

Abstract
Hypoxia-induced pulmonary arterial hypertension (PAH) is a deadly disease characterized by progressive remodeling and persistent vasoconstriction of the pulmonary arterial (PA) system. Remodeling of the PA involves smooth muscle cell (SMC) proliferation, hypertrophy, migration and elevated extracellular matrix (ECM) production elicited by mitogens and oxidants produced in response to hypoxic insult. We previously reported that the transcription factor CREB is depleted in medial PA SMCs in remodeled, hypertensive vessels in rats or calves exposed to chronic hypoxia. In culture, CREB loss can be induced in PA SMCs by exogenous oxidants or PDGF. Forced depletion of CREB with siRNA in PA SMCs is sufficient to induce their proliferation, hypertrophy, migration, dedifferentiation and ECM production. This suggests that oxidant and/or mitogen-induced loss of CREB in medial SMCs is, in part, responsible for PA thickening. Here we tested whether oxidant scavengers could prevent loss of CREB in PA SMCs, and inhibit SMC proliferation, migration and ECM production using in vitro and in vivo models. Exposure of PA SMCs to hypoxia induced H2O2 production and loss of CREB. Treatment of SMCs with exogenous H2O2 or a second oxidant, Sin-1, elicited CREB depletion under normoxic conditions. Exogenous H2O2 also induced SMC proliferation, migration and increased elastin levels as did forced depletion of CREB. In vivo, hypoxia-induced thickening of PA wall was suppressed by the superoxide dismutase mimetic, Tempol, which also prevented loss of CREB in medial SMCs. Tempol also reduced hypoxia-induced SMC proliferation and elastin deposition in the PA. The data indicate that CREB levels in the arterial wall are regulated in part by oxidants produced in response to hypoxia, and that CREB plays a crucial role in regulating SMC phenotype and PA remodeling.
INTRODUCTION
Pulmonary arterial hypertension (PAH) as a result of intermittent or chronic hypoxia is frequently observed in interstitial lung disease and chronic obstructive pulmonary diseases like chronic bronchitis and emphysema. Hypoxia-induced PAH is also linked to reduced ventilatory capacity and sleep-related hypoventilation associated with obesity. Unfortunately, this deadly disease is generally unresponsive to current treatments and therapies.
Hypoxia-induced PAH is characterized by sustained vasoconstriction of the pulmonary vasculature and remodeling or thickening of pulmonary arterial (PA) system1. PA remodeling is characterized by proliferation and hypertrophy of medial smooth muscle cells (SMCs), migration of SMCs to previously non-muscularized arterioles, and increased production of extracellular matrix (ECM). These changes are largely elicited by cytokines and growth factors including platelet-derived growth factor (PDGF), insulin-like growth factor-1, fibroblast growth factor 2, serotonin and endothelin, that are produced in the arterial wall in response to hypoxic insult and act as potent SMC mitogens2,3,4,5. In addition it is now widely recognized that production of reactive oxygen species (ROS) in the pulmonary vasculature in response to hypoxia also plays a critical role in PA remodeling. Both chronic and intermittent hypoxia stimulate superoxide production in the PA wall in rodent models6,7,8, concomitant with increased expression of superoxide producing enzymes like xanthine oxidase and NADPH oxidase9,10,11. Superoxide scavengers like the compound Tempol, xanthine oxidase inhibition with allopurinol, and general anti-oxidants like N-acetyl cysteine suppress superoxide and hydrogen peroxide levels in the hypoxic PA wall, and inhibit PA remodeling, decrease right ventricular (RV) hypertrophy and lower PA or RV systolic pressure in these models6,7,10. Similar results are observed in mice deficient in the NADPH oxidase subunit gp91phox 12,13. These experiments indicate that oxidant production in the arterial wall promotes PA remodeling, and suggest that antioxidants may be an important therapeutic strategy for hypoxia-induced PAH. However, the mechanisms by which oxidants promote arterial thickening have not been resolved.
We previously reported that levels of the transcription factor CREB are diminished in medial SMCs in remodeled, hypertensive PAs from rats and calves with chronic hypoxia-induced PAH. The loss of CREB can be recapitulated in primary PA SMCs in culture by prolonged exposure to PDGF-BB or oxidant stress. These treatments also stimulate SMC proliferation, hypertrophy, migration and ECM production14,15,16,17. These results suggested that CREB might play an active role in regulating SMC phenotype and PA remodeling. To test this concept, we employed gain and loss-of-function strategies to assess the impact of CREB on SMC phenotype. Forced loss or inhibition of CREB, in the absence of mitogens or exogenous oxidants, was sufficient to stimulate SMC proliferation and migration, inhibit SM-myosin and calponin expression, and alter extracellular matrix production. Alternately, overexpression of wild type or constitutively active forms of CREB suppressed mitogen-induced SMC growth and migration. These data demonstrate that forced depletion of CREB alone reproduces the same changes in SMC phenotype that occur with oxidant or mitogen exposure.
Here we investigated whether superoxide scavengers could prevent CREB depletion and block SMC proliferation, migration and ECM production in PA SMCs exposed to exogenous oxidants in vitro. We also explored the ability of oxidant scavenging in vivo to suppress CREB loss in medial SMCs and inhibit PA remodeling in rats exposed to chronic hypoxia. We found that superoxide scavengers prevent CREB depletion in vitro and in vivo. These agents also repress oxidant-induced switching of SMCs to a synthetic, proliferative phenotype in vitro, and reduce PA remodeling in vivo. The results demonstrate that ROS production in the PA wall in response to hypoxia is a fundamental mechanism for CREB loss in medial SMCs.
MATERIALS AND METHODS
Materials
Hydrogen peroxide (H2O2), 3-morpholinosydnonimine hydrochloride (Sin-1), 4,-dihydroxyl-1,3-benzededisulfonic acid (Tiron), and 4-Hydroxy-2,2,6,6-tetramethylpiperidine 1-oxyl (Tempol) were purchased from Sigma-Aldrich (St. Louis, MO). Fetal bovine serum, glutamine, and penicillin/streptomycin were purchased from Gemini Bio Products (Sacramento, CA). Minimal Essential Eagle Medium (MEM) were from Hyclone (Logan, UT). Oligofectamine was obtained from Invitrogen (Carlsbad, CA). SuperSignal West Pico Chemiluminescent Substrate and NE-PER Nuclear and Cytoplasmic Extract Reagents were from Pierce (Rockford, IL). Amplex Red fluorescence and CyQuant cell proliferation assays were purchased from Invitrogen (Carlsbad, CA). QCM 24-well colorimetric cell migration assay kit was from Millipore (Temecula, CA). Rat monoclonal anti-BrdU antibody was from Abcam (Cambridge, MA). Polyclonal antibody to α-smooth muscle (α-SM) actin was purchased from Sigma (St. Louis, MO). Rabbit polyclonal antibody to CREB was from Cell Signaling Technology, Inc (Beverly, MA). Mouse monoclonal antibodies to gp91phox and p67phox were purchased from BD Transduction laboratories (BD Biosciences, San Jose, CA). Nitrotyrosine polyclonal antibody was obtained from Upstate/Millipore (Billerica, MA). Mouse monoclonal anti-PCNA (PC10) and rabbit polyclonal anti-actin (C-11) were from Santa Cruz Biotechnology (Santa Cruz, CA). Three double stranded siRNA oligonucleotides to CREB (284-AAGATTCAACAGGAGTCTGTGG-305, 528-AATACAGCTGGCTAACAATGG-549, and 670-AACCAAGTTGTTGTTCAAGCT-691) and a control siRNA oligonucleotide to firefly luciferase (CGTACGCGGAATACTTCGA) were from Dharmacon (Lafayette, CO). Single stranded DNA primers for PCR amplification of elastin (forward-TGGAGCCCTGGGATATCAAG and reverse-GAAGCACCAACATGTAGCAC),β-actin (forward-CCAACCGTGAAAAGATGACC and reverse-TCTAGGGCAACATAGCACAGC), and CREB (forward-AATGACCATGGAATC and reverse-CAAAATTTTCCTGTAGGAAGG) were obtained from Integrated DNA Technologies, Inc (Coralville, CA). Picrosirius red stain was obtained from Electron Microscopy Sciences (Hatfield, PA). Vector NovaRed Peroxidase Staining Kits, VectaShield Mounting Medium with DAPI, and peroxidase conjugated-goat anti-rabbit secondary antibody were from Vector Laboratories (Burlingame, CA).
PA SMC Isolation and Cell Culture
Five hundred-micrometer PAs were recovered from adult rat lungs. Segments of the PAs were cut open and mechanically stripped of adventitia and endothelium. The segments were then placed lumen side down into individual wells of a six-well plate. Tissue explants were maintained in complete modified Eagle medium (MEM) supplemented with200 U/ml penicillin, 0.2 mg/ml streptomycin, and 10% FCS. Although these vessels are larger than those evaluated by morphometric analysis (50–250 μm), techniques to isolate cells from smaller vessels have not been developed. Since our goal was to obtain pure subpopulations of SMCs, we selectively isolated individual cell colonies with a distinct, although uniform, morphological appearance from primary culture by using cloning cylinders. Expression of SMC-specific markers(α-SM actin and SM-myosin heavy chains) in each isolated cell subpopulation was examined. Only cell subpopulations with uniform morphological appearance and uniform patterns of expression of SMC markers were selected for further experimentation. Individual cell colonies growing from tissue explants in primary culture were isolated by placing cloning cylinders (5–10 mm in diameter, greased on the bottom) over each cell colony of interest. Cells within the ring were trypsinized and transferred to a 24-multiwell plate for expansion. For low serum conditions, cells were transferred to MEM containing 0.1% fetal calf serum. During experiments, the cells were placed in sealed Plexiglas chambers filled with either normoxic (80% nitrogen, 20% oxygen) or hypoxic (97% nitrogen, 3% oxygen) gas mixtures. The gas mixture in each chamber was replaced every24 h. All studies were carried out using cells at passages 1–8. Cell cultures were tested for mycoplasma contamination with the use of a GenProbe Mycoplasma T. C. rapid detection system(San Diego, CA).
H2O2 production by SMCs
The Amplex Red fluorescence assay (Invitrogen, Carlsbad, CA) was used to measure basal H2O2 production by PA SMCs. H2O2 standards (0–0.125 μM) and SMCs were transferred to a 96-well plate. Fluorescence was determined in 100-μl solutions containing 15 μM Amplex Red reagent (10-acetyl-3,7-dihydroxyphenoxazine) and 0.1 U/ml horseradish peroxidase. Fluorescence was measured in a fluorimeter (Flexstation; Molecular Devices) using an excitation filter of 530 nm and an emission filter of 590 nm. Background fluorescence was subtracted and H2O2 (micromoles) accumulation was normalized for cellular DNA content (CyQuant Assay, Invitrogen, Carlsbad, CA).
Subcellular fractionation and western blots
Cytosolic and nuclear fractions were prepared from SMCs using NE-PER reagents following the supplier’s instructions. After correcting for protein concentrations, cell lysates were mixed with an equal volume of Laemmli SDS loading buffer, resolved on 10% polyacrylamide-SDS gels and transferred to PVDF membranes. The blots were blocked with phosphate buffered saline (PBS) containing 5% dry milk and 0.1% Tween 20, and then treated with antibodies that recognize the target proteins indicated in each figure overnight at 4°C. The blots were washed and subsequently treated with appropriate secondary antibodies conjugated to horseradish peroxidase. After the blots are washed, specific immune complexes will visualized with SuperSignal West Pico Chemiluminescent Substrate.
SiRNA treatments
SMCs were plated at 30–50% confluency on 6-well plates in complete medium. Twenty-four later the cells were transferred to serum-free MEM for transfection. Double stranded CREB siRNA oligonucleotides (equal amounts of 3 CREB siRNAs were mixed together) or the control luciferase-specific siRNA were complexed with Oligofectamine reagent and applied to the cells according to the manufacturer’s recommendations. After 3 hours, and equal volume of MEM containing 30% FCS was added to the wells. Cells were allowed to recover for at least 48 hours before subsequent manipulations.
Cell migration assay
SMC migration was measured with QCM 24-well colorimetric cell migration assay kit according to the manufacturer’s direction.
Animal care and treatment
All animal procedures were performed with approval and in accordance with the guidelines of the University of Colorado Denver for Laboratory Animal Care. Adult male Wistar-Kyoto rats (6wk old) were randomized to normoxic (Nx; 1,600 m, 630 mmHg) or hypobaric hypoxia (Hx; 5,500 m, 410 mmHg) and treated with and without the SOD mimetic Tempol at 86 mg·kg−1·day−1 for 21 days in their drinking water (n = 6 animals/treatment group)6. Fresh food, water, and clean cages with fresh bedding were provided every other day. Light was maintained on a 12 hour cycle, and humidity was 40–45% with a temperature of 25–27°C. The animals were monitored daily, and weight was checked once a week. At the end of 21 days, the rats were anesthetized and catheterized and the following measurements were obtained: body weight, hematocrit, mean PA pressure. After the rats were killed, right ventricular (RV) hypertrophy and pulmonary artery (PA) wall morphometry data were collected.
Hemodynamic measurements and tissue procurement
Rats were anesthetized with intramuscular ketamine (100 mg/kg) and xylazine (15 mg/kg) for placement of catheters in the right jugular vein, right ventricle, and right carotid artery. Anesthetized rats were placed in a ventilated plastic box, and RV systolic pressure and mean PA pressure were measured with pressure transducers. Cardiac output (CO) was determined by a standard dye-dilution method, as previously described18. Following hemodynamic measurements, the rats were then euthanized by exsanguination. Lungs were fixed with4% paraformaldehyde in PBS containing 5 mM EDTA by airway inflation at 30 cmH2O pressure. Pulmonary arteries were perfused with 4% paraformaldehyde-PBS-5 mM EDTA (to maintain vasodilation) at a pressure similar to that measured in vivo. The heart and lungs were then removed en bloc. The heart was removed for assessment of right ventricular hypertrophy (Right Ventricle/Left Ventricle+Septum) (RV/LV+S), and the lungs were prepared for morphometric analysis.
Immunostaining and microscopy
Five micrometer sections of paraformaldehyde-fixed, paraffin-embedded lung tissue were deparaffinized with Hemo-D, and rehydrated in a graded ethanol/water series. Sections were subjected to antigen retrieval in citrate buffer in a microwaveable pressure cooker for 20 minutes. Sections were blocked with PBS containing 5% horse serum for 30 minutes at room temperature. The sections were incubated overnight in PBS/5% FBS at 4°C with the primary antibodies indicated in the figure legends. The sections were then washed and incubated with the indicated Alexa Fluor-conjugated secondary antibodies for 1 hour at room temperature. Pentachrome staining was conducted by the University of Colorado Histology Core Laboratory. Picrosirius red staining was performed according to supplier’s instructions.
Distal pulmonary vessels (outside diameter 10–50 μm)were assessed by a blinded observer for degrees of circumferential α-SM actin-positive staining indicative of muscularization. Vessels smaller than 10 μm were considered capillaries and excluded from further consideration. Proximal vessels (outside diameter 50–250 μm) were analyzed for medial wall thickness at four points around the vessel circumference and for lumen diameter along two axes.
Staining for BrdU incorporation was performed on cells fixed in PBS/4%paraformaldehyde. Slides were incubated in 0.5% hydrogen peroxide for 10 minutes to quench endogenous catalase and briefly rinsed in tap water. The slides were then incubated in 2 N HCl at 37 C for 30 minutes and then transferred to 0.1 N disodium tetraborate for 10 minutes. The slides were rinsed and immunostaining was conducted as described above using a rat monoclonal anti-BrdU primary antibody and an anti-rat secondary antibody conjugated to horse radish peroxidase. Slides were then developed with Vector NovaRed reagents.
Microscopy was performed on a Nikon TE2000-U inverted epifluorescent microscope. Brightfield, phase contrast, and fluorescent digital deconvolution images were captured to a computer with either a Spot RT/KE monochrome camera or Spot Insight color camera (Diagnostic Imaging, Sterling Heights, MI). Images were analyzed and processed with MetaMorph 6.1 Software (Molecular Devices, Sunnyvale, CA).
Statistics
Statistical analysis was performed using the Super ANOVA software program (Abacus Concepts, Berkeley, CA). Comparisons were performed using two-way analysis of variance followed by the Scheffé’s multiple comparison test for individual comparisons within and between groups of data points. Data were considered statistically significant with a P value ≤0.05.
RESULTS
Hypoxia induces ROS production in PA SMCs
We previously reported that exposure of PA SMCs to chronic hypoxia19 or H2O2 under normoxic conditions17 elicited loss of CREB. Figure 1A shows decreased CREB levels in hypoxia-treated PA SMCs. As a first step in determining whether CREB depletion due to these two stimuli are related we measured H2O2 production by SMCs exposed to hypoxia using an Amplex Red assay. We found that hypoxia increased H2O2 generation in PA SMCs for at least 72 hours (Fig. 1B). Pretreatment of the cells with 10 mM of the intracellular free radical scavenger, Tiron, abolished the hypoxia-induced H2O2 generation (Fig. 1C) pointing to the activity of both NAD(P)H oxidase and/or mitochondrial respiratory chain as source of ROS in PA SMCs.
Figure 1. Hypoxia induces CREB loss and H2O2 production in PA SMCs in culture.

Rat PA SMCs were maintained under ambient normoxia (room air plus 5% CO2) or isobaric hypoxia for 48 hours (A and C) or as for the times indicated (B). A) Ten μg of cell lysate protein was resolved on SDS-polyacrylamide gels and transferred to PVDF membranes. The membranes were blocked and probed with antibodies to CREB and β-actin. A representative blot is shown. B) H2O2 was measured by Amplex Red Fluorescence Bioassay. Values are given as means ± SE (micromoles per hour per nanogram DNA, n = 6). Normoxia = crosshatched bars, hypoxia = solid bars. * indicates p≤0.05. C) PA SMCs were treated with Tiron (10 mM, solid bars) and exposed to normoxia and hypoxia for 48 hours. H2O2 was measured by Amplex Red Fluorescence Bioassay. * indicates p≤0.05.
ROS stimulates loss of CREB in PA SMCs
Next, we tested whether exogenous H2O2 was sufficient to induce CREB depletion in cultured rat PA SMCs. Addition of H2O2 to the culture medium for 48 hours induced complete CREB loss at doses as low as 50 μM (Fig. 2A). H2O2-stimulated CREB depletion occurs as early as 4 hours after treatment (Fig. 2B). We also tested a second oxidant Sin-1, a molecule that has been shown to decompose in two steps, thereby releasing superoxide anions and NO leading to formation of peroxynitrite (ONOO−). Peroxynitrite is a very reactive molecule and there are no methods to measure ONOO− as such in physiological conditions. It can be assayed indirectly by either measuring nitrotyrosine or using SIN-1 as a peroxynitrite donor20. Sin-1 also induced CREB depletion in a dose dependent manner when added to the culture media for 48 hours (Fig. 2C). However, Tiron blocked H2O2-induced CREB loss in a dose-dependent manner (Fig. 2D) confirming that depletion of CREB is mediated by ROS.
Figure 2. Oxidant-induced CREB loss in PA SMCs in culture is blocked by Tiron.
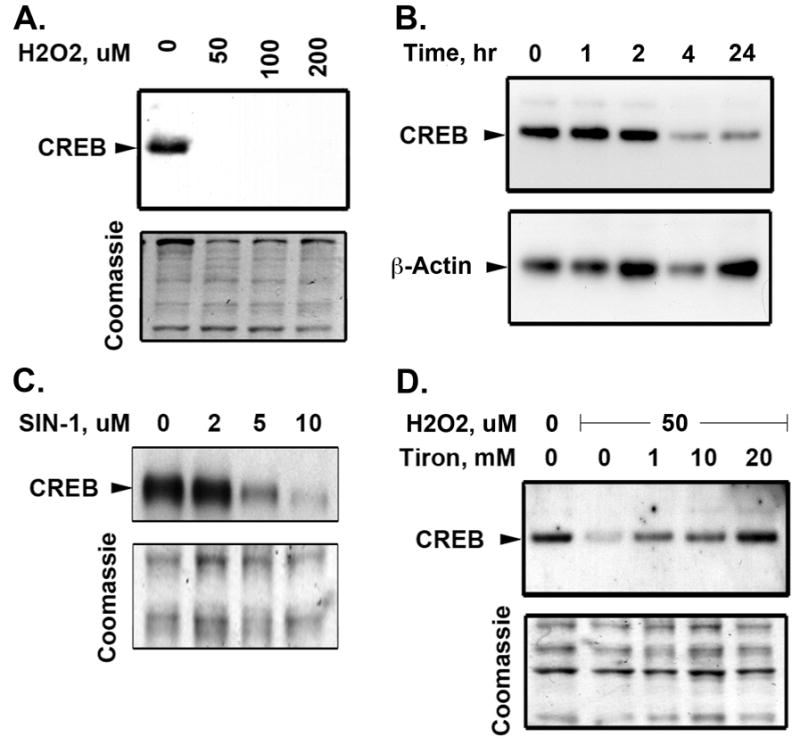
Rat PA SMCs grown in complete medium were transferred to MEM containing 0.1% FCS for 24 hours. The cells were then treated with increasing dose of H2O2 for 48 hours (A) or stimulated with H2O2 (50 μM) at the times indicated above the lanes (B) or treated with increasing dose of the peroxynitrite inducer, Sin-1 for 48 hours (C). Nuclear extracts were prepared and separated on 10% polyacrylamide-SDS gels and transferred to PVDF membranes. Western blotting was performed with a CREB-specific antibody. A representative Coomassie Blue stained blot and β-actin western blot are shown as loading control. Nuclear expression of CREB is downregulated in response to H2O2 (A) or Sin-1 (C) in a dose dependent manner. A detailed time course of H2O2-stimulated PA SMCs demonstrates that decreased CREB occurs as early as 4 hours after treatment (B). D) SMCs were treated with increasing dose of Tiron in presence of H2O2 (50 μM) for 48 hours. Tiron restored nuclear CREB levels in H2O2-treated cells. The blots are representative of 3 separate experiments
ROS or CREB depletion independently stimulate PA SMC proliferation, migration and elastin expression
Given that forced depletion of CREB induces SMC proliferation and other features of a detrimental SMC phenotype19, we next tested whether ROS would elicit similar changes. PA SMCs were grown on slides in the presence or absence of H2O2 with or without Tiron for 48 hours and proliferation was measured by BrdU incorporation. Immunolabeling for BrdU showed that H2O2 increases SMC proliferation as did transfection of cells with CREB-specific siRNA (Fig. 3A and 3B). Exogenous H2O2 or CREB knockdown also stimulated elastin gene expression (Fig. 3C). Concomitant treatment with the Tiron, markedly reduced the H2O2-triggered SMC proliferation and elastin production. Similarly, SMC migration was elevated by H2O2 treatment and was also higher in CREB-deficient SMCs (Fig. 3D). The data show that H2O2 elicits the same detrimental changes in SMC phenotype as forced CREB loss. This suggests that the negative impact of ROS on SMC phenotype is mediated, in part, by CREB depletion.
Figure 3. H2O2 or CREB loss both stimulate PA SMC proliferation, migration and elastin production.

Rat PA SMCs were plated at 20% confluency in complete medium. Medium was replaced with MEM containing 0.1% FCS 24 hours prior to treatments. The cells were treated with 50 μM H2O2 with or without Tiron (20 mM) for 48 hours. SiRNA knockdown of CREB was achieved by transfection of CREB-specific siRNA oligonucleotides 48 hours prior to medium replacement with MEM/0.1% FCS. Cells transfected with a non-specific siRNA oligonuleotides were used as controls (Cntrl). BrdU was added to the wells for the last 6 hours of the experiment. The SMCs were then fixed and stained for BrdU incorporation. The number of total and BrdU-positive nuclei (red) were counted in 10 microscope fields in three triplicate wells. Figure A) shows the average percent BrdU-positive nuclei for each treatment. * p≤0.05 compared to NA and Cntrl siRNA groups. Figure B) shows representative bright field micrographs of cells exposed to each treatment. BrdU-positive nuclei are red. C) RNA was recovered from the cells and subjected to semi-quantitative RT-PCR for elastin, CREB, or β-actin messages. A representative gel of the resolved PCR products is shown. D) Migration was measured by Boyden chamber assay. The graph shows the average of 2 replicates in 4 separate experiments (n=4). *p≤0.05 compared to NA and Cntrl siRNA groups.
To confirm this model, we attempted to inhibit H2O2-induced SMC proliferation, migration and elastin production by overexpressing CREB. However, CREB levels were not restored in SMCs transfected with a CMV-promoter driven CREB expression vector following treatment with H2O2. We believe that this due to the fact that CREB depletion occurs post-translationally by proteasomal degradation rather than at the level of gene transcription21.
Chronic hypoxia induces ROS production in the lung and PA wall, and expression of oxidant enzymes in the lung
Reduction of ROS production or levels with agents like the superoxide scavenger Tempol has been shown to reduce PA thickening in response to chronic hypoxic exposure6. This beneficial affect might be related the ability of these agents to prevent CREB loss in medial SMCs. The first step in testing this idea was to verify that exposure to chronic hypoxia stimulates ROS production in the PA wall and lung of rats, and determine whether this was abrogated by simultaneous treatment with the superoxide dismutase (SOD) mimetic, Tempol. During hypoxia the formation of both ROS and reactive nitrogen species (RNS) results in widespread lipid peroxidation, protein oxidative and nitrosative modifications and because nitrotyrosine is a stable marker for nitric oxide (NO)-derived oxidants, we measured nitrotyrosine levels in whole lung homogenates to determine if changes consistent with nitrosative stress occur in response to hypoxia. Immunohistochemical analysis of lung sections obtained from rats exposed to hypoxia for 21 days revealed positive staining for nitrotyrosine (Fig. 4A). Nitrotyrosine was not detected in sections of lung tissue from rats maintained under normoxic conditions. Likewise, no positive staining for nitrotyrosine was found in the lung tissues of hypoxic-Tempol rats. The data demonstrate ROS production in the lungs of rats exposed to prolonged hypoxia, and the inhibition of this response by Tempol. We also examined levels of the membrane-bound NADPH oxidase subunit gp91phox as well as the cytosolic subunit p67phox in lung homogenates from normoxic and hypoxic rats. The levels of both subunits were elevated in the hypoxic homogenates (Fig. 4B), suggesting that these and similar enzymes may be responsible for ROS production in response to hypoxia.
Figure 4. Hypoxia stimulates peroxynitrate formation, decreases CREB levels and increases NAD(P)H oxidase subunits gp91phox and p67phox in rat lung.

Adult male Wistar-Kyoto rats (6wk old) were exposed to normoxia (1,600 m; 630 mmHg) or to hypobaric hypoxia (5,500 m, 410 mmHg) for 21 days. A) Some adult rats were also treated with the SOD mimetic Tempol at 86 mg.kg−1.day−1 in their drinking water as indicated. Paraffin sections of lung tissue were de-paraffinized, blocked and incubated with an antibody against nitrotyrosine. The sections were then rinsed and incubated sequentially with biotinylated secondary antibody followed by horseradish peroxidase-conjugated streptavidin, followed by incubation with the peroxidase substrate solution, diaminobenzidine. Bar = 75 μm. B) Lung tissue from 4 normoxic or 4 hypoxic rats was homogenized and equal quantities of protein were separated on 10% polyacrylamide-SDS gels. Proteins were transferred to PVDF membranes. The blots were subjected to western blot analysis with the antibodies to the proteins indicated on the left side of each panel. Equal protein loading was verified prior to antibody exposure by Ponceau Red staining. Representative blots are shown.
Tempol attenuates chronic hypoxia-induced pulmonary arterial remodeling and loss of CREB in medial SMCs
We next examined whether Tempol would prevent CREB loss in medial SMCs and thickening of the PA in rats exposed to chronic hypoxia. Lung sections from normoxic, hypoxic and hypoxic-Tempol treated rats were subjected to immunohistochemical staining for CREB and SM-actin. CREB was easily detected (red/CREB + blue/DAPI = magenta) in nuclei throughout the lungs of normoxic animals (Fig. 5). In hypoxic animals, CREB was detected in nuclei in the lung parenchyma, PA endothelial cells and PA adventitia. However, no CREB signal (blue/DAPI only) was observed in nuclei of medial cells. CREB was present throughout the lung and PA media hypoxic-Tempol rats. Thus, reduction of ROS levels in the lungs of hypoxic animals inhibited CREB depletion in medial PA SMCs.
Figure 5. Tempol prevents loss of CREB in PA medial SMCs and PA remodeling in chronic hypoxic animals.

Adult rats were treated with SOD mimetic Tempol at 86 mg.kg−1.day−1 in their drinking water and maintained under normoxic or hypoxic conditions for 21 days. Five-micrometer sections of lung tissue were subjected to immunohistochemical staining for 4–6-diamidino-2-phenylindole (DAPI; nuclei), CREB and, α-SM actin. Data were obtained for at least 5 vessels per animal from 6 animals. Representative fluorescent digital deconvolution photomicrographs show that CREB is depleted in medial SMCs (SM-actin-positive) and medial thickness is increased in rats exposed to hypoxia. CREB loss and medial thickening are not apparent in hypoxic animals that also received Tempol.
Examination of the sections also revealed a marked increase in PA wall thickness (area of SM-actin staining) in hypoxic animals, whereas PA thickness was less in hypoxic-Tempol rats. The ability of Tempol to attenuate remodeling was confirmed by examination of hematoxylin and eosin-stained lung tissue sections, which showed a profound increase in PA wall thickness in chronically hypoxic compared with normoxic animals (Fig. 6A). Tempol treatment markedly decreased PA remodeling. Attenuation of remodeling was confirmed by morphometric analysis, which revealed a statistically significant decrease in PA wall thickness in the hypoxic-Tempol rats compared to hypoxic rats (Fig. 6B).
Figure 6. Tempol markedly decreased hypoxia-induced pulmonary arterial (PA) remodeling.

Adult rats were treated with SOD mimetic Tempol at 86 mg.kg−1.day−1 in their drinking water and maintained under normoxic or hypoxic conditions for 21 days. A) Representative bright-field photomicrographs of hematoxylin and eosin-stained lung sections show that Tempol inhibited hypoxia-induced PA remodeling. Bar, 100 μm. B) Morphometric analysis confirms that Tempol attenuates (solid bars) hypoxia-induced PA wall thickening compared with control (hatched bars). Data are average values obtained from at least 30 vessels with diameters between 50 and 250 μm from 6 animals per group; error bars represent SD. *p≤0.05 compared to hypoxia/untreated group.
Finally, since both H2O2 and forced CREB loss in cultured SMCs stimulates proliferation and elastin/matrix production, we examined these features in lung sections from normoxic, hypoxic and hypoxic-Tempol animals. Immunohistochemical staining of lung sections showed few proliferating cell nuclear antigen (PCNA)-positive cells in the PA wall of normoxic rats (Fig. 7A). Many PCNA-positive cells were present in the media and adventitia of vessels in the lungs of hypoxic rats. However, PCNA staining was substantially lower, particularly in the media, of PAs in animals exposed to hypoxia and Tempol. We assessed PCNA protein expression in homogenized whole lung tissue from animals subjected to normoxia or hypoxia by western blotting. Similar to the results described with immunohistochemical staining, we found a significant increase of PCNA expression in whole lung homogenates under hypoxic conditions (Fig 7B). Likewise, the number of elastin fibers (gray fibers in pentachrome-stained sections) and collagen (yellow-staining material) was elevated in hypoxic vessel walls. Elastin and collagen levels in hypoxic/Tempol animals were greatly reduced although not normalized.
Figure 7. Tempol inhibits proliferation and elastin production in the PA wall in response to chronic hypoxia.

Adult rats were treated with SOD mimetic Tempol at 86 mg.kg−1.day−1 in their drinking water and maintained under normoxic or hypoxic conditions for 21 days. A) Five-micrometer sections of fixed lung tissue were subjected to immunohistochemistry for PCNA or pentachrome staining for elastin production as indicated. Data were obtained for at least 5 vessels per animal from 6 animals. Representative bright-field photomicrographs show that Tempol inhibited proliferation and elastin deposition in rats exposed to hypoxia. Bar, 50 μm. B) Western blots of homogenates of whole lung tissue from animals subjected to normoxia or hypoxia were probed with antibodies to PCNA or β-actin as indicated. Representative of blots are shown.
DISCUSSION
In the present study, we have provided evidence that hypoxia stimulates oxidant production by PA SMCs in vitro, and increases expression of oxidant-generating enzymes and oxidant levels throughout the lung in vivo. Elevated lung oxidant levels were associated with loss of CREB in medial PA SMCs in vivo with concomitant remodeling of the PA wall. Exogenous H2O2 treatment of PA SMCs in culture also elicited CREB depletion and increased cell proliferation, migration and altered production of ECM components. Oxidant scavengers effectively inhibited loss of CREB in response to H2O2 in vitro and chronic hypoxia in vivo, while simultaneously attenuating SMC proliferation, migration and PA remodeling. Given our previous results14,19 demonstrating that CREB directly controls SMC growth, migration, differentiation and ECM production, the current data suggest that the detrimental impact of ROS on PA remodeling and SMC phenotype are largely mediated by oxidant-induced CREB loss. We are currently generating a SMC-targeted CREB loss-of-function mouse model to directly test the involvement of CREB in hypoxia/oxidant-induced PA remodeling and SMC function.
In the present study we show that oxidants elicit CREB loss in PA SMCs, and that superoxide scavengers completely inhibit this phenomenon. We previously reported that PDGF-BB, a mitogen produced in the arterial wall in response to hypoxia, also stimulates CREB depletion in SMCs via the PI3K/Akt signaling pathway14. In this study, we demonstrated that activation of this signaling system promotes phosphorylation of CREB by casein kinase 2, which targets CREB for polyubiquitination and proteasomal degradation. Since inhibition of PDGF/PI3K/Akt signaling and oxidant scavenging both prevent CREB loss in SMCs, a common signaling mechanism may be involved in both processes. One potential candidate is the serine/threonine protein kinase Akt, which is activated by both oxidant stress22 and PDGF14 in vascular SMCs. Another candidate is the PDGF receptor itself. Oxidants stimulate PDGF receptor phosphorylation and activity13,23, and non-PDGF growth factors have been shown to stimulate PDGF receptor activity via a mechanism involving ROS and src-family kinases24. Likewise, antioxidants reduce PDGF receptor signaling in cultured SMCs and in restenotic lesions25. In our hands H2O2 alone induces the entire signaling cascade from PDGF receptor phosphorylation to activation of PI3K/Akt to increased casein kinase 2 catalytic subunit expression that we previously showed was responsible for CREB degradation in response to PDGF-BB14 (unpublished data). We propose that ROS and PDGF, both produced in the hypoxic arterial wall, may induce CREB loss via PDGF receptor activation, and are exploring this concept in greater detail.
Studies from our laboratory have demonstrated that CREB depletion in vascular SMCs elicited changes consistent with those observed in SMCs from pathologically remodeled arteries in vivo. Such changes include decreased expression of SMC markers and contractile factors like SM-myosin, calponin, and fibronectin, increases in proliferation and proliferation-related factors like cyclin D1, and increases in ECM production14. Leonard et al26 recently reported in vivo lung-selective phosphorylation of CREB on serine-133 and activation of CREB in the absence of any change in total CREB levels in response to hypoxia. These results likely differ from ours because they were obtained in studies in which mice were exposed for shorter periods of time to modest levels of hypoxia, rather than our studies with rats exposed for 21 days to ~11% O2. Furthermore, rodents respond moderately to altitude exposure27 and therefore require longer exposure to higher levels of hypoxia to induce PH and vascular remodeling. This may explain why we observe a decrease of total CREB levels in our models.
A somewhat surprising aspect of our experiments was that Tempol did not have any effect on the PA pressure elevation in hypoxic rats. Tempol is a stable nitroxide radical, which has also a low molecular weight and permeates biological membranes. Tempol scavenges both intra-and extracellular deleterious ROS and therefore mimics the benefits of the enzyme superoxide dismutase (SOD)28. In addition tempol has been shown to effectively suppress pathological conditions associated with marked oxidative stress in vivo29,30,31,32. Many studies have evaluated the capability of Tempol to prevent the increase in blood pressure in various rat models of hypertension. It has been reported that Tempol normalized right ventricular systolic pressure and reduced right ventricular hypertrophy in chronic hypoxic rats6. In the majority of animal studies Tempol was administered in the drinking water with doses ranging from 1 to 3 mM. In our study the hypoxic group received 86 mg.kg−1.day−1 corresponding to 1 mM. Thus, the inability of Tempol to lower PA pressure and RV hypertrophy is probably not related to the dosage used in our study nor to an accumulation of ROS because immunohistochemical analysis of lung sections obtained from rats treated with Tempol and exposed to hypoxia revealed no positive staining for nitrotyrosine compared to hypoxic untreated rats. There are other studies in which Tempol failed to decrease arterial pressure. For example, Elmarakby et al30 have reported that Tempol did not have any effect of the blood pressure elevation in angiotensin-II induced systemic hypertension in rats. It is not clear whether there are other factors that limit Tempol’s activity.
Finally, our data suggest that pulmonary arterial wall remodeling or thickening may not contribute to the development of the hypoxic hypertension itself, since blockade of remodeling with Tempol had no beneficial affect on PA pressure. This concept is supported by studies employing methods to prevent or compensate for fixation method-dependent changes in lumen area. These studies showed that when the pulmonary vascular bed was maximally vasodilated during lung fixation, there was no reduction in vessel luminal area directly due to the medial and adventitial thickening33,34. Other experiments have demonstrated that angiotensin-converting enzyme inhibitors and PPARg agonists prevent PA remodeling in rats exposed to chronic hypoxia but do not attenuate the development of PH or RV hypertrophy35,36. Likewise, Nagaoka et al37 have reported that acute inhibition of RhoA/Rhokinase signaling almost completely reverses PH in rats exposed to chronic hypoxia, although the brief exposure to the inhibitors would not be expected to have any effect on the structural thickening of the PA wall. However, hypoxia-induced PH in rodents fails to mirror the progressive intimal remodeling observed in PH in cows and humans27. Thus, antioxidant therapies that block remodeling may still prove useful in treating human PH, particularly in combination with the many vasodilators currently in use.
In conclusion, oxidative stress contributes to PA remodeling by promoting SMC growth and ECM deposition; characteristic features of the SMC phenotype in PH. We have demonstrated that chronic hypoxia induced ROS production is associated with an increase of NADPH oxidase expression. H2O2 leads to depletion of CREB and increased SMC proliferation. The development of chronic hypoxic pulmonary vascular remodeling was attenuated in rats treated with the superoxide dismutase mimetic, Tempol. While CREB is not the sole mediator of SMC growth, our study supports a preeminent role for this factor in controlling SMC functions in response to ROS.
Acknowledgments
We thank Dr Ivan F. McMurtry (University of South Alabama School of Medicine, Mobile, AL) for the critical review of this manuscript.
This work was supported by a grant from American Heart Association (Scientist Development Grant 073512N to CVG), an NIH/NHLBI Program Project Grant (P01-HL014985 to DJK).
References
- 1.Mandegar M, Fung YC, Huang W, Remillard CV, Rubin LJ, Yuan JX. Cellular and molecular mechanisms of pulmonary vascular remodeling: role in the development of pulmonary hypertension. Microvasc Res. 2004;68:75–103. doi: 10.1016/j.mvr.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 2.Schultz K, Fanburg BL, Beasley D. Hypoxia and hypoxia-inducible factor-1 alpha promote growth factor-induced proliferation of human vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 2006;290:H2528–H2534. doi: 10.1152/ajpheart.01077.2005. [DOI] [PubMed] [Google Scholar]
- 3.Perkett EAD, Badesch B, Roessler MK, Stenmark KR, Meyrick B. Insulin-like growth factor-1 and pulmonary hypertension induced by continuous air embolism in sheep. Am Rev Respir Cell Mol Biol. 1992;6:82–87. doi: 10.1165/ajrcmb/6.1.82. [DOI] [PubMed] [Google Scholar]
- 4.Fanburg B, Lee SL. A role for the serotonin transporter in hypoxia-induced pulmonary hypertension. J Clin Invest. 2000;105:1521–1523. doi: 10.1172/JCI10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hassoun PM, Thappa V, Landman MJ, Fanburg BL. Endothelin 1: mitogenic activity on pulmonary artery smooth muscle cells and release from hypoxic endothelial cells. Proc Soc Exp Biol Med. 1992;199:165–170. doi: 10.3181/00379727-199-43342. [DOI] [PubMed] [Google Scholar]
- 6.Elmedal B, de Dam MY, Mulvany MJ, Simonsen U. The superoxide dismutase mimetic, Tempol, blunts right ventricular hypertrophy in chronic hypoxic rats. Br J Pharmacol. 2004;141:105–113. doi: 10.1038/sj.bjp.0705580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoshikawa Y, Ono S, Suzuki S, Tanita T, Chida M, Song C, Noda M, Tabata T, Voelkel NF, Fujimura S. Generation of oxidative stress contributes to the development of pulmonary hypertension induced by hypoxia. J Appl Physiol. 2001;90:1299–1306. doi: 10.1152/jappl.2001.90.4.1299. [DOI] [PubMed] [Google Scholar]
- 8.Lachmanová V, Hnilicková O, Povýsilová V, Hampl V, Herget J. N-acetylcysteine inhibits hypoxic pulmonary hypertension most effectively in the initial phase of chronic hypoxia. Life Sc. 2005;77:175–182. doi: 10.1016/j.lfs.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 9.Brennan LA, Steinhorn RH, Wedgwood S, Mata-Greenwood E, Roark EA, Russell JA, Black SM. Increased superoxide generation is associated with pulmonary hypertension in fetal lambs: a role for NADPH oxidase. Circ Res. 2003;92:683–691. doi: 10.1161/01.RES.0000063424.28903.BB. [DOI] [PubMed] [Google Scholar]
- 10.Jankov RP, Kantores C, Pan J, Belik J. Contribution of xanthine oxidase-derived superoxide to chronic hypoxic pulmonary hypertension in neonatal rats. Am J Lung Cell Mol. 2008;294:L233–L245. doi: 10.1152/ajplung.00166.2007. [DOI] [PubMed] [Google Scholar]
- 11.Marshall C, Mamary AJ, Verhoeven AJ, Marshall BE. Pulmonary artery NADPH-oxidase is activated in hypoxic pulmonary vasoconstriction in pulmonary arteries from control and pulmonary hypertensive rats. Br J Pharmacol. 1996;119:917–930. [Google Scholar]
- 12.Liu JQ, Zelko IN, Erbynn EM, Sham JSK, Folz RJ. Hypoxic pulmonary hypertension: role of superoxide and NADPH oxidase (gp91phox) Am J Physiol Lung Cell Mol Physiol. 2006;290:L2–L10. doi: 10.1152/ajplung.00135.2005. [DOI] [PubMed] [Google Scholar]
- 13.Nisbet RE, Graves AS, Kleinhenz DJ, Rupnow HL, Reed AL, Fan THM, Mitchell PO, Sutliff RL, Hart MC. The role of NADPH oxidase in chronic intermittent hypoxia-induced pulmonary hypertension in mice. Am J Respir Cell mol Biol. 2009;40:601–609. doi: 10.1165/rcmb.2008-0145OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garat CV, Fankel D, Erickson PF, Reusch JE, Bauer NN, McMurtry IF, Klemm DJ. Platelet-derived growth factor BB induces nuclear export and proteosomal degradation of CREB via phosphatidylinositol 3-kinase/Akt signaling in pulmonary artery smooth muscle cells. Mol Cell Biol. 2006;26:4934–4948. doi: 10.1128/MCB.02477-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jambal P, Masterson S, Nesterova A, Bouchard R, Bergman B, Hutton JC, Boxer LM, Reusch JE, Pugazhenti S. Cytokine-mediated down-regulation of the transcription factor cAMP-response element-binding protein in pancreatic beta cells. J Biol Chem. 2003;278:23055–23065. doi: 10.1074/jbc.M212450200. [DOI] [PubMed] [Google Scholar]
- 16.Pugazhenti S, Nesterova A, Sable C, Heidenreich KA, Boxer LM, Heasley LE, Reusch JE. Akt/protein kinase B up-regulates Bcl-2 expression through cAMP-response element-binding protein. J Biol Chem. 2000;275:10761–10766. doi: 10.1074/jbc.275.15.10761. [DOI] [PubMed] [Google Scholar]
- 17.Watson PA, Vinson C, Nesterova A, Reusch JE. Content and activity of cAMP response element-binding protein regulate platelet-derived growth factor receptor-alpha content in vascular smooth muscles. Endocrinology. 2002;143:2922–2929. doi: 10.1210/endo.143.8.8959. [DOI] [PubMed] [Google Scholar]
- 18.Stevens T, Morris K, McMurtry IF, Zamora M, Tucker A. Pulmonary and systemic vascular responsiveness to TNF-alpha in conscious rats. J Appl Pysiol. 1993;74:1905–1910. doi: 10.1152/jappl.1993.74.4.1905. [DOI] [PubMed] [Google Scholar]
- 19.Klemm DJ, Watson PA, Frid MG, Dempsey EC, Schaack J, Colton LA, Nesterova A, Stenmark KR, Reusch JEB. cAMP response element-binding protein content is a molecular determinant of smooth muscle cell proliferation and migration. J Biol Chem. 2001;276:46132–46141. doi: 10.1074/jbc.M104769200. [DOI] [PubMed] [Google Scholar]
- 20.Hogg N, darley-Usmar VM, Wilson MT, Moncada S. Production of hydroxyl radicals from the simultaneous generation of superoxide and nitric oxide. Biochem J. 1992;281:419–424. doi: 10.1042/bj2810419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ozgen N, Guo J, Gertsberg Z, Danilo P, Jr, Rosen MR, Steinberg SF. Reactive oxygen species decrease cAMP response element binding protein expression in cardiomyocytes via a protein kinase D1-dependent mechanism that does not require Ser133 phosphorylation. Mol Pharmacol. 2009;76:896–902. doi: 10.1124/mol.109.056473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ushio-Fukai M, Alexander RW, Akers M, Yin Q, Fujio Y, Walsh K, Griendling KK. Reactive oxygen species mediate the activation of Akt/protein kinase B by angiotensin II in vascular smooth muscle cells. J Biol Chem. 1999;274:22699–22704. doi: 10.1074/jbc.274.32.22699. [DOI] [PubMed] [Google Scholar]
- 23.Shimizu H, Hirose Y, Nishijima F, Tsubakihara Y, Miyazaki H. ROS and PDGF-{beta} receptors are critically involved in indoxyl sulfate actions that promote vascular smooth muscle cell proliferation and migration. Am J Physiol Cell Physiol. 2009;297:389–396. doi: 10.1152/ajpcell.00206.2009. [DOI] [PubMed] [Google Scholar]
- 24.Lei H, Kazlauskas A. Growth factors outside of the platelet-derived growth factor (PDGF) family employ reactive oxygen species/Src family kinases to activate PDGF receptor alpha and thereby promote proliferation and survival of cells. J Biol Chem. 2009;284:6329–6336. doi: 10.1074/jbc.M808426200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kappert K, Sparwel J, Sandin A, Seiler A, Siebolts U, Leppänen O, Rosenkranz S, Ostman A. Antioxidants relieve phosphatase inhibition and reduce PDGF signaling in cultured VSMCc and in restenosis. Arterioscler Thromb Vasc Biol. 2006;26:2644–2651. doi: 10.1161/01.ATV.0000246777.30819.85. [DOI] [PubMed] [Google Scholar]
- 26.Leonard MO, Howell K, Madden SF, Costello CM, Higgins DG, Taylor CT, McLoughlin P. Hypoxia selectively activates the CREB family of transcription factors in the in vivo lung. Am J Respir Crit Care Med. 2008;178:977–983. doi: 10.1164/rccm.200712-1890OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tucker A, McMurtry IF, Reeves JT, Alexander AF, Will DH, Grover RF. Lung vascular smooth muscle as a determinant of pulmonary hypertension at high altitude. Am J Physiol. 1975;228:762–767. doi: 10.1152/ajplegacy.1975.228.3.762. [DOI] [PubMed] [Google Scholar]
- 28.Krishna MC, Russo A, Mitchell JB, Goldstein S, Dafni H, Samuni A. Do nitroxide antioxidants act as scavengers of O2− or SOD mimics? J Biol Chem. 1996;271:26026–26031. doi: 10.1074/jbc.271.42.26026. [DOI] [PubMed] [Google Scholar]
- 29.Lavina B, Gracia-Sancho J, Rodriguez-Vilarrupla A, Chu Y, Heistad DD, Bosch J, Garcia-Pagan JC. Superoxide dismutase gene transfer reduces portal pressure in CCl4 cirrhotic rats with portal hypertension. Gut. 2009;58:118–125. doi: 10.1136/gut.2008.149880. [DOI] [PubMed] [Google Scholar]
- 30.Elmarakby AA, Williams JM, Imig JD, Pollock JS, Pollock DM. Synergistic actions of enalapril and Tempol during chronic angiotensin II-induced hypertension. Vascul Pharmacol. 2007;46:144–151. doi: 10.1016/j.vph.2006.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karmeli F, Eliakim R, Okon E, Samuni A, Rachmilewitz D. A stable nitroxide radical effectively decreases mucosal damage in experimental colitis. Gut. 1995;37:386–393. doi: 10.1136/gut.37.3.386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gelvan D, Saltman P, Powell SR. Cardiac reperfusion damage prevented by a nitroxide free-radical. Proc Nat Acad Sci USA. 1991;88:4680–4684. doi: 10.1073/pnas.88.11.4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hyvelin JM, Howell K, Nichol A, Cosello CM, Preson RJ, McLoughlin P. Inhibition of Rho kinase attenuates hypoxia-induced angiogenesis in the pulmonary circulation. Circ Res. 2005;97:185–191. doi: 10.1161/01.RES.0000174287.17953.83. [DOI] [PubMed] [Google Scholar]
- 34.Van Suylen RJ, Smits JF, Daemen MJ. Pulmonary artery remodeling differs from hypoxia- and monocrotaline-induced pulmonary hypertension. Am J Respir Crit Care Med. 1998;157:1423–1428. doi: 10.1164/ajrccm.157.5.9709050. [DOI] [PubMed] [Google Scholar]
- 35.Clozel JP, Saunier C, Hartemann D, Fischli W. Effects of cilazapril, a novel angiotensin converting enzyme inhibitor, on the structure of pulmonary arteries exposed to chronic hypoxia. J Cardiovasc Pharmacol. 1991;17:36–40. doi: 10.1097/00005344-199101000-00006. [DOI] [PubMed] [Google Scholar]
- 36.Crossno JT, Jr, Garat CV, Reusch JE, Morris KG, Dempsey EC, McMurtry IF, Stenmark KR, Klemm DJ. Rosiglitazone attenuates hypoxia-induced pulmonary arterial remodeling. Am J Physiol Lung Cell Mol Physiol. 2007;292:L885–L897. doi: 10.1152/ajplung.00258.2006. [DOI] [PubMed] [Google Scholar]
- 37.Nagaoka T, Fagan KA, Gebb SA, Morris KG, Suzuki T, Shimokawa H, McMurtry IF, Oka M. Inhaled Rho kinase inhibitors are potent and selective vasodilators in rat pulmonary hypertension. Am J Respir Crit Care Med. 2005;171:494–499. doi: 10.1164/rccm.200405-637OC. [DOI] [PubMed] [Google Scholar]