genetically predisposed individuals (reviewed in 10).
Mouse models have been successfully used in the field of IBD research to understand pathogenic mechanisms of the human disease condition. In fact, the introduction of the first gene deletion models revealed the critical role of several molecules, including cytokines, in the pathogenesis of chronic intestinal inflammation (9, 11–15). In addition, chemically-induced and immunologically-manipulated animal models have allowed specific questions to be answered, including the role of the hematopoietic versus non-hematopoietic compartments, in experimental colitis. However, since single defects underlie many of these models, which are often not present in patients with IBD, there remains a great need for spontaneous animal models that resemble the multi-factorial nature of the disease in humans.
The SAMP1/YitFc mouse model of CD-like ileitis is particularly suitable for investigating pathogenic mechanisms of experimental CD. In fact, the ileal phenotype occurs spontaneous without genetic, chemical or immunological manipulation and displays a time course that allows investigation of the pre-disease, early, and chronic phases of ileitis. In addition, SAMP1/YitFc mice possess remarkable similarities to CD in regard to disease location, histologic features, extra-intestinal manifestations, as well as response to therapies that are effective in patients with CD, including treatment with steroids and anti-TNF. Work from several laboratories have utilized the SAMP1/YitFc model to further understand the role of genetics, mucosal immunity, leukocyte trafficking, cytokine biology, and epithelial barrier function in the pathogenesis of chronic intestinal inflammation. In the following review, we summarize important contributions made by our group and others to undercover potential pathogenic mechanisms leading to IBD using this unique murine model of spontaneous CD-like ileitis.
Derivation and Histopathology of the SAMP1/YitFc Strain
SAMP1/YitFc mice represent a sub-strain of AKR/J mice produced through a program of selective breeding. Originally, a colony of AKR/J mice was developed at Kyoto University in Japan from several pairs of founder mice purchased from The Jackson Laboratory (Bar Harbor, MN). After a number of generations of brother-sister breeding, litters of mice were produced that spontaneously expressed an accelerated senescent phenotype, likely due to an accidental outcross with a non-AKR strain, and were referred to as senescence accelerated mice (SAM) (16). In 1975, the propagation of ten lines of "senescence-prone" mice (SAMP1–10) resulted from brother-sister mating with selection of breeders with accelerated senescence, shortened life span, and other signs of pathology. Among the clinical phenotypes noted in these senescence-prone mice were severe skin lesions and other autoimmune manifestations (17). A SAMP1/Yit substrain was subsequently generated from the SAMP1 line by selective breeding of littermates that exhibited skin lesions and ileitis (18). By the 20th generation of brother-sister mating, this substrain had lost the premature senescent phenotype, but consistently developed terminal ileitis. The initial development of the SAMP1/Yit mouse was carried out in the laboratory of Dr. Satoshi Matsumoto at the Yakult Central Institute of Microbiological Research in Tokyo (18). These mice were described to develop both acute and chronic intestinal inflammation that primarily localized to the ileum and cecum with a discontinuous pattern. The inflammatory infiltrate consisted of lymphocytes, macrophages, and neutrophils, with crypt microabscesses in older mice, and was accompanied by progressive disruption of the epithelium, tissue atrophy, and crypt elongation (18).
Following the acquisition of two breeding pairs provided by Dr. Matsumoto in 1996, a colony of SAMP1/Yit mice was established at the University of Virginia (UVA). Four years later, the colony began to display new and unique features of chronic intestinal inflammation, including perianal disease with fistulae (5–10% of mice), an earlier emergence of ileitis (10 weeks compared to the 30–40 weeks originally described by Matsumoto’s group), high IFNγ production by four weeks of age preceding the onset of ileitis, and chronic ileitis with prominent muscular hypertrophy and focal collagen deposition in inflamed segments, with intestinal stricture formation in approximately 50% of mice greater than 40 weeks of age (19, 20). As a result of these phenotypic changes, the UVA colony was registered as a distinct substrain (SAMP1/YitFc) according to the guidelines of the International Committee on Standardized Genetic Nomenclature for Mice.
The SAMP1/Yit-SAMP1/YitFc mouse has maintained a relatively stable phenotype over the last several years, with several different colonies established throughout the U.S. and Japan. Within the GI tract, the most severe inflammatory changes remain in the terminal ileum that are transmural and discontinuous in nature, with the presence of normal areas of gut mucosa alternating with inflamed regions (skip areas) (Figure 1). The earliest histopathologic alterations occur in the epithelial compartment, with crypt elongation, villous blunting and an overall transition from cells of absorptive function (enterocytes) to that of cells derived from secretory lineages (i.e., Paneth cells, goblet cells, etc.) as the severity of inflammation increases (21, 22). Both active and chronic inflammatory infiltrates characterize the ileal mucosa, with the presence of polymorphonuclear and mononuclear cells in the lamina propria and submucosa, and subsequent occurrence of cryptitis, crypt microabscess formation, focal granulomatous inflammation, basal plasmacytosis, and neural hyperplasia as disease severity progresses (19, 20). In the later stages of established disease, thickening of the bowel wall occurs with stricture formation occurring in the vast majority of SAMP1/YitFc mice over 40 weeks of age (20). In addition, although the current review will provide a comprehensive overview regarding the ileitis phenotype in the SAMP1/YitFc strain, other GI-related pathologies have been previously reported, including chronic, Helicobacter-negative Crohn’s-like gastritis (23) and autoimmune hepatitis (24).
Figure 1. Histologic features of SAMP ileitis.
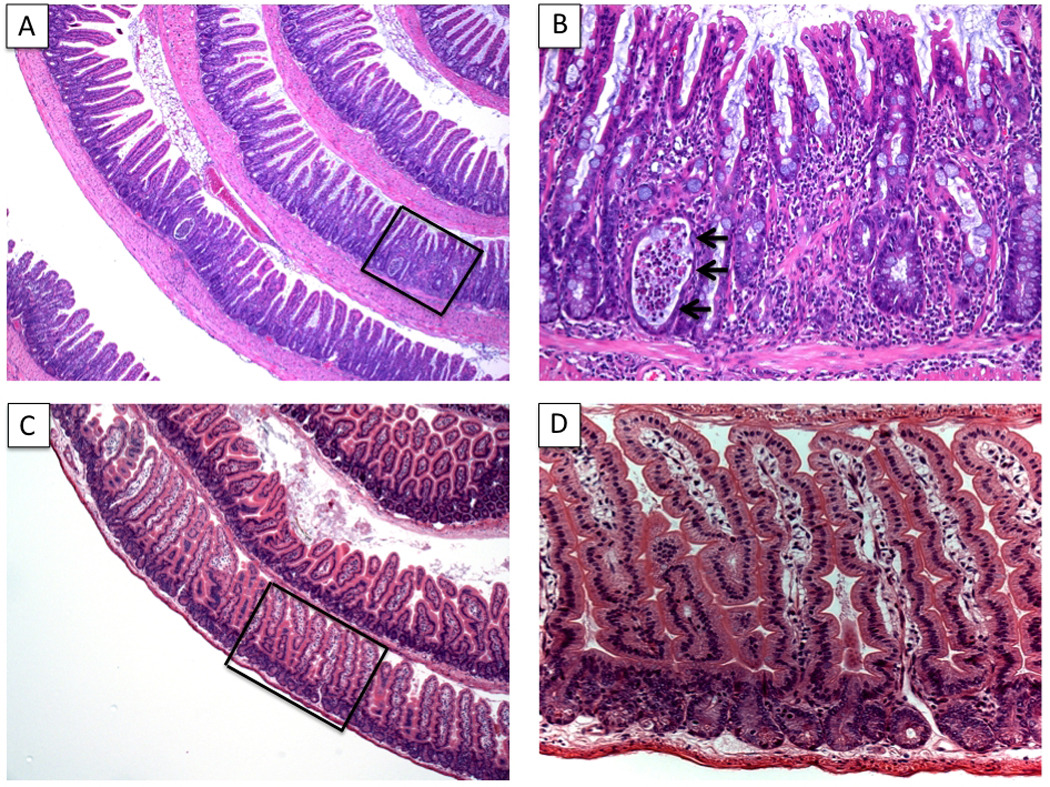
Ileal sections from 20 week-old SAMP and AKR control mice were prepared using the Swiss-roll technique. SAMP mice develop spontaneous, transmural inflammation of the terminal ileum characterized by discontinuous inflammatory infiltrates, villous architecture alterations and bowel wall thickening, with hypertrophy of the muscular layers (A, 4X). Higher magnification of panel A (framed area) shows marked villous blunting and distortion, infiltration of acute and chronic immune cells, and the presence of a crypt abscess (arrows) (B, 20X). Uninflamed, control AKR sections are also shown (C, 4X & D, 20X). Panels A and B originally published in Ann N Y Acad Sci, 2009; 1165:301–7.
Genetic Control of SAMP1/YitFc Ileitis
In contrast to the genetic models of murine intestinal inflammation in which the inherent defect (i.e. suppression or overexpression of certain genes) is predetermined and thus clearly defined, ileitis in SAMP1/YitFc mice develops spontaneously (25). In this regard, the SAMP1/YitFc model truly resembles the human condition and accordingly, the search for genotype-phenotype associations is similar to and carries the same difficulties as the exploration of genes responsible for human IBD. In addition, as mentioned earlier, although the original SAMP lines were derived from AKR/J mice, inadvertent cross to a non-AKR strain resulted in genetic contamination which, in the case of the parental SAMP1/Yit strain, accounted for 20–30% of the total genomic material (16).
To determine the genetic composition of SAMP1Yit/Fc mice in more detail, B6SF1 mice (SAMP1Yit/Fc × C57BL/6J) were tested for a set of 206 mapped microsatellite loci which can efficiently detect fragment size differences between AKR and B6 mice (26). Surprisingly, polymorphisms with a distinct pattern of two alleles were seen in only 43% of the loci in B6SF1 mice (27). More than 50% of loci were found to be different between SAMP1/YitFc and AKR mice based on precise microsatellite size determinations (27). Allelic size determinations on multiple control strains for non-AKR alleles predicted no similarity between genetic material of the unidentified strain in SAMP1/YitFc mice and any of the known inbred strains. Therefore, SAMP1/YitFc represents a truly “recombinant” mouse, resulting from a genetic cross between AKR/J and a not yet identified strain(s).
Neither AKR nor B6 mice develop intestinal inflammation after co-housing with SAMP1/YitFc mice under normal specific pathogen-free (SPF) vivarium conditions. Therefore, the original approach taken for the identification of ileitis-associated loci was the outcross of the SAMP1/YitFc mouse (ileitis-prone) to the parental (AKR/J) and the unrelated C57BL/6J (B6) strains (both ileitis-resistant) (27). The aim of this strategy was to reveal the genetic components of the strain that determine the disease pattern in SAMP1/YitFc mice. Outcross of SAMP1/YitFc to AKR/J mice resulted in complete elimination of ileitis, clearly pointing to dominant suppressive effects of AKR-derived alleles on the disease phenotype. On the other hand, backcross to B6 mice showed that both B6SF1 and F2 generations developed ileitis with characteristics of SAMP1/YitFc disease, demonstrating that the disease phenotype was due to inheritance of both dominant and recessive alleles. Although major differences were detected in the MHC components between the SAMP1/YitFc strain, which carries the AKR-derived H2k haplotype, and B6 mice, which display the H2b haplotype, the MHC pattern did not affect susceptibility to ileitis (27).
Initially, to identify ileitis-associated alleles, genome-wide scans were performed in the cohorts that were produced by the aforementioned outcrosses. These scans were able to reveal chromosomal loci that were strongly linked to the presence of inflammatory changes (described in detail below). The strongest associations were then confirmed through the generation of interval-specific congenic strains. Subsequently, genes contained in each locus were identified through a genetic database search. Finally, the most suitable regional candidates were selected and further studied by both sequence analysis as well as by expression and functional studies.
Identification of Ileitis-Susceptibility Loci
An initial genome-wide scan was performed in the two cohorts of F2 mice representing the extremes of the phenotype. Equal numbers of mice with a total ileitis score of >8 (SAMP-like) or <0.5 (B6-like) were compared for a panel of 103 informative microsatellite loci spanning the entire genome. Analysis of single-point quantitative trait loci (QTL) for total inflammatory scores showed a single SAMP-derived susceptibility locus on chromosome 9 (Chr9) (D9Mit123, maximal likelihood ratio statistic (LRS)=19.0; P=0.000074). Regression analysis after stratifying by genotype at D9Mit123 showed evidence suggestive of additional linkage to loci on Chr6, 17, and X (D6Mit39, LRS=11.3, P=0.0036; D17Mit130, LRS=12.9, P= 0.0016; DXMit117, LRS=11.1, P=0.0038). The respective loci conferring susceptibility to ileitis were thereafter designated Ibdq1 (Chr9), Ibdq2 (Chr6), Ibdq3 (Chr8) Ibdq4 (ChrX), respectively.
Ibdq1
In the (SAMP1/YitFc × B6)F2 cohort selected for extreme phenotypes, indices for each of the three primary traits (active inflammation, chronic inflammation, and epithelial architecture) were highly correlated and did not allow testing for linkage to individual components of the histology score. On the other hand, in F2 mice with ileitis of intermediate severity, these individual components varied in their contributions to the total inflammatory score. Single-point QTL analysis for each of the primary trait indices for the entire F2 population showed that when analysis was limited to the severity of inflammation-associated epithelial changes, it strengthened evidence of linkage to loci on Chr9, with a peak LRS of 14.2 at D9Mit48. In addition, additive effects of SAMP1/YitFc-AKR alleles on Chr9 were demonstrated since the median total inflammatory score for heterozygotes (SAMP1/YitFc-B6) was intermediate between SAMP1/YitFc and B6 homozygotes, suggesting a dose-dependent effect for the Chr9–linked, Ibdq1 susceptibility locus.
The Ibdq1 region contains several genes that encode for proteins that are selectively expressed on intestinal epithelial and/or hematopoietic cells involved in the immune response (for detailed analysis see (27)) (Table I, Figure 2). Of extreme interest is that the chemically-induced colitis susceptible locus (both Dss and Tnbs1) was also predicted on Chr9 and overlaps with Ibdq1 (28, 29). On Ibdq1, the genes encoding IL-10 receptor alpha (Il10ra) and IL-18 (Il18) serve as attractive regional candidates based on their ability to regulate inflammatory responses that might result in epithelial damage. Mice with induced null mutations in Il10 develop significant colitis (11) and tissue-specific deletion of a major signaling target of the IL-10 receptor, Stat3, in macrophages and neutrophils produces a similar phenotype (30). Recently, mutations in genes encoding the IL10R subunits were associated with early-onset IBD-like enterocolitis with familial segregation (31). Comparative sequencing in AKR/J, C57BL6/J, and SAMP1/YitFc strains detected two single-nucleotide insertions at positions 732 and 3098 of introns 1 and 3 of the Il10ra gene, in the latter. Based on their locations, none of these polymorphisms are predicted to influence the signaling event, but a possible long-range transcriptional effect in this haplotype cannot be ruled out. Despite allelic differences between the Il10ra for B6 and SAMP1/YitFc/AKR mice, no differences were evident for IL-10 signaling in bone-marrow derived macrophages from SAMP1/YitFc versus B6 mice, indicating no differences for the expression and function for IL-10ra in the two strains.
Table I.
Candidate genes for SAMP1/YitFc ileitis.
| Chromosome location | Gene (Loci) | Mouse IBD Designation |
|---|---|---|
| Chr 6 | Pparγ (D6Mit 155-D6Mit288) | Ibdq2 |
| Chr 6 | Prok2 (D6Mit 149, 3p13) | Ibdq2 |
| Chr 6 | Itpr1 (D6Mit149, 3p26.1) | Ibdq2 |
| Chr 6 | Ogg1, Hrh1 (D6Mit149, 3p25.3) | Ibdq2 |
| Chr 6 | Mdb4 (D6Mit149, 3q21.3) | Ibdq2 |
| Chr 6 | Grin2b, Emp1 (D6Mit 149, 12p 13.1) | Ibdq2 |
| Chr 6 | Cdkn1b, Prh1, Klrd1 (D6Mit 149, 12p 13.2) | Ibdq2 |
| Chr 6 | Tnfrsf1a, Il5ra,Cd9, Nf3, Cd69, Cd4, Eno2,Ltbr1,Bcap37,Tnfrsf7,Klrg1,Apobec1(D6Mit149, 12p13.31) | Ibdq2 |
| Chr 6 | Cxcl12, Ret-C (D6Mit149, 10q11.21) | Ibdq2 |
| Chr 6 | Alox5 (D6Mit149, 10q21.1) | Ibdq2 |
| Chr 6 |
Ccnd2 (D6Mit149,12p13.32) Wnt5b (D6Mit149,12p13.33) |
Ibdq2 |
| Chr 6 | Atp6v1e1, Il17r (D6Mit149, 22q11.21) | Ibdq2 |
| Chr 6 | Bid (D6Mit149, 22q11.22–23) | Ibdq2 |
| Chr8 | Nod2 (DMit215) | Ibdq3 |
| Chr 9 | Il-10rα, Il-18 (DMit 297-DMit123) | Ibdq1 |
| ChrX | Foxp3, wasp (A1.1) | Ibdq4 |
| ChrX | Nemo, Irak1 (A7.3) | Ibdq4 |
| ChrX | Tlr7/Tlr8 (F7) | Ibdq4 |
| ChrX | Tlr13 (D) | Ibdq4 |
Bold indicates genes experimentally tested to date in SAMP1/YitFc mice
Figure 2. Mapping of potential chromosomal loci and genes for the susceptibility to SAMP ileitis.
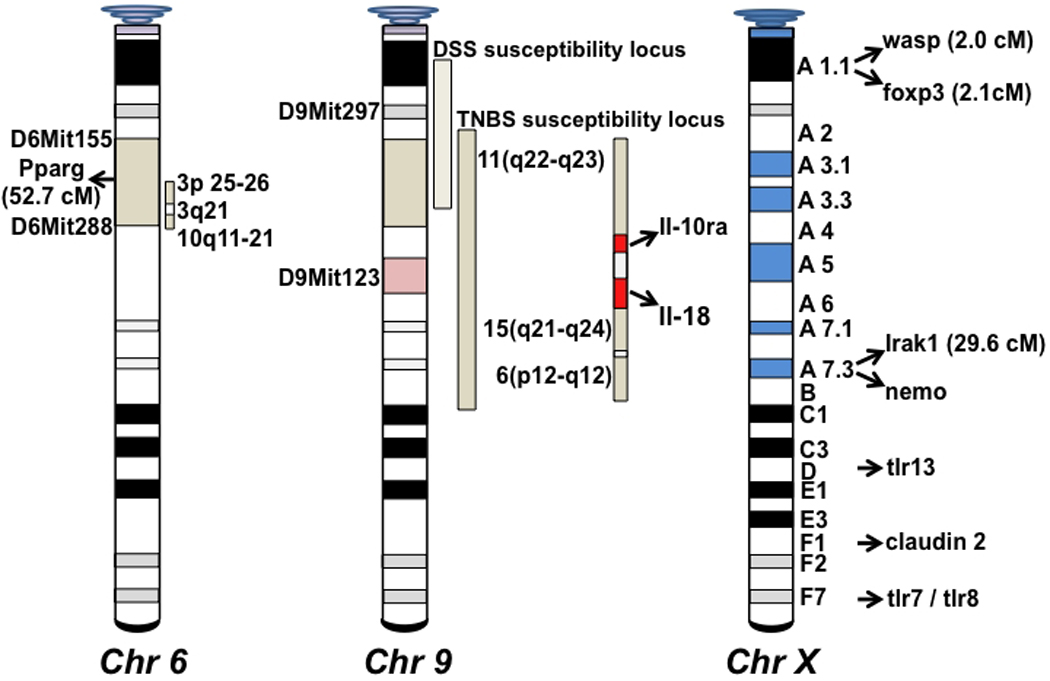
Indicated genes include information from both published (27, 42) and preliminary (not yet confirmed) data. Many of the depicted genes are associated with epithelial barrier as well as immune regulatory functions. Portions of the figure originally published in Gastroenterol, 2003; 125:477–90.
The experimental evidence for the role of Il18 comes from studies showing protective effects of IL-18 blockade on chemically-induced murine colitis (32). Furthermore, increased intestinal expression in CD patients has been shown for both IL-18 and IL-1 converting enzyme, which is required for processing of proIL-18 to its active form (33). Similar to Il10ra, transcribed sequences of exons 1–5 and of 3’ untranslated region (UTR) for Il18 seemed identical among the three mouse strains studied (AKR, SAMP1/YitFc, B6). In addition, no polymorphisms were detected within the 1500 base pairs (bp) immediately upstream of the transcription start site or the terminal 700 bp of intron 1. IL-18 immunoreactivity, however, was present at markedly increased levels in serum and mesenteric lymph nodes (MLNs) from young (4 week-old) SAMP1/YitFc mice relative to age-matched B6 mice, that is, before the development of overt ileitis. This is compatible with a role for this cytokine in the very earliest stages of intestinal inflammation. In all, it appears that enhanced IL-18 expression in SAMP1/YitFc mice may result from differences at other genetic loci that can upregulate expression in SAMP1/YitFc mice rather than from differences in the Il18 locus itself. Interestingly, an association between CD in a human population and a silent allelic variant in the coding region of IL-18 has been reported by another group (34). If this association can be confirmed, it suggests that long-range transcriptional control of IL-18 expression in certain haplotypes may alter susceptibility to CD in humans. Additionally, previous studies have confirmed association of polymorphisms in the promoter region of IL-18 (−137 G/C) and the IL-18 gene haplotype-2 (−607A, −137C) with IBD (35, 36). These findings increase the likelihood that Ibdq1 reflects a yet undetected difference at the Il18 locus in the SAMP1/YitFc strain.
Ibdq2
Kozaiwa et al. showed significant evidence for linkage of ileitis at Chr6, with a peak LRS of 15.3 (P=0.0001) at D6Mit288 (Table I, Figure 2) (27). This locus appears to originate from non-AKR genetic material and was designated as Ibdq2 showing no primary linkage to any other chromosome. Included in this locus is a homolog to the human Chr3(p21–p26) region previously suggested to encode a susceptibility locus for human CD in four independent genome-wide scans (37–41).
Several candidate genes are located within the Ibdq2 region (Table I). Among these, the role of peroxisome proliferator-activated receptor-gamma (PPARγ) in the pathogenesis of SAMP1/YitFc ileitis was studied in depth (42). PPARγ is a nuclear receptor highly expressed in intestinal epithelial cells (IEC) (43) and to a lesser extent, in immune cells (44, 45), and is a key molecule in the regulation of insulin metabolism (46), cell proliferation and in the downstream signaling of innate immune pathways (47). It was selected as an obvious candidate due of its role in suppressing inflammatory responses (48). In both human and mice, two gene isoforms, Pparγ1 and Pparγ2, were found to be regulated differentially and show small differences in N-terminal sequences resulting from the use of alternative exons (46, 49). Several important findings, pointing to a complex, tissue-specific regulation of Pparγ in SAMP1/YitFc mice were demonstrated (42). First, PPARγ mRNA levels were notably higher in both the ileum and jejunum of AKR and B6 as compared with SAMP1/YitFc mice. Second, expression patterns for PPARγ immunostaining were clearly affected by the stage of ileitis development. In young mice (4-weeks of age, before the development of ileitis), PPARγ staining was remarkably weaker in SAMP1/YitFc mice as compared to AKR controls. Conversely, differences were less evident in older mice with established inflammation (10–30 weeks of age). Third, staining was absent in the epithelial crypt region of SAMP1/YitFc mice, extending only from the base to the tip of villi. This was not the case in AKRs in which persistent expression in epithelial crypts was seen. PPARγ staining was consistently absent in the crypts of SAMP1/YitFc mice, independent of age. In fact, expression was limited to epithelial cells adjacent to the gut lumen, which is consistent with the reported localization of PPARγ in colonic epithelium (43). Interestingly, when SAMP1/YitFc mice were treated with the synthetic PPARγ agonist, rosiglitazone, no beneficial effects were observed in the severity of ileitis. This suggests that specific expression in the crypts of the small intestine is required for a protective effect of enhanced PPARγ activation. Finally, in all strains examined, the colon displayed weaker staining for PPARγ in comparison to the small intestine. This is in line with the report of Dubuquoy et al. showing no genetic association between Pparγ and ulcerative colitis (UC) (50).
PPARγ inhibits NF-κB-mediated proinflammatory signaling (51). In this perspective, it is interesting to note that PPARγ is expressed in the same mucosal compartment (epithelium) as the intracellular pathogen receptor NOD2/CARD15. The latter is a human CD susceptibility gene and functions by regulation of NF-κB activity. Concomitant expression of NOD2 and PPARγ is suggestive of co-operative control of NF-κB signaling in IEC. Alternatively, genetically-determined defective regulation of NF-κB by PPARγ may contribute to genetic risk in a subset of individuals with CD who do not carry causative mutations in NOD2 (42).
Ibdq3
This susceptibility locus was discovered during a B6 backcross which found statistical evidence for the existence of a QTL on Chr8. Interestingly, the effects of this potential QTL were restricted to a cohort of young mice (10–12 wk olds). Therefore, this locus may be particularly related to early-onset disease. As more severe disease was seen in heterozygous mice, these results are suggestive of the presence of a dominant susceptibility allele in the B6 strain that functions to accelerate the development of ileitis. Similar to Ibdq1, this locus may also be associated with epithelial changes more than with inflammatory responses. Of note, the mouse homolog of the nod2/card15 gene maps to Chr8, which originally raised the possibility of NOD2/CARD15 defects in SAMP1/YitFc mice. However, upon further analysis it became evident that no significant association was evident for the Nod2 gene and ileitis-like disease in SAMP1/YitFc mice (42).
Ibdq4
Weaker evidence of linkage to epithelial abnormalities was also seen on ChrX in the primary analysis, and this linkage persisted after controlling for the effects of the Chr9 locus in a composite analysis. This locus was eventually designated Ibdq4 and was shown to be AKR-related. It is of interest that total inflammatory scores in SAMP1/YitFc female mice are usually significantly higher than those in males. Indeed, median total inflammatory scores of 11.5 (range, 5.0 –20.5) and 9.25 (range, 1.25–26.0) were found for female and male mice, respectively. This result is consistent with the X-linked effects on disease severity. Moreover, ChrX has been shown to be involved in gender differences exhibited in various autoimmune diseases (52). ChrX includes some critical candidate genes in SAMP1/YitFc mice with immunoregulatory and epithelial barrier function, such as foxp3, wasp, Irak1, nemo, claudin-2, as well as the tlr7/8 locus (Figure 2, Table I), that has recently been reported to have high-frequency haplotypes associated with both female CD and UC patients (53).
Conclusively, selecting interval-specific recombinations involving the backcross between congenic generations with the parental SAMP1/YitFc strain gives substantial confirmation of precise genetic mapping and helps to identify true interval-specific candidate genes. The congenic approach is a powerful tool to understand the genetic mechanism of disease, like IBD, where even a slight difference can mask/expose the disease by virtue of additional environmental effects. Having differences only at limited intervals, the congenic mouse lines should allow direct confirmation of local gene effects on the disease phenotype even in the presence of minor changes. Most of the linkage analysis done thus far in SAMP1/YitFc mice suggests that the major susceptibility loci are associated with epithelial changes. This highlights the importance of the epithelial compartment in disease pathogenesis and is consistent with published data pointing to the central role of the epithelial barrier for the progression of ileitis in SAMP1/YitFc mice (54).
Contribution of the Intestinal Epithelium
Increasing evidence supports a pivotal role of the intestinal epithelium in maintaining gut homeostasis. The intestinal epithelial monolayer has the ability to act as a selective barrier against harmful molecules, as well as food and bacterial antigens, while simultaneously allowing the absorption of nutrients from the intestinal lumen. As such, dysregulation of epithelial barrier function has been proposed as a potential etiologic factor in the development of spontaneous intestinal inflammation. In support of this concept, previous studies on IBD patients with active disease report impaired intestinal permeability and alterations of epithelial intercellular junction proteins, which persists during disease remission and can predict the onset of recurrence (55–57).
Remarkably, SAMP1/YitFc (referred to from this point forward as SAMP) mice show striking alterations in epithelial morphology and function that are likely an important pathogenic factor for the onset of disease. In fact, as early as 4 weeks of age, before the onset of histologic evidence of gut inflammation, SAMP mice display a dramatic expansion of epithelial cells of the secretory lineage, including goblet, Paneth, and intermediate cells (21) (Figure 3A–H). These alterations persist throughout the duration of ileitis in this model and are also accompanied by expansion of the crypt stem cell population. Moreover, in inflamed areas, SAMP mice show an increase in IEC apoptosis, which is restored to normal levels following the administration of anti-TNF antibodies (58). Importantly, it has been clearly demonstrated that the primary defect that triggers ileitis in this model originates from a non-hematopoietic source (54), suggesting that epithelial morphologic features characteristic of SAMP may represent the initiating factor(s) leading to downstream mucosal immune dysregulation. In fact, bone marrow chimeras (BMCs), consisting of irradiated SAMP receiving uninflamed control AKR BM, developed severe ileitis, which was minimal to absent in reciprocal BMCs consisting of AKR recipients receiving SAMP BM. Functionally, increased epithelial paracellular permeability was demonstrated, both in vivo and ex vivo, in SAMP ilea compared to control AKR mice as early as 3 weeks of age, again, before the onset of ileal inflammation (Figure 3I). The inherent permeability defect appears to be independent of bacterial colonization in the gut as 3 week-old SAMP raised under germ-free (GF) conditions showed the same defect in small intestinal barrier function, and subsequent ileitis, as those raised under SPF conditions; however, the gut microflora did appear to modulate the severity of disease by exacerbating inflammation (Figure 3J). No alteration of colonic permeability was detected, suggesting a pathogenic link between an intestinal epithelial barrier defect and the development of ileal-specific inflammation.
Figure 3. Morphologic and functional epithelial alterations in SAMP mice.
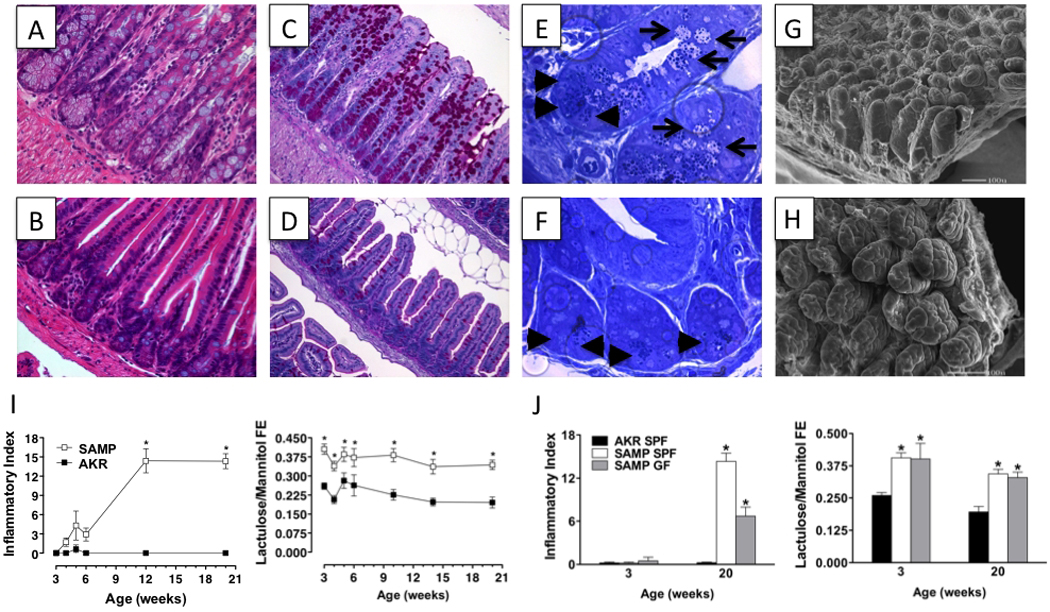
Epithelial hyperplasia, primarily of cells derived from secretory cell lineages, including Paneth (A & B, 40X) and goblet (C & D, PAS staining, 20X) cells, as well as an altered villous:crypt ratio is observed in the ileum of SAMP (A & C) compared to AKR control mice (B & D). Representative semi-thin sections (□200 nm) of ileal crypt region in SAMP mice (E, 100X) demonstrate abnormal Paneth cell number (arrowheads), and presence of “intermediate cells” (Paneth/goblet hybrid cells, arrows) compared to AKR mice (F, 100X). Scanning electron micrographs show severe villous expansion and blunting, as well as abundant mucus production in SAMP (G) versus AKR controls (H). (A–H, originally published in Ann N Y Acad Sci, 2009; 1165:301–7). (I) Time course of SAMP ileitis versus AKR controls measured by total inflammatory scores (left panel), compared to small intestinal permeability, measured in vivo by Fractional Excretion (FE) of regional-specific sugar probes (lactulose:mannitol) (right panel) demonstrate that small intestinal paracellular permeability is impaired prior to the onset of ileitis in SAMP mice. (J) Early, pre-inflammatory alterations in small intestinal barrier function are present in the absence of the enteric microflora. Small intestinal permeability and ileal inflammation were measured in 3- and 20-week-old GF SAMP mice. N≥4 per group; *P<0.05 vs. age-matched AKR and SPF SAMP. Data originally published in J Exp Med, 2006; 203:541–52 and in Ann N Y Acad Sci, 2009; 1165:301–7.
Epithelial barrier dysfunction in SAMP mice is also accompanied by intrinsic alterations of tight junction (TJ) protein expression and localization, characterized by an 8-fold increase in claudin-2 (CLDN2) and a decrease in occludin (OCLDN) mRNA transcript levels, in young mice, prior to the onset of inflammation. Overexpression of CLDN2 in MDCK I cells leads to increased epithelial paracellular permeability due to aberrant CLDN2-CLDN2 pairing on adjacent cells that results in a leakier epithelial barrier compared to pairing with other CLDN(1–4) proteins (59). Conversely, overexpression of OCLDN in MDCK cells results in an increased number and width of the TJ network and a decrease in epithelial paracellular permeability, indicative of a tighter barrier (60). These aforementioned trends in CLDN2 and OCLN expression have also been reported in patients with both CD and UC (61, 62). In SAMP mice, increased CLDN2 and decreased OCLN mRNA and protein levels remain sustained throughout the course of ileitis, with increase in CLDN2 aberrantly distributed beyond the crypt-villous junction and up onto the villous tips. Interestingly, the CLDN2 gene is located on the telomeric end (135.15–135.16 Mb) of ChrX; however, future studies will resolve whether the genetic contribution of CLDN2 is critical for the development of SAMP ileitis.
In this same line of thought, several susceptibility loci, located on Chr6, 8, 9, 17 and X (27, 42), with significant linkage to the development of SAMP ileitis, contain genes involved in the regulation of intestinal epithelial functions. In fact, E-cadherin (Cdh1) on Chr8, JAM-C (Jam3), cingulin-like 1 (Cgnl1), nectin-1 (Pvrl1) and β-catenin (Ctnnb1) on Chr9, afadin (Mllt4) on Chr17, as well as Cldn2 on ChrX are all potential candidate susceptibility genes as they are related to the structural formation of the apical junctional complex (22). Indeed, further analysis is needed in order to assess their actual role(s) in genetically transmitting the SAMP phenotype.
Interactions between the epithelium and intestinal microflora are also known to have a major impact on the modulation of chronic intestinal inflammation. In SAMP, the administration of antibiotics significantly dampened the severity of ileitis with greater effects in younger mice (9 weeks at the end of a 6-wk treatment period) than in older mice (44 weeks at the end of treatment) (63). In addition, as mentioned earlier, GF SAMP develop a much less severe disease phenotype compared to SPF-raised mice (64). Interestingly, the administration of probiotic-derived products, such as Bifidobacterium breve, Bifidobacterium bifidum and Lactobacillus acidophilus fermented milk or heat killed Lactobacillus casei substrain Shirota, reduced ileal inflammatory and proinflammatory cytokine production in treated SAMP mice (65, 66). In fact, our group recently reported that the probiotic mixture, VSL#3 (a mixture of 8 different probiotic strains), whose efficacy in the treatment of human UC and pouchitis has been previously reported (67, 68), is effective in preventing the onset of ileal inflammation in SAMP mice, however is not effective in mice with established disease (69). VSL#3 appears to exert its preventive effects by directly influencing epithelial barrier function; in fact, the inherent permeability defect characteristic of SAMP mice is almost completely reversed by VSL#3. Paradoxically, VSL#3 increases TNF expression in IEC and anti-TNF antibody treatment in young SAMP abolished VSL#3’s preventive effects on ileitis (69). Indeed, the mechanism of action of VSL#3 appears to be related to an increase in epithelial production of TNF, which is likely defective in young SAMP. It is postulated that local TNF production by IEC may be pivotal in maintaining proper innate immune defenses and therefore, an early impairment of its expression may lead to an increased bacterial penetrance with more extensive exposure of underlying mucosal immune cells to commensal antigens, thus triggering the onset of intestinal inflammation. In support of this concept, SAMP ileitis is characterized by different immunological stages: an early stage of innate immunodeficiency, wherein epithelial-derived TNF expression is deficient and cannot be enhanced in order to prevent the onset of disease, and a late stage of chronic and hyperactivation of adaptive immunity when activated immune cells produce high levels of TNF that sustains inflammation, which can benefit from the selective blockade of TNF (58).
Apart from permeability defects, several other epithelial alterations have been described in SAMP mice. As mentioned and discussed in detail earlier, PPARγ expression and localization is also significantly altered in ileal IEC from SAMP mice (42). In addition, resistin-like molecule-β (RELMβ), whose mRNA is normally expressed in undifferentiated colonic, but not ileal, crypt epithelial cells and whose protein is detected in goblet cell mucin granules, is highly expressed at both the mRNA and protein levels in SAMP ileal epithelial cells (70). This protein is thought to have potent proinflammatory qualities as RELMβ KO mice do not develop colonic inflammation after DSS challenge (71). RELMβ expression in SAMP is also observed at the onset of ileitis, and the magnitude of its expression is related to the extent of histological damage. Despite the reported physiologic release of RELMβ in response to bacterial load, SAMP also express the protein after antibiotic treatment and in mice housed in a GF environment. Thus, RELMβ overexpression in SAMP mice appears to be a primary event, not related to bacterial colonization (70).
SAMP mice also present with dysregulated Defcr-rs gene expression, a gene encoding the microbicidal cysteine-rich sequence 4C (CRS4C) peptides. In fact, CRS4C production is 1000-fold increased before the onset of ileal inflammation in Paneth cells from 4 week-old SAMP compared to age-matched AKR and B6 control mice, and remains elevated throughout the SAMP’s lifespan (72). In addition, increased expression of STAT3, a critical mediator of IL-6 signaling, has also been described in the nuclei of IEC from SAMP mice (73). Overexpression of STAT3 in SAMP IEC suggests hyper-responsiveness to IL-6 activation that may result in amplification of IL-6-dependent proinflammatory events in SAMP ileitis.
In addition to TNF, IEC from SAMP mice have been reported to be potent producers of other pro- and anti-inflammatory cytokines, including the IL-1 receptor antagonist (IL-1Ra), IL-18, and most recently, IL-33 (74). IL-33 is the newest member of the IL-1 family of cytokines and appears to play an important role in several chronic inflammatory disorders, including IBD (reviewed in Pastorelli L, De Salvo C, Cominelli M et al.). However, in contrast to other IL-1 family member, including IL-1 and IL-18, which predominantly promote Th1-type responses, IL-33 mainly induces Th2 cytokines (i.e., IL-5 and IL-13) (75), and specifically in the gut, promotes epithelial hyperplasia and eosinophil/neutrophil infiltration (75). In SAMP mice, IEC were found to be a potent producer of IL-33, which was increased compared to uninflamed AKR controls and correlated to ileal disease severity (74). IL-33 also had the ability to induce IL-5, IL-6 and IL-17 from MLN cells, suggesting its role as a primary, initiating mediator in the development of ileitis, promoting downstream Th2 immune responses found in later stages of SAMP ileitis (76), as well as proinflammatory and Th17 effects. These results are consistent with, and confirm recent findings emphasizing, the important role of IL-33 in patients with IBD (74, 77–79).
However, despite the relevance and aforementioned results suggesting that defects in the gut epithelium and cells of epithelial lineage may be a primary etiologic factor in the development of SAMP ileitis (summarized in Figure 4), it should be noted that this mouse strain also displays significant alterations in more than one component of normal mucosal immune responses. In fact, backcross of the RAG-2 knockout mutation onto the SAMP background, resulting in SAMP mice that lack mature, functional T and B lymphocytes, suppress the development of ileitis (unpublished results), and indicate that the adaptive arm of the mucosal immune system is required for intestinal inflammation to occur. In addition, our group has recently begun to investigate the interaction between the intestinal epithelium and innate immune cells, such as mucosal dendritic cells (DCs) in SAMP mice. Our preliminary findings show an expanded population of lamina propria CD11c+ cells with DC morphology (Figure 5) and increased “protrusions” between IEC in SAMP compared to AKR mice, potentially indicating active luminal antigen sampling of DC dendrites (unpublished results). Therefore, it is likely that the SAMP mouse strain represents a “multiple hit” model of chronic intestinal inflammation that develops after several interacting components of host mucosal immune responses, of both innate and adaptive origin, are put into motion.
Figure 4. Summary of the epithelial contribution to SAMP ileitis.
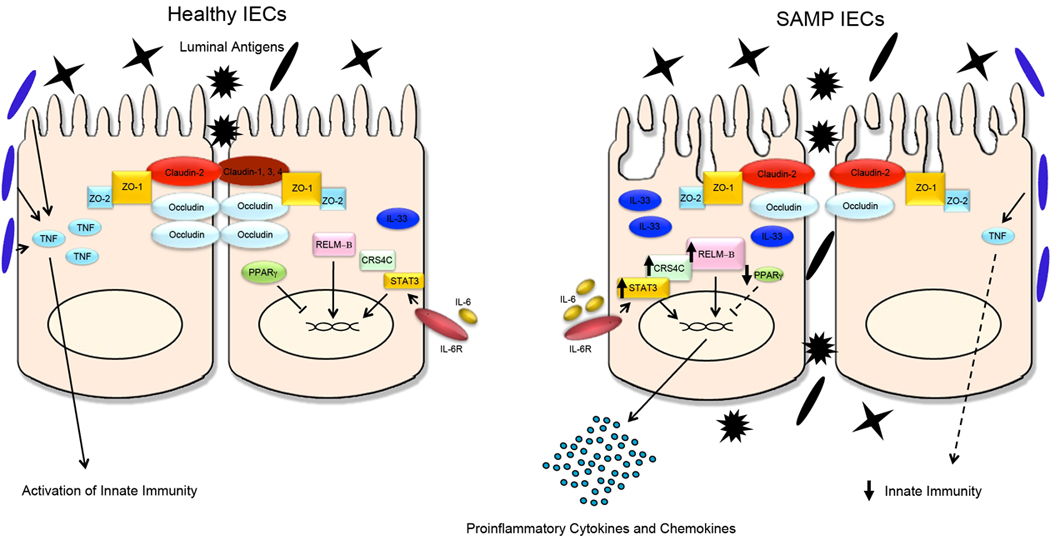
Schematic representation of IEC biology from healthy, uninflamed gut and from SAMP ileitis. Several molecular aberrations in SAMP IEC may exist in initiating and sustaining chronic intestinal inflammation characteristic of these mice. A primary dysregulation of TJ protein expression is characterized by marked upregulation of CLDN2 and downregulation of OCCLDN, leading to increased epithelial paracellular permeability and penetration of luminal antigens into the underlying gut mucosa. During health, probiotics have the ability to augment epithelial-derived TNF, which is pivotal for maintaining epithelial barrier function and innate immune defenses; however, this mechanism is dysregulated in SAMP mice. Increased levels of epithelial-derived pro-inflammatory mediators, including IL-33, CRS4C, STAT3 and RELM®, and a decrease in anti-inflammatory molecules, such as PPARγ, have the ability to initiate and activate inflammatory cascades leading to chronic ileitis. ZO-1, -2, zonula occludens-1, -2; TNF, tumor necrosis factor: IL-6, IL-6R, interleukin-6, IL-6 receptor; IL-33, interleukin-33; CRS4C, cysteine-rich sequence 4C; STAT-3, signal transducer and activator of transcription-3; RELM®, resistin-like molecule-®; PPARγ, peroxisome proliferator-activated receptor-γ.
Figure 5. Epithelial-DC morphology is significantly altered in SAMP mice.
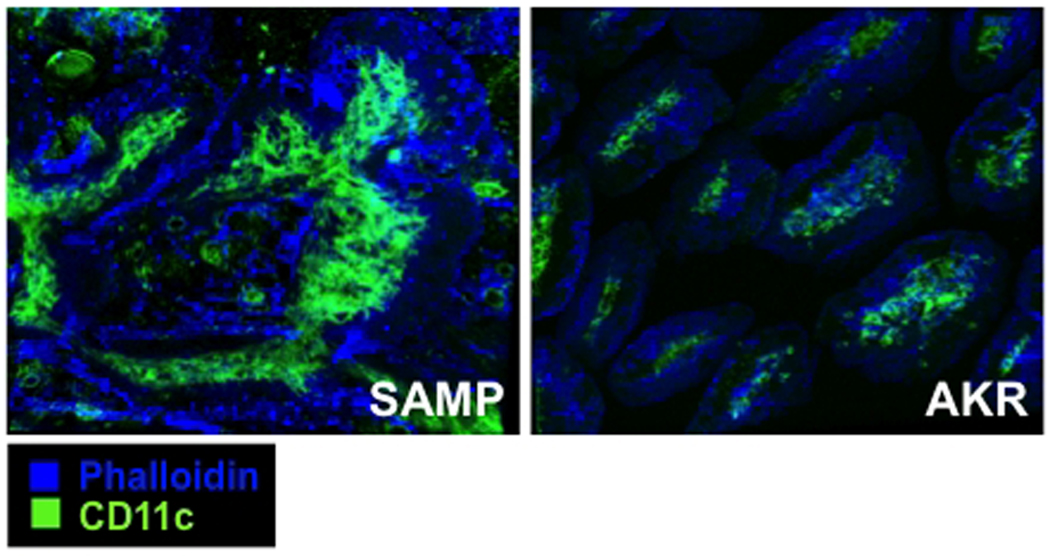
Massive recruitment of CD11c+ DCs and villous expansion in SAMP (left panel) ileum vs. AKR (right panel) mice occurs early during the onset of inflammation, and increases as disease progresses. 40 week-old SAMP and AKR ilea were fixed in 1% PFA solution and embedded in OCT. Frozen sections were stained for CD11c (green) and phalloidin (blue), and images acquired using a Leica SP5 confocal microscope and analyzed by Bitplane Imaris.
Mucosal Cytokine Profile and Role of the Commensal Flora
One of the unique characteristics of the SAMP mouse is the spontaneous development of inflammation in the terminal ileum. From a histological standpoint, ileitis is fully established when mice reach 9–10 weeks of age with a penetrance of almost 100% (19, 20). This uniform distribution and precise temporal identification of histologic disease in SAMP mice offers the exceptional opportunity to compare the immunological phenomena that take place before and after the appearance of histologically-detectable intestinal inflammation. Our studies so far have clearly demonstrated that SAMP ileitis consists of two immunologically-distinct phases (summarized in Figure 6).
Figure 6. Mixed Th1/Th2 cytokine phenotype characteristic of SAMP mice.
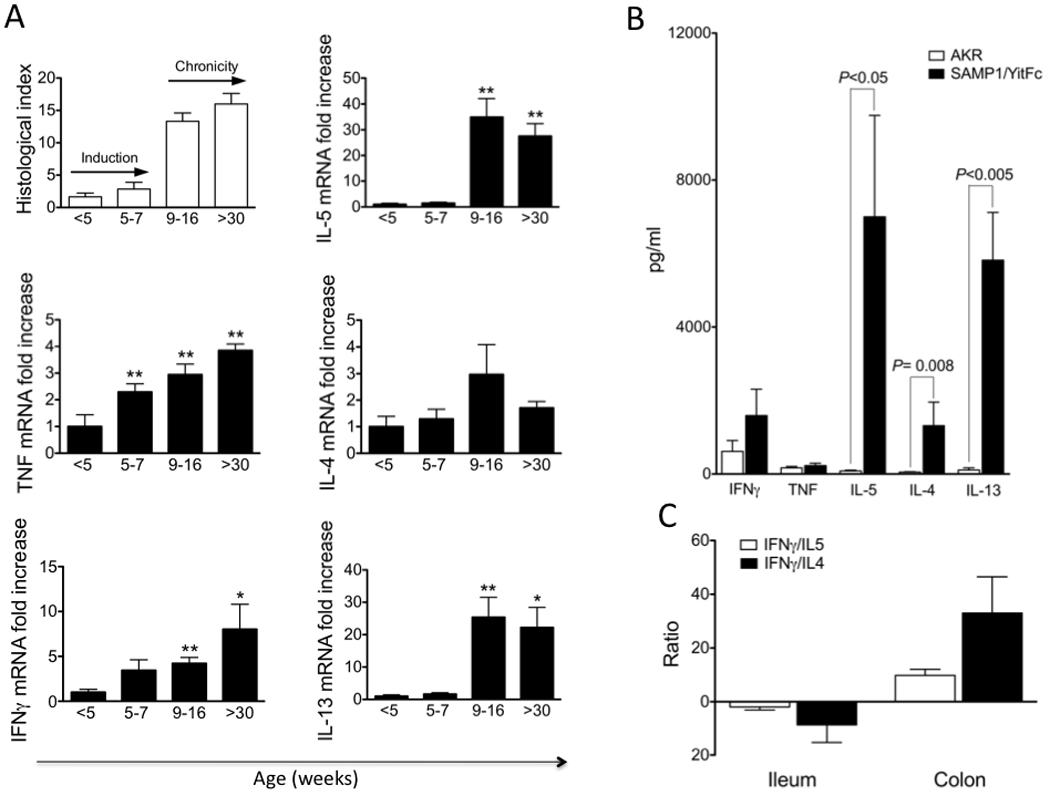
Cytokine profiles from gut-associated lymphoid tissues of SAMP mice change from a Th1 predominant (early/inductive phase) to a mixed Th1/Th2 (chronic phase) phenotype as ileitis severity progresses. (A) Total RNA was extracted from SAMP at different ages. Expression of cytokine mRNA levels were measure by real-time PCR and normalized to the 18S ribosomal RNA. All values are expressed relative to the mean value from the <5-wk group, and the y-axis indicates relative mRNA expression. Total inflammatory index for each time point is also indicated; *P<0.05 and **P<0.01. (B) Cytokine production from anti-CD3 stimulated LPL from SAMP vs. AKR ilea. (C) Comparative study of ratios of IFN-γ/IL-4 and IFN-γ/IL-5 secretion by ileal vs. cecal LPL from SAMP mice. Cytokine levels were measured by Cytometric Bead Array and IL-13 by ELISA. Data are expressed as mean±SEM. Data was originally published in Gastroenterol, 2005; 128:654–66.
Early immune responses in SAMP1/YitFc ileitis
The first stage in the natural history of SAMP ileitis occurs between 4 to 7-wk of age. Although minimal to no inflammatory lesions are detected in the terminal ileum histologically at this stage, very intense immunological responses take place, which culminate in the development of ileitis shortly thereafter. This period has therefore been referred to as the inductive phase. The major characteristic of this stage is a strong mucosal Th1 response. First, stimulated MLN cells from 4 week-old SAMP secrete high amounts of IFNγ. This initial IFNγ (and also TNF) production represents one of the earliest signs of mucosal dysregulation in SAMP mice, and precedes changes in both immune cell activation marker expression and the histologic onset of ileitis (20). Second, this original Th1 polarization is further amplified before the appearance of histologic inflammatory lesions, as it was clearly shown when cytokine mRNA expression was studied in the small intestinal mucosa (76). During the early inductive phase (4–7-wk of age), the relative expression of IFNγ and TNF increased by 3.5- and 2.3-fold, respectively, whereas no significant changes were detected for IL-4, IL-5 or IL-13 (Figure 6A). Third, early (from week 3) blocking of IFNγ with a monoclonal neutralizing antibody displayed a strong anti-inflammatory effect, significantly suppressing the severity of SAMP ileitis by week 10 (76). These studies provide convincing evidence that Th1-mediated pathways are pivotal for the initial induction of small intestinal inflammation in SAMP mice.
Subsequent work from our group indicated that the early, Th1-mediated immunological activation of the small intestinal mucosa in SAMP mice is, in fact, bacteria driven (Figure 7). In fact, continuous administration of broad-spectrum antibiotics in a preventive manner (from 3 weeks of age) significantly ameliorated the severity of ileitis by week 9 and completely prevented it in half of the treated mice (63). The histologic improvement was paralleled with a significant decrease in both IFNγ and TNF production by stimulated MLNs, whereas no effect was seen in IL-4. Interestingly, when SAMP mice were raised under GF conditions, they developed ileitis that, compared to SPF littermates, had delayed onset (later than 13 weeks of age), lower penetrance (around 70%), and decreased severity (1/3 of mice had severe ileitis, Figure 7A) (64). Taken together, these findings suggest that intraluminal commensal bacteria provide the antigens that exacerbate, and perhaps trigger, dysregulated activation of the mucosal immune system, eventually leading to ileitis in SAMP mice.
Figure 7. Impact of bacterial flora on the development of SAMP ileitis.
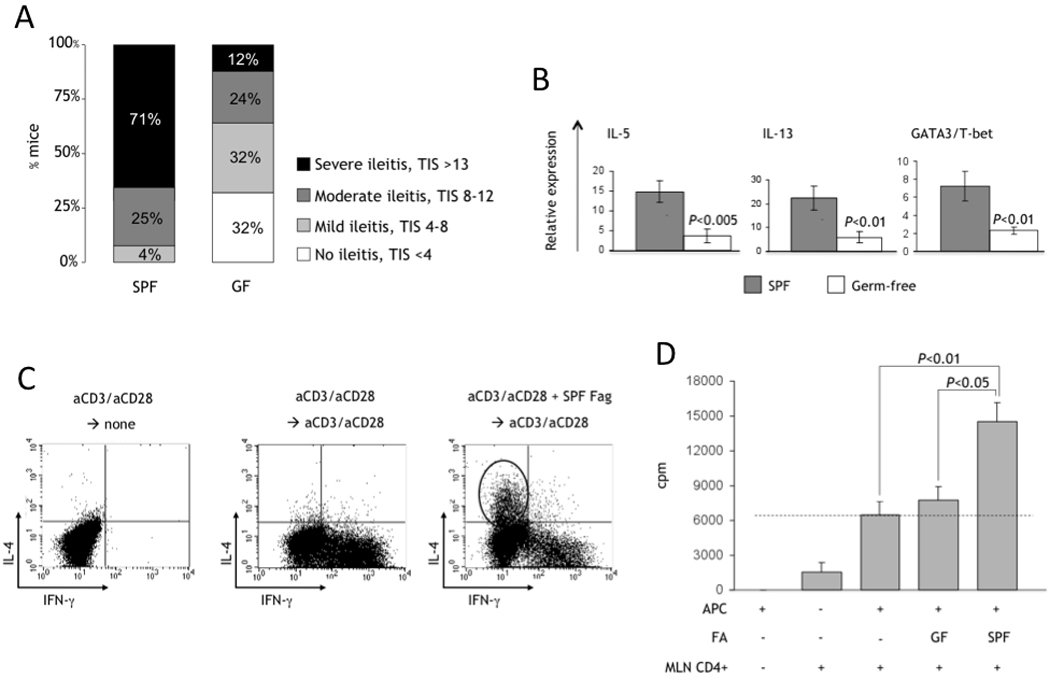
(A) Distribution of ileitis severity in GF vs. SPF SAMP mice (>13 wks of age); TIS, total inflammatory score. (B) Total RNA was extracted from the ileum of GF and SPF SAMP, and mRNA quantified by real-time PCR and normalized to 18S ribosomal RNA. GATA3:T-bet ratio represents the ratio of GATA3 and T-bet mRNA after normalization. Data are expressed as mean±SEM. (C) Activated (anti-CD3/anti-CD28) splenocytes from SPF SAMP cultured in the presence/absence of fecal antigens (Fag). IL-4 or IFNγ were produced by CD4+ lymphocytes, detected by intracellular staining. (D) Reactivity of mucosal lymphocytes from inflamed SPF SAMP mice to stimulation with fecal antigens (FA) prepared from GF or SPF mice. CD4+ MLN T cells represent the effector population. Syngeneic splenocytes were pulsed with FA overnight, irradiated and used as antigen-presenting cells (APC) as indicated. Proliferation of effector lymphocytes was estimated at 72h by measuring [3H]thymidine incorporation for the last 18h of culture. Data originally published in J Immunol, 2007; 178:1809–18.
Copyright 2007. The American Association of Immunologists, Inc.
Late immune responses in SAMP1/YitFc ileitis
The appearance of inflammatory lesions in the terminal ileum of SAMP mice at 9–10 weeks of age signifies a dramatic change in the mucosal cytokine milieu and identifies the second phase of SAMP ileitis. This period has therefore been referred to as the chronic inflammatory phase. During this stage, the already established Th1 response is coupled with a striking increase in Th2 effector pathways. Indeed, between week 9 and 16, the mucosal expression of IL-13 and IL-5 increases >25-fold when compared to the baseline expression in young mice, and is maintained thereafter (Fig. 6A) (76). Within the same period, there is also a very small increase in the mucosal expression of IFNγ and TNF. The Th2 predominance in the ileum of SAMP mice during established chronic inflammation is also evident at the level of the master polarizing transcription factors for the induction of Th1 or Th2 responses, that is T-bet and GATA3, respectively (80, 81). An elevated GATA3/T-bet ratio was detected in the terminal ileum of inflamed SAMP mice and was significantly higher than in either AKR ileum or in SAMP cecum. In accordance to the mRNA pattern, lamina propria lymphocytes (LPL) from SAMP mice were found to secrete greater quantities of IL-5 and IL-13 compared to their AKR counterparts, whereas the secretion of IFNγ and TNF did not differ substantially (Figure 6B) (76). In striking contrast, in cecal LPL cultures, IFNγ secretion far exceeded that of both IL-4 and IL-5 (Figure 6C). Therefore, the induction of Th2 responses does not appear to be an inherent predisposition of the SAMP strain, but more likely relates to the presence of small intestinal inflammation.
Mucosal Th2 responses in SAMP mice appear to be accelerated by bacterial flora (64). Indeed, in the absence of bacterial flora, the mucosal immune system is unable to mount a vigorous Th2 effector response, as indicated by a dramatic decrease in the mRNA expression for IL-5, IL-13 and in the ratio of GATA3:T-bet in GF SAMP mice (Figure 7B). This may explain the amelioration of disease severity in GF mice. To further test this hypothesis, immunoreactivity of SAMP lymphocytes against proteinic material collected from fecal antigen samples (FA) was investigated. Long-term cultures of spleen-derived cells were used in order to test the ability of FA to specifically expand effector lymphocytes. When non-specific stimulation (anti-CD3/CD28 or PMA) was used, the default pathway for lymphocytes was towards a Th1-type response, with the generation of large numbers of IFNγ-positive, but not of IL-4-positive, lymphocytes. In sharp contrast, when FA were added to the cultures, expansion of a distinct population of IL-4-positive lymphocytes was observed, in addition to the IFNγ-positive population, indicating a mixed Th1/Th2 response (Figure 7C). These data indicate that IL-4-secreting lymphocytes were specifically generated in response to FA stimulation. On the other hand, when mucosal MLN CD4+ lymphocytes from SAMP mice were used as the effector cells and stimulated with autologous FA, they proliferated robustly (Figure 7D), and secreted generous amounts of cytokines that included Th1- (IFNγ), Th2- (IL-4, IL-13) and regulatory- (IL-10) type cytokines. This result is indicative of the broad effects that the bacterial flora has on the development of effector mucosal immune responses, and emphasizes that additional factors, likely genetically determined, are required for the final shaping of the terminal pathway that predominates in the pathogenesis of IBD.
Although, bacterial-driven mechanisms participate in the pathogenesis of SAMP ileitis, it is important to keep in mind that inflammation develops in this model even in the absence of the bacteria microflora, as ileitis develops in a substantial percentage of GF mice (64). As such, the data presented thus far point to the co-existence of bacterial-dependent and bacterial-independent pathways during chronic intestinal inflammation. This is of great importance since the SAMP mouse is a model that closely resembles the human condition. The dissection of diverse components of the inflammatory response will, undoubtedly, offer unique diagnostic and therapeutic opportunities for CD.
Proinflammatory Function of Th2 Cytokines
The co-existence of both Th1 and Th2 pathways in SAMP ileitis represents somewhat of an “immunological paradox” since IL-4 is often referred to as anti-inflammatory cytokine, and Th1 and Th2 pathways are most often considered mutually exclusive. Nevertheless, during the chronic stage of SAMP ileitis, Th2 pathways contribute to and further amplify the inflammatory process. In support of this concept, administration of anti-IL-4 monoclonal antibodies to SAMP mice with established disease (>15-wk-old) resulted in significant amelioration of ileitis severity (76). Anti-IL-5 antibody administration also had the ability to significantly attenuate SAMP ileitis as IL-5 was shown to be markedly increased in Peyer’s patches, MLNs and inflamed mucosa of SAMP mice, in which numerous infiltrating eosinophils are present (82). Further evidence for a pathogenic role of Th2 cytokines in SAMP ileitis is also provided by studies wherein an enriched population of CD4+IL-4+ T cells from SAMP MLN were injected into naïve SCID recipients and tested for their ability to induce small intestinal inflammation. The results clearly showed that IL-4-enriched and IL-4-depleted populations were both effective in inducing ileitis of similar severity to recipient mice (76), and are in agreement with recent reports that both Th1 and Th2 bacterial antigen–specific lymphocytes from C3H/HeJBir colitic mice have the ability to transfer disease to SCID recipients (83).
How the proinflammatory effect of Th2 cytokines is achieved is not yet clear, but studies performed by our group provide some important insights into the potential interaction between Th1 and Th2 pathways. In particular, the addition of recombinant murine IL-4 to cultured SAMP MLN cells did not block, but further induced, the secretion of IFNγ (76). In addition, the in vivo blockade of IFNγ/Th1 or IL-4/Th2 pathways did not result in augmentation, but rather downregulation, of the opposing pathway. Finally, in the aforementioned adoptive transfer studies, increased mucosal IFNγ expression was observed in SCID recipients after the transfer of IL-4-producing lymphocytes, and similarly, upregulation of Th2 cytokines resulted after transfer of IL-4–depleted CD4+ cells. As such, it appears that, in the setting of chronic mucosal inflammation in SAMP mice, IFNγ and IL-4 can act synergistically. A similar effect has been reported in the CD4+CD45RBhi adoptive transfer model of colitis; although this is considered a typical Th1-mediated colitic model, the in vivo administration of IL-4 caused the exacerbation of colitis (84).
Novel Cytokine Pathways
Recently, additional immunologic effector pathways have been described. The IL-23/IL-17 axis that defines the Th17 effector, pro-inflammatory pathway is rapidly evolving as a central pathogenetic component in chronic intestinal inflammation (85). The precise involvement of this cascade in SAMP ileitis is not yet known, but is currently under intense research from our group. We recently reported a marked upregulation of IL-17 from activated MLN cells from SAMP versus AKR mice that have been stimulated with IL-33 (74), with preliminary findings indicating the source of differential IL-17 expression in SAMP are likely T regulatory cells as well as mucosal macrophages (unpublished results).
The TL1A/DR3/DcR3 system of the TNF/TNFR superfamily has also been reported to be upregulated during chronic clinical as well as experimental ileitis (86, 87). TL1A and its functional receptor, DR3, are elevated in the inflamed mucosa of mice with ileitis, including the SAMP strain (87). The role of these pathways in the development of SAMP ileitis has not yet been clarified, is also currently under investigation in our group.
The distinction between early and late immunologic pathways is of critical importance, primarily due to the vastly different therapeutic approaches it offers (88). Elucidation of pathways that precede the clinical phenotype may lead to preventive, and therefore more efficacious, intervention with the natural history of the disease. Alternatively, after disease is fully developed, therapeutic measures inevitably are directed against chronic, existing pathways and may be less efficient in ameliorating established disease. Similar diversity between early and late cytokine responses has been shown in experimental and clinical IBD. When long-term studies were performed in IL-10−/− colitic mice, a transition from early Th1 to late Th2 responses was found (89). Earlier studies in humans showed that different cytokines predominate in early as opposed to late CD (90). In a more recent study, it was found that at the onset of CD, mucosal T cells have a typical Th1 phenotype, but this is lost as disease progresses (91). Therefore, patients with early IBD may serve as better candidates for responding optimally to immunomodulatory therapies. This has, in fact, been shown in pediatric patients where immunomodulation results in longer periods of remission than those reported for adults (92). Our findings using the SAMP model clearly support early immunologic intervention for intestinal inflammation since it appears that the inductive phase is dominated by much more defined pathways, mainly a bacterial-driven Th1 response. This explains why treatments targeted at flora manipulation with broad-spectrum antibiotics or probiotics are extremely effective for disease prevention (63, 69). Such therapeutic intervention, however, does not have the same efficacy during the late phase of SAMP ileitis since at this advanced stage of inflammation, there exists an expanded and multifaceted dysregulation of mucosal homeostasis (93). The effector pathways show broad redundancy and cannot be effectively controlled with single-targeted therapies.
Role of B Cells
To date, the majority of studies investigating the immunological aspects of chronic gut inflammation, specifically the adaptive arm of mucosal immunity, have focused on the role of T lymphocytes. However, emerging evidence supports B lymphocytes as pivotal players in shifting the gut homeostatic balance towards an inflammatory state characteristic of IBD (94, 95). Accordingly, SAMP ileitis is not only driven by T cells, but B cells have been reported to promote chronic intestinal inflammation in this particular model of chronic intestinal inflammation. In fact, SAMP MLN have been shown to display a marked expansion of the B cell population, which was highly activated and produced elevated levels of IgG1κ, IgAκ, IgAλ and IgMλ (96). MLN B cell numbers correlated with the severity of ileitis in native SAMP mice, and co-transfer with CD4+ T cells augmented disease severity in naïve SCID recipients compared to CD4+ T cell transfer alone. The precise role of B cells in the development of SAMP ileitis, however, has not been fully elucidated. Nonetheless, several lines of evidence suggests that SAMP B cells have the ability to interrupt or override anti-inflammatory or regulatory signals, perhaps through a GITRL-dependent mechanism that involves abrogation of αE+CD4+ T cell regulatory function. Alternatively, SAMP B cells may also contribute to ileitis by producing immunoglobulins that recognize specific antigenic epitopes, which lead to the formation of immune complexes and that can ultimately result in increased adhesion, leukocyte homing, and infiltration of immune cells into the gut mucosa (96).
Leukocyte Trafficking and Adhesion Molecule Expression
Trafficking of lymphocytes during normal gut homeostasis and under normal physiological conditions has been relatively well-characterized; however, during chronic intestinal inflammation, such as that observed in IBD, less in known regarding specific mechanisms of lymphocyte recruitment into the gut. Of paramount importance is the differential, and perhaps preferential, trafficking to the small intestine versus the colon that has important implications in the characteristics of disease phenotypes. One of the distinguishing features of human CD is the massive infiltration of immune cells into the intestinal lamina propria compartment, which leads to overly aggressive local immune responses, intestinal inflammation, loss of function, and tissue damage (10). Histological assessment of intestinal tissues from patients with CD ileitis reveals patchy transmural inflammation, often involving granuloma, and a marked increased number of T and B lymphocytes, monocytes, neutrophils, and eosinophils present in the mucosa (10). Similar to the human disease, SAMP ileitis is also characterized by patchy transmural inflammation with a massive presence of immune cells within the intestinal lamina propria compartment (19). The continued recruitment of such a large population of immune cells to the intestinal mucosa suggests that a key mechanism underlying disease pathogenesis could involve dysregulation of leukocyte homing to the gut. In particular, three members of the immunoglobulin family have been implicated in leukocyte adhesion pathways during chronic ileitis and IBD pathogenesis: ICAM-1, VCAM-1 and MAdCAM-1 (97). The SAMP mouse has played an important role in efforts to dissect the relative contribution of redundant adhesion pathways that may be involved in promoting and maintaining CD-like ileitis.
SAMP ileitis is mediated by CD4+ T cells, as demonstrated by the SAMP adoptive transfer model in which donor CD4+ MLN cells from SAMP mice induced severe ileitis in recipient MHC-matched SCID mice (19). Recipient SCIDs displayed increased levels of several adhesion molecules within the small intestinal microvasculature, including ICAM-1, MAdCAM-1 and VCAM-1, consistent with the human disease (98, 99). Building on the discovery in the late 1990s that monoclonal antibody blockade of TNF can induce a therapeutic response in up to 70% of patients with CD, the SAMP adoptive transfer model has been used in pre-clinical investigations to explore the therapeutic effects of monoclonal antibody blockade and involvement of specific endothelial adhesion molecules that are expressed during small intestinal inflammation, namely ICAM-1 and VCAM-1, as well as their ligand, the α4 integrin, expressed on circulating leukocytes. Although pre-treatment of recipient SCID mice with individual monoclonal antibodies targeting ICAM-1, V-CAM-1, or α4 integrin had no effect on ileitis, adoptive transfer mice treated with combination antibody therapy (anti-ICAM-1 + anti-α4 integrin or anti-ICAM-1 + anti-VCAM-1) experienced a 70% reduction in intestinal inflammation compared to untreated mice (99) (Figure 8). This therapeutic effect was not observed in 40 week-old SAMP mice with established disease, highlighting the redundant nature of adhesion pathways in the gut under conditions of chronic inflammation, and providing important insight for the clinical development of an anti-α4 monoclonal antibody therapy in human CD, as well as the potential need to target multiple adhesion pathways in order to achieve optimal clinical benefits.
Figure 8. Summary of response to therapies in the SAMP model of CD-like ileitis.

Efficacy of different therapies to treat SAMP ileitis before the onset of chronic intestinal inflammation (pre-treatment), during established disease (treatment), or using the adoptive transfer model utilizing SAMP MLN cells transferred into naïve SCID recipients. Bars represent total inflammatory scores of various treatment modalities expressed as the percentage of placebo-treated SAMP (set as 100%) (i.e., ileal inflammation in SAMP mice was decreased by 40% following a pretreatment strategy of metronidazole + ciprofloxacine). NS, not significant.
Interestingly, blockade of the physiologically-dominant α4β7/MAdCAM-1 adhesion pathway, with antibodies blocking β7 subunit or α4β7 did not attenuate ileitis in the SAMP adoptive transfer model, despite the marked increase of both β7 on CD4+ cells and MAdCAM-1 in lamina propria endothelial cells (98). However, when the β7 KO mutation was backcrossed onto the SAMP background (SAMP X β7 KO strain), ileitis was reduced by 30–50% compared to native SAMP mice (100). In fact, SAMP X β7 KO MLN showed a dramatic decrease in size, which was due to a significant drop in lymphocyte numbers and a reduction in short-term B cell homing. As mentioned earlier, co-transfer of SAMP B cells with CD4+ SAMP T cells exacerbated the severity of ileitis in MHC-matched SCID recipients compared to transfer of CD4+ SAMP T cells alone (96); however, when B cells from SAMP X β7 KO mice were co-transferred, ileitis severity in recipient mice was unchanged compared to the group receiving the CD4+ SAMP T cells alone (100). Taken together, these data suggest that β7 integrins play an important role in SAMP ileitis, perhaps by promoting the homing of pathogenic B cells into the gut mucosa and further exacerbating chronic ileal inflammation.
MAdCAM-1-independent pathways have also been reported to play a critical role for leukocyte recruitment in CD-like ileitis. Gut-homing CD4+ T cells from SAMP mice co-express the α4β7 and α4β1 integrins, as well as L-selectin (98). Although blockade of the MAdCAM-1 pathway alone did not attenuate SAMP ileitis, combination blockade of both MAdCAM-1 and L-selectin together caused amelioration of both acute and chronic inflammatory infiltrates, as well as partial restoration of villous and crypt architecture (98). These results suggest that L-selectin, in conjunction with α4β7 and α4β1, may function as a marker for small intestinal-specific leukocyte homing to the gut. Additional studies in the SAMP adoptive transfer model have identified PSGL-1 as the endothelial ligand for L-selectin during conditions of chronic ileitis (101). Blockade of PSGL-1 not only attenuated acute and chronic ileitis in SCID recipients 6 weeks after adoptive transfer of SAMP CD4+ MLN cells, but interestingly, unlike other single or combination anti-adhesion therapies, PSGL-1 blockade also reduced active and chronic inflammation as well as villous distortion in 10 week-old SAMP mice (101, 102). Unlike in other models of colitis, no other single or combination antibody therapy has been proven efficacious for the treatment of chronic CD-like ileitis in SAMP mice. PSGL-1 also binds to P-selectin and E-selectin; however, simultaneous blockade of all three selectins did not replicate the therapeutic effects of anti-PSGL-1 treatment in SAMP mice, discounting the possibility that the effects are due to a generalized selectin antagonism.
The SAMP mouse also represents an ideal model for studying site-specific leukocyte homing to the small intestine versus colon during conditions of chronic inflammation, since few mouse models develop spontaneous ileitis. Under homeostatic conditions, the small intestinal-specific chemokine receptor, CCR9, is expressed on the cell surface of memory/effector CD4+ T cells and selectively binds to the small intestinal lymphocyte chemoattractant CCL25 (or TECK) (103). Blockade of the CCR9/CCL25 interaction inhibits lymphocyte infiltration into the small intestinal mucosa and thereby limits inflammation and disease at this site. A large-scale phase III clinical trial is currently underway to evaluate the efficacy of CCR9 antagonism for the treatment of CD, however pre-clinical data from animal models of intestinal inflammation is limited. Recent studies utilized the SAMP mouse to study the site-specific effects of the CCR9/CCL25 system on lymphocyte recruitment to areas of small intestinal inflammation and how these effects change as a function of disease progression. SAMP mice displayed a higher fraction of CCR9+ lymphocytes, along with a unique subset of CCR9high lymphocytes (mostly CD8+), during the early stages of ileitis (104). However, CCR9 expression subsequently decreased with disease progression. As in humans, CCL25 expression in SAMP mice was found to be limited to the small intestine. Blockade of either CCR9 or CCL25 significantly decreased ileitis during the early stages of disease, but had no therapeutic effect during established disease when CCR9 expression had abated and the CCR9high lymphocyte population was no longer detectable.
Other chemokine receptors that are expressed in SAMP mice include the Th1 receptors CCR2 and CCR5, as well as the Th2 receptors CCR3, CCR4, and CCR8 (82). In particular, CCR5 showed the highest increase in expression in the inflamed ilea of SAMP mice (105). CCR5 is expressed on CD8+ and CD4+ cells and chemotactically recruits Foxp3+ T regulatory cells to areas of active inflammation within the SAMP ileum. Blockade of CCR5 resulted in significant exacerbation of ileitis in both native SAMP mice as well as the SAMP adoptive transfer model, most likely due to restricted homing of T regulatory cells to areas of inflammation.
We have also used the SAMP model as a tool for evaluating new technologies for noninvasive imaging of intestinal fibrosis and inflammation in patients with CD. The current standard for imaging studies of the intestine includes colonoscopy, upper endoscopy, and small bowel follow-through with barium enema. Each of these techniques are relatively safe and efficacious for IBD patients, but less invasive strategies could greatly improve safety and quality of life. Our group has developed an image-enhancing ultrasound contrast agent consisting of encapsulated gaseous microbubbles conjugated with anti-MAdCAM-1 antibodies that to bind MAdCAM-1, a gut-restricted marker of intestinal inflammation (106). When tested in SAMP mice, these MAdCAM-1-specific microbubbles selectively accumulated in focal areas of ileal inflammation and their measured video intensity of ultrasonic acoustic echos correlated strongly with total ileal inflammatory scores (R=0.92). This unique study highlights the utility of a well-characterized spontaneous model of intestinal inflammation beyond the traditional scope of mechanistic and preclinical experimental studies.
Finally, one of the most exciting aspects of the SAMP model is the predictive value in testing the efficacy of novel treatment modalities for chronic intestinal inflammation and IBD. In fact, SAMP mice appear to respond to conventional anti-inflammatory and immunosuppressive treatments, such as steroids and anti-TNF (42, 58, 99, 107), which are the most common therapies currently used by gastroenterologists to treat patients with IBD. A variety of experimental treatments have also been tested by our group and others using the SAMP model, with both negative and positive results (summarized in Figure 8 and in Table S1). It would be interesting in future years to compare the results of phase II clinical trials using these experimental therapies with results initially obtained in preclinical studies using SAMP mice. This indeed would allow the determination of the true predictive value of the SAMP model in preclinical testing of novel compounds for their efficacy in treating patients with IBD, specifically CD.
Supplementary Material
Acknowledgements
The authors would like to acknowledge Dr. Satoshi Matsumoto and the Yakult Central Institute of Microbiological Research for providing the original breeding pairs of SAMP1/Yit mice and initial collaborations with our group. We also acknowledge members of our individual research teams and collaborators, as well as past and continued support from the National Institutes of Health: DK057880 (TTP, KL, & FC), DK056762 (TTP), DK042191 (FC & TTP), DK066880 (FC), DK055812 (FC), and the Crohn’s & Colitis Foundation of America: Senior Investigator Award (TTP), Career Development Award (BKR), and Research Fellowship Awards (GB, KOA & RRG).
References
- 1.Peyrin-Biroulet L, Loftus EV, Jr, Colombel JF, et al. The natural history of adult Crohn's disease in population-based cohorts. Am J Gastroenterol. 2010;105:289–297. doi: 10.1038/ajg.2009.579. [DOI] [PubMed] [Google Scholar]
- 2.Hugot JP, Chamaillard M, Zouali H, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 3.Ogura Y, Bonen DK, Inohara N, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. 2001;411:603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 4.Peltekova VD, Wintle RF, Rubin LA, et al. Functional variants of OCTN cation transporter genes are associated with Crohn disease. Nat Genet. 2004;36:471–475. doi: 10.1038/ng1339. [DOI] [PubMed] [Google Scholar]
- 5.Stoll M, Corneliussen B, Costello CM, et al. Genetic variation in DLG5 is associated with inflammatory bowel disease. Nat Genet. 2004;36:476–480. doi: 10.1038/ng1345. [DOI] [PubMed] [Google Scholar]
- 6.Duerr RH, Taylor KD, Brant SR, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hampe J, Franke A, Rosenstiel P, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–211. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 8.Rioux JD, Xavier RJ, Taylor KD, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaser A, Lee AH, Franke A, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008;134:743–756. doi: 10.1016/j.cell.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med. 2009;361:2066–2078. doi: 10.1056/NEJMra0804647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuhn R, Lohler J, Rennick D, et al. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 12.Mombaerts P, Mizoguchi E, Grusby MJ, et al. Spontaneous development of inflammatory bowel disease in T cell receptor mutant mice. Cell. 1993;75:274–282. doi: 10.1016/0092-8674(93)80069-q. [DOI] [PubMed] [Google Scholar]
- 13.Panwala CM, Jones JC, Viney JL. A novel model of inflammatory bowel disease: mice deficient for the multiple drug resistance gene, mdr1a, spontaneously develop colitis. J Immunol. 1998;161:5733–5744. [PubMed] [Google Scholar]
- 14.Kontoyiannis D, Pasparakis M, Pizarro TT, et al. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity. 1999;10:387–398. doi: 10.1016/s1074-7613(00)80038-2. [DOI] [PubMed] [Google Scholar]
- 15.Nenci A, Becker C, Wullaert A, et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature. 2007;446:557–561. doi: 10.1038/nature05698. [DOI] [PubMed] [Google Scholar]
- 16.Higuchi K. Genetic characterization of senescence-accelerated mouse (SAM) Exp Gerontol. 1997;32:129–138. doi: 10.1016/s0531-5565(96)00060-5. [DOI] [PubMed] [Google Scholar]
- 17.Takeda T, Hosokawa M, Takeshita S, et al. A new murine model of accelerated senescence. Mech Ageing Dev. 1981;17:183–194. doi: 10.1016/0047-6374(81)90084-1. [DOI] [PubMed] [Google Scholar]
- 18.Matsumoto S, Okabe Y, Setoyama H, et al. Inflammatory bowel disease-like enteritis and caecitis in a senescence accelerated mouse P1/Yit strain. Gut. 1998;43:71–78. doi: 10.1136/gut.43.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kosiewicz MM, Nast CC, Krishnan A, et al. Th1-type responses mediate spontaneous ileitis in a novel murine model of Crohn's disease. J Clin Invest. 2001;107:695–702. doi: 10.1172/JCI10956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rivera-Nieves J, Bamias G, Vidrich A, et al. Emergence of perianal fistulizing disease in the SAMP1/YitFc mouse, a spontaneous model of chronic ileitis. Gastroenterology. 2003;124:972–982. doi: 10.1053/gast.2003.50148. [DOI] [PubMed] [Google Scholar]
- 21.Vidrich A, Buzan JM, Barnes S, et al. Altered epithelial cell lineage allocation and global expansion of the crypt epithelial stem cell population are associated with ileitis in SAMP1/YitFc mice. Am J Pathol. 2005;166:1055–1067. doi: 10.1016/S0002-9440(10)62326-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reuter BK, Pizarro TT. Mechanisms of tight junction dysregulation in the SAMP1/YitFc model of Crohn's disease-like ileitis. Ann N Y Acad Sci. 2009;1165:301–307. doi: 10.1111/j.1749-6632.2009.04035.x. [DOI] [PubMed] [Google Scholar]
- 23.Reuter BK, Scott KG, Meddings JB, et al. The SAMP1/YitFc mouse displays spontaneous gastric inflammation and represents a viable model to study Crohn's gastritis. Gastroenterology. 2005;128:A209. [Google Scholar]
- 24.Brogi M, Cominelli F, Sachedina MA, et al. Autoimmune hepatitis precedes the onset of terminal ileitis in the SAMP1/YitFc mouse model of Crohn's disease. Hepatology. 2009;50:1009A. [Google Scholar]
- 25.Pizarro TT, Arseneau KO, Bamias G, et al. Mouse models for the study of Crohn's disease. Trends Mol Med. 2003;9:218–222. doi: 10.1016/s1471-4914(03)00052-2. [DOI] [PubMed] [Google Scholar]
- 26.Dietrich W, Katz H, Lincoln SE, et al. A genetic map of the mouse suitable for typing intraspecific crosses. Genetics. 1992;131:423–447. doi: 10.1093/genetics/131.2.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kozaiwa K, Sugawara K, Smith MF, Jr, et al. Identification of a quantitative trait locus for ileitis in a spontaneous mouse model of Crohn's disease: SAMP1/YitFc. Gastroenterology. 2003;125:477–490. doi: 10.1016/s0016-5085(03)00876-x. [DOI] [PubMed] [Google Scholar]
- 28.Mahler M, Bristol IJ, Sundberg JP, et al. Genetic analysis of susceptibility to dextran sulfate sodium-induced colitis in mice. Genomics. 1999;55:147–156. doi: 10.1006/geno.1998.5636. [DOI] [PubMed] [Google Scholar]
- 29.Bouma G, Kaushiva A, Strober W. Experimental murine colitis is regulated by two genetic loci, including one on chromosome 11 that regulates IL-12 responses. Gastroenterology. 2002;123:554–565. doi: 10.1053/gast.2002.34752. [DOI] [PubMed] [Google Scholar]
- 30.Takeda K, Clausen BE, Kaisho T, et al. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 1999;10:39–49. doi: 10.1016/s1074-7613(00)80005-9. [DOI] [PubMed] [Google Scholar]
- 31.Glocker EO, Kotlarz D, Boztug K, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. 2009;361:2033–2045. doi: 10.1056/NEJMoa0907206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ten Hove T, Corbaz A, Amitai H, et al. Blockade of endogenous IL-18 ameliorates TNBS-induced colitis by decreasing local TNF-alpha production in mice. Gastroenterology. 2001;121:1372–1379. doi: 10.1053/gast.2001.29579. [DOI] [PubMed] [Google Scholar]
- 33.Pizarro TT, Michie MH, Bentz M, et al. IL-18, a novel immunoregulatory cytokine, is up-regulated in Crohn's disease: expression and localization in intestinal mucosal cells. J Immunol. 1999;162:6829–6835. [PubMed] [Google Scholar]
- 34.Tamura K, Fukuda Y, Sashio H, et al. IL18 polymorphism is associated with an increased risk of Crohn's disease. J Gastroenterol. 2002;37 Suppl 14:111–116. doi: 10.1007/BF03326428. [DOI] [PubMed] [Google Scholar]
- 35.Haas SL, Andreas Koch W, Schreiber S, et al. −137 (G/C) IL-18 promoter polymorphism in patients with inflammatory bowel disease. Scand J Gastroenterol. 2005;40:1438–1443. doi: 10.1080/00365520510023738. [DOI] [PubMed] [Google Scholar]
- 36.Takagawa T, Tamura K, Takeda N, et al. Association between IL-18 gene promoter polymorphisms and inflammatory bowel disease in a Japanese population. Inflamm Bowel Dis. 2005;11:1038–1043. doi: 10.1097/01.mib.0000182868.67025.b9. [DOI] [PubMed] [Google Scholar]
- 37.Duerr RH, Barmada MM, Zhang L, et al. Evidence for an inflammatory bowel disease locus on chromosome 3p26: linkage, transmission/disequilibrium and partitioning of linkage. Hum Mol Genet. 2002;11:2599–2606. doi: 10.1093/hmg/11.21.2599. [DOI] [PubMed] [Google Scholar]
- 38.Cho JH, Nicolae DL, Gold LH, et al. Identification of novel susceptibility loci for inflammatory bowel disease on chromosomes 1p, 3q, and 4q: evidence for epistasis between 1p and IBD1. Proc Natl Acad Sci U S A. 1998;95:7502–7507. doi: 10.1073/pnas.95.13.7502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hampe J, Lynch NJ, Daniels S, et al. Fine mapping of the chromosome 3p susceptibility locus in inflammatory bowel disease. Gut. 2001;48:191–197. doi: 10.1136/gut.48.2.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paavola P, Helio T, Kiuru M, et al. Genetic analysis in Finnish families with inflammatory bowel disease supports linkage to chromosome 3p21. Eur J Hum Genet. 2001;9:328–334. doi: 10.1038/sj.ejhg.5200626. [DOI] [PubMed] [Google Scholar]
- 41.Satsangi J, Parkes M, Louis E, et al. Two stage genome-wide search in inflammatory bowel disease provides evidence for susceptibility loci on chromosomes 3, 7 and 12. Nat Genet. 1996;14:199–202. doi: 10.1038/ng1096-199. [DOI] [PubMed] [Google Scholar]
- 42.Sugawara K, Olson TS, Moskaluk CA, et al. Linkage to peroxisome proliferator-activated receptor-gamma in SAMP1/YitFc mice and in human Crohn's disease. Gastroenterology. 2005;128:351–360. doi: 10.1053/j.gastro.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 43.Lefebvre M, Paulweber B, Fajas L, et al. Peroxisome proliferator-activated receptor gamma is induced during differentiation of colon epithelium cells. J Endocrinol. 1999;162:331–340. doi: 10.1677/joe.0.1620331. [DOI] [PubMed] [Google Scholar]
- 44.Spiegelman BM. PPARgamma in monocytes: less pain, any gain? Cell. 1998;93:153–155. doi: 10.1016/s0092-8674(00)81567-6. [DOI] [PubMed] [Google Scholar]
- 45.Tontonoz P, Nagy L, Alvarez JG, et al. PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell. 1998;93:241–252. doi: 10.1016/s0092-8674(00)81575-5. [DOI] [PubMed] [Google Scholar]
- 46.Fajas L, Auboeuf D, Raspe E, et al. The organization, promoter analysis, and expression of the human PPARgamma gene. J Biol Chem. 1997;272:18779–18789. doi: 10.1074/jbc.272.30.18779. [DOI] [PubMed] [Google Scholar]
- 47.Rousseaux C, Lefebvre B, Dubuquoy L, et al. Intestinal antiinflammatory effect of 5-aminosalicylic acid is dependent on peroxisome proliferator-activated receptor-gamma. J Exp Med. 2005;201:1205–1215. doi: 10.1084/jem.20041948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Daynes RA, Jones DC. Emerging roles of PPARs in inflammation and immunity. Nat Rev Immunol. 2002;2:748–759. doi: 10.1038/nri912. [DOI] [PubMed] [Google Scholar]
- 49.Zhu Y, Qi C, Korenberg JR, et al. Structural organization of mouse peroxisome proliferator-activated receptor gamma (mPPAR gamma) gene: alternative promoter use and different splicing yield two mPPAR gamma isoforms. Proc Natl Acad Sci U S A. 1995;92:7921–7925. doi: 10.1073/pnas.92.17.7921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dubuquoy L, Jansson EA, Deeb S, et al. Impaired expression of peroxisome proliferator-activated receptor gamma in ulcerative colitis. Gastroenterology. 2003;124:1265–1276. doi: 10.1016/s0016-5085(03)00271-3. [DOI] [PubMed] [Google Scholar]
- 51.Dubuquoy L, Dharancy S, Nutten S, et al. Role of peroxisome proliferator-activated receptor gamma and retinoid X receptor heterodimer in hepatogastroenterological diseases. Lancet. 2002;360:1410–1418. doi: 10.1016/S0140-6736(02)11395-X. [DOI] [PubMed] [Google Scholar]
- 52.Fish EN. The X-files in immunity: sex-based differences predispose immune responses. Nat Rev Immunol. 2008;8:737–744. doi: 10.1038/nri2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saruta M, Targan SR, Mei L, et al. High-frequency haplotypes in the X chromosome locus TLR8 are associated with both CD and UC in females. Inflamm Bowel Dis. 2009;15:321–327. doi: 10.1002/ibd.20754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Olson TS, Reuter BK, Scott KG, et al. The primary defect in experimental ileitis originates from a nonhematopoietic source. J Exp Med. 2006;203:541–552. doi: 10.1084/jem.20050407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.D'Inca R, Di Leo V, Corrao G, et al. Intestinal permeability test as a predictor of clinical course in Crohn's disease. Am J Gastroenterol. 1999;94:2956–2960. doi: 10.1111/j.1572-0241.1999.01444.x. [DOI] [PubMed] [Google Scholar]
- 56.Kucharzik T, Walsh SV, Chen J, et al. Neutrophil transmigration in inflammatory bowel disease is associated with differential expression of epithelial intercellular junction proteins. Am J Pathol. 2001;159:2001–2009. doi: 10.1016/S0002-9440(10)63051-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hollander D, Vadheim CM, Brettholz E, et al. Increased intestinal permeability in patients with Crohn's disease and their relatives. A possible etiologic factor. Ann Intern Med. 1986;105:883–885. doi: 10.7326/0003-4819-105-6-883. [DOI] [PubMed] [Google Scholar]
- 58.Marini M, Bamias G, Rivera-Nieves J, et al. TNF-alpha neutralization ameliorates the severity of murine Crohn's-like ileitis by abrogation of intestinal epithelial cell apoptosis. Proc Natl Acad Sci U S A. 2003;100:8366–8371. doi: 10.1073/pnas.1432897100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Furuse M, Furuse K, Sasaki H, et al. Conversion of zonulae occludentes from tight to leaky strand type by introducing claudin-2 into Madin-Darby canine kidney I cells. J Cell Biol. 2001;153:263–272. doi: 10.1083/jcb.153.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McCarthy KM, Skare IB, Stankewich MC, et al. Occludin is a functional component of the tight junction. J Cell Sci. 1996;109(Pt 9):2287–2298. doi: 10.1242/jcs.109.9.2287. [DOI] [PubMed] [Google Scholar]
- 61.Zeissig S, Burgel N, Gunzel D, et al. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn's disease. Gut. 2007;56:61–72. doi: 10.1136/gut.2006.094375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Heller F, Florian P, Bojarski C, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology. 2005;129:550–564. doi: 10.1016/j.gastro.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 63.Bamias G, Marini M, Moskaluk CA, et al. Down-regulation of intestinal lymphocyte activation and Th1 cytokine production by antibiotic therapy in a murine model of Crohn's disease. J Immunol. 2002;169:5308–5314. doi: 10.4049/jimmunol.169.9.5308. [DOI] [PubMed] [Google Scholar]
- 64.Bamias G, Okazawa A, Rivera-Nieves J, et al. Commensal bacteria exacerbate intestinal inflammation but are not essential for the development of murine ileitis. J Immunol. 2007;178:1809–1818. doi: 10.4049/jimmunol.178.3.1809. [DOI] [PubMed] [Google Scholar]
- 65.Matsumoto S, Watanabe N, Imaoka A, et al. Preventive effects of Bifidobacterium-and Lactobacillus-fermented milk on the development of inflammatory bowel disease in senescence-accelerated mouse P1/Yit strain mice. Digestion. 2001;64:92–99. doi: 10.1159/000048846. [DOI] [PubMed] [Google Scholar]
- 66.Matsumoto S, Hara T, Hori T, et al. Probiotic Lactobacillus-induced improvement in murine chronic inflammatory bowel disease is associated with the down-regulation of pro-inflammatory cytokines in lamina propria mononuclear cells. Clin Exp Immunol. 2005;140:417–426. doi: 10.1111/j.1365-2249.2005.02790.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bibiloni R, Fedorak RN, Tannock GW, et al. VSL#3 probiotic-mixture induces remission in patients with active ulcerative colitis. Am J Gastroenterol. 2005;100:1539–1546. doi: 10.1111/j.1572-0241.2005.41794.x. [DOI] [PubMed] [Google Scholar]
- 68.Gionchetti P, Rizzello F, Venturi A, et al. Oral bacteriotherapy as maintenance treatment in patients with chronic pouchitis: a double-blind, placebo-controlled trial. Gastroenterology. 2000;119:305–309. doi: 10.1053/gast.2000.9370. [DOI] [PubMed] [Google Scholar]
- 69.Pagnini C, Saeed R, Bamias G, et al. Probiotics promote gut health through stimulation of epithelial innate immunity. Proc Natl Acad Sci U S A. 2010;107:454–459. doi: 10.1073/pnas.0910307107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Barnes SL, Vidrich A, Wang ML, et al. Resistin-like molecule beta (RELMbeta/FIZZ2) is highly expressed in the ileum of SAMP1/YitFc mice and is associated with initiation of ileitis. J Immunol. 2007;179:7012–7020. doi: 10.4049/jimmunol.179.10.7012. [DOI] [PubMed] [Google Scholar]
- 71.McVay LD, Keilbaugh SA, Wong TM, et al. Absence of bacterially induced RELMbeta reduces injury in the dextran sodium sulfate model of colitis. J Clin Invest. 2006;116:2914–2923. doi: 10.1172/JCI28121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shanahan MT, Vidrich A, Shirafuji Y, et al. Elevated expression of Paneth cell CRS4C in ileitis-prone SAMP1/YitFc mice: regional distribution, subcellular localization, and mechanism of action. J Biol Chem. 2010;285:7493–7504. doi: 10.1074/jbc.M109.083220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mitsuyama K, Matsumoto S, Rose-John S, et al. STAT3 activation via interleukin 6 trans-signalling contributes to ileitis in SAMP1/Yit mice. Gut. 2006;55:1263–1269. doi: 10.1136/gut.2005.079343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pastorelli L, Garg RR, Hoang SB, et al. Epithelial-derived IL-33 and its receptor ST2 are dysregulated in ulcerative colitis and in experimental Th1/Th2 driven enteritis. Proc Natl Acad Sci U S A. 2010;107:8017–8022. doi: 10.1073/pnas.0912678107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schmitz J, Owyang A, Oldham E, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–490. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 76.Bamias G, Martin C, Mishina M, et al. Proinflammatory effects of TH2 cytokines in a murine model of chronic small intestinal inflammation. Gastroenterology. 2005;128:654–666. doi: 10.1053/j.gastro.2004.11.053. [DOI] [PubMed] [Google Scholar]
- 77.Beltran CJ, Nunez LE, Diaz-Jimenez D, et al. Characterization of the novel ST2/IL-33 system in patients with inflammatory bowel disease. Inflamm Bowel Dis. 2010;16:1097–1107. doi: 10.1002/ibd.21175. [DOI] [PubMed] [Google Scholar]
- 78.Kobori A, Yagi Y, Imaeda H, et al. Interleukin-33 expression is specifically enhanced in inflamed mucosa of ulcerative colitis. J Gastroenterol. 2010;45:999–1007. doi: 10.1007/s00535-010-0245-1. [DOI] [PubMed] [Google Scholar]
- 79.Seidelin JB, Bjerrum JT, Coskun M, et al. IL-33 is upregulated in colonocytes of ulcerative colitis. Immunol Lett. 2010;128:80–85. doi: 10.1016/j.imlet.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 80.Afkarian M, Sedy JR, Yang J, et al. T-bet is a STAT1-induced regulator of IL-12R expression in naive CD4+ T cells. Nat Immunol. 2002;3:549–557. doi: 10.1038/ni794. [DOI] [PubMed] [Google Scholar]
- 81.Ouyang W, Lohning M, Gao Z, et al. Stat6-independent GATA-3 autoactivation directs IL-4-independent Th2 development and commitment. Immunity. 2000;12:27–37. doi: 10.1016/s1074-7613(00)80156-9. [DOI] [PubMed] [Google Scholar]
- 82.Takedatsu H, Mitsuyama K, Matsumoto S, et al. Interleukin-5 participates in the pathogenesis of ileitis in SAMP1/Yit mice. Eur J Immunol. 2004;34:1561–1569. doi: 10.1002/eji.200324680. [DOI] [PubMed] [Google Scholar]
- 83.Iqbal N, Oliver JR, Wagner FH, et al. T helper 1 and T helper 2 cells are pathogenic in an antigen-specific model of colitis. J Exp Med. 2002;195:71–84. doi: 10.1084/jem.2001889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fort M, Lesley R, Davidson N, et al. IL-4 exacerbates disease in a Th1 cell transfer model of colitis. J Immunol. 2001;166:2793–2800. doi: 10.4049/jimmunol.166.4.2793. [DOI] [PubMed] [Google Scholar]
- 85.Bamias G, Cominelli F. Immunopathogenesis of inflammatory bowel disease: current concepts. Curr Opin Gastroenterol. 2007;23:365–369. doi: 10.1097/MOG.0b013e3281c55eb2. [DOI] [PubMed] [Google Scholar]
- 86.Bamias G, Martin C, 3rd, Marini M, et al. Expression, localization, and functional activity of TL1A, a novel Th1-polarizing cytokine in inflammatory bowel disease. J Immunol. 2003;171:4868–4874. doi: 10.4049/jimmunol.171.9.4868. [DOI] [PubMed] [Google Scholar]
- 87.Bamias G, Mishina M, Nyce M, et al. Role of TL1A and its receptor DR3 in two models of chronic murine ileitis. Proc Natl Acad Sci U S A. 2006;103:8441–8446. doi: 10.1073/pnas.0510903103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cominelli F. Early and late gut immune responses in IBD. J Pediatr Gastroenterol Nutr. 2008;46 Suppl 1:E20. doi: 10.1097/01.mpg.0000313833.47207.47. [DOI] [PubMed] [Google Scholar]
- 89.Etling MR, Davies S, Campbell M, et al. Maturation of the mucosal immune system underlies colitis susceptibility in interleukin-10-deficient (IL-10−/−) mice. J Leukoc Biol. 2007;82:311–319. doi: 10.1189/jlb.0606396. [DOI] [PubMed] [Google Scholar]
- 90.Desreumaux P, Brandt E, Gambiez L, et al. Distinct cytokine patterns in early and chronic ileal lesions of Crohn's disease. Gastroenterology. 1997;113:118–126. doi: 10.1016/s0016-5085(97)70116-1. [DOI] [PubMed] [Google Scholar]
- 91.Kugathasan S, Saubermann LJ, Smith L, et al. Mucosal T-cell immunoregulation varies in early and late inflammatory bowel disease. Gut. 2007;56:1696–1705. doi: 10.1136/gut.2006.116467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kugathasan S, Levy MB, Saeian K, et al. Infliximab retreatment in adults and children with Crohn's disease: risk factors for the development of delayed severe systemic reaction. Am J Gastroenterol. 2002;97:1408–1414. doi: 10.1111/j.1572-0241.2002.05784.x. [DOI] [PubMed] [Google Scholar]
- 93.Bamias G, Nyce MR, De La Rue SA, et al. New concepts in the pathophysiology of inflammatory bowel disease. Ann Intern Med. 2005;143:895–904. doi: 10.7326/0003-4819-143-12-200512200-00007. [DOI] [PubMed] [Google Scholar]
- 94.Noronha AM, Liang Y, Hetzel JT, et al. Hyperactivated B cells in human inflammatory bowel disease. J Leukoc Biol. 2009;86:1007–1016. doi: 10.1189/jlb.0309203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Brandtzaeg P, Carlsen HS, Halstensen TS. The B-cell system in inflammatory bowel disease. Adv Exp Med Biol. 2006;579:149–167. doi: 10.1007/0-387-33778-4_10. [DOI] [PubMed] [Google Scholar]
- 96.Olson TS, Bamias G, Naganuma M, et al. Expanded B cell population blocks regulatory T cells and exacerbates ileitis in a murine model of Crohn disease. J Clin Invest. 2004;114:389–398. doi: 10.1172/JCI20855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rivera-Nieves J, Gorfu G, Ley K. Leukocyte adhesion molecules in animal models of inflammatory bowel disease. Inflamm Bowel Dis. 2008;14:1715–1735. doi: 10.1002/ibd.20501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rivera-Nieves J, Olson T, Bamias G, et al. L-selectin, alpha 4 beta 1, and alpha 4 beta 7 integrins participate in CD4+ T cell recruitment to chronically inflamed small intestine. J Immunol. 2005;174:2343–2352. doi: 10.4049/jimmunol.174.4.2343. [DOI] [PubMed] [Google Scholar]
- 99.Burns RC, Rivera-Nieves J, Moskaluk CA, et al. Antibody blockade of ICAM-1 and VCAM-1 ameliorates inflammation in the SAMP-1/Yit adoptive transfer model of Crohn's disease in mice. Gastroenterology. 2001;121:1428–1436. doi: 10.1053/gast.2001.29568. [DOI] [PubMed] [Google Scholar]
- 100.Gorfu G, Rivera-Nieves J, Hoang S, et al. Beta7 integrin deficiency suppresses B cell homing and attenuates chronic ileitis in SAMP1/YitFc mice. J Immunol. 2010;185:5561–5568. doi: 10.4049/jimmunol.0903938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rivera-Nieves J, Burcin TL, Olson TS, et al. Critical role of endothelial P-selectin glycoprotein ligand 1 in chronic murine ileitis. J Exp Med. 2006;203:907–917. doi: 10.1084/jem.20052530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Inoue T, Tsuzuki Y, Matsuzaki K, et al. Blockade of PSGL-1 attenuates CD14+ monocytic cell recruitment in intestinal mucosa and ameliorates ileitis in SAMP1/Yit mice. J Leukoc Biol. 2005;77:287–295. doi: 10.1189/jlb.0204104. [DOI] [PubMed] [Google Scholar]
- 103.Arseneau KO, Tamagawa H, Pizarro TT, et al. Innate and adaptive immune responses related to IBD pathogenesis. Curr Gastroenterol Rep. 2007;9:508–512. doi: 10.1007/s11894-007-0067-3. [DOI] [PubMed] [Google Scholar]
- 104.Rivera-Nieves J, Ho J, Bamias G, et al. Antibody blockade of CCL25/CCR9 ameliorates early but not late chronic murine ileitis. Gastroenterology. 2006;131:1518–1529. doi: 10.1053/j.gastro.2006.08.031. [DOI] [PubMed] [Google Scholar]
- 105.Kang SG, Piniecki RJ, Hogenesch H, et al. Identification of a chemokine network that recruits FoxP3(+) regulatory T cells into chronically inflamed intestine. Gastroenterology. 2007;132:966–981. doi: 10.1053/j.gastro.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 106.Bachmann C, Klibanov AL, Olson TS, et al. Targeting mucosal addressin cellular adhesion molecule (MAdCAM)-1 to noninvasively image experimental Crohn's disease. Gastroenterology. 2006;130:8–16. doi: 10.1053/j.gastro.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 107.Odashima M, Bamias G, Rivera-Nieves J, et al. Activation of A2A adenosine receptor attenuates intestinal inflammation in animal models of inflammatory bowel disease. Gastroenterology. 2005;129:26–33. doi: 10.1053/j.gastro.2005.05.032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.