

Summary
Background
Hyperthermia is one of the main disturbances of homeostasis occurring during sepsis or hypermetabolic states such as cancer. Platelets are important mediators of the inflammation that accompany these processes, but very little is known about the changes in platelet function that occur at different temperatures.
Objectives
To explore the effect of higher temperatures on platelet physiology.
Methods
Platelet responses including adhesion, spreading (fluorescence microscopy), αIIbbeta;3 activation (flow cytometry), aggregation (turbidimetry), ATP release (luminescence), thromboxane A2 generation, alpha-granule protein secretion (ELISA), and protein phosphorylation from different signaling pathways (immunoblotting) were studied.
Results
Preincubation of platelets at temperatures higher than 37°C (38.5°–42°C) inhibited thrombin-induced haemostasis including platelet adhesion, aggregation, ATP release, and thromboxane A2 generation. The expression of P-selectin and CD63, as well as vascular endothelial growth factor (VEGF) release were completely inhibited by hyperthermia, whereas von Willebrand factor (vWF) and endostatin levels remained substantially increased at high temperatures. This suggested that release of proteins from platelet granules is modulated not only by classical platelet agonists but also by microenvironmental factors. The observed gradation of response involved not only antiangiogenesis regulators, but also other cargo proteins. Some signaling pathways were more stable than others. While ERK1/2 and AKT phosphorylation were resistant to changes in temperature, Src, Syk, p38 phosphorylation as well as IkappaB degradation were decreased in a temperature-dependent fashion.
Conclusions
Higher temperatures, such as those observed with fever or tissue invasion, inhibit the haemostatic functions of platelets and selectively regulate the release of alpha-granule proteins.
Keywords: hyperthermia, temperature, platelets, inflammation, alpha-granule secretion, platelet aggregation
Introduction
The primary function of platelets is to initiate and maintain haemostasis. However, their function is not limited to thrombosis, and extends into physiological processes such as tissue repair, wound remodeling and antimicrobial host defense, or pathologic processes such as atherosclerosis, chronic ischemia or cancer [1–4]. In all these processes, platelets act as sentinels capable of responding instantly to chemical changes in their environment. The effect of platelets is protective and their absence can further exacerbate the injury to tissues [5]. A great variety of molecules are released from platelet granules upon activation, and these include prothrombotic substances, growth factors, chemokines and proteases. Many of the substances contained in platelets have physiologically opposing functions. It is not surprising that platelets would secrete these proteins selectively, and only in response to tissue signals.
The role of platelets in inflammation is strongly influenced by reciprocal interaction between the cellular and humoral components of the microenvironment. As activated leukocytes and endothelial cells express adhesion molecules on their luminal surfaces, and as more inflammatory mediators (cytokines, chemokines, nitric oxide, and reactive oxygen species) are secreted into the microenvironment, the sum of the various stimuli leads to either a positive platelet response (an enhancement of inflammation) or a negative one (an inhibition of inflammation) [6, 7]. Although the typical attributes of the inflammatory microenvironment include acidosis, hypoxia, and hyperthermia, there is little information as to the effect of these environmental stimuli on platelet function. It is generally accepted that hypothermia is associated with an increased risk of bleeding and may contribute to the morbidity and mortality in trauma and during complicated surgical procedures [8, 9]. Nevertheless, the exact manner in which hypothermia affects platelet function remains controversial because low temperatures have been shown to both inhibit [8, 9] or stimulate [10, 11] thrombosis. In terms of platelet transfusion, lowering storage temperature has several benefits among which a bacteriostatic effect. However, platelet refrigeration remains impossible, because platelets desialyted due to chilling or sepsis are cleared in the liver by macrophages [12, 13]. New evidence shows that galactosylation restores the circulation of short-term chilled murine platelets, but not of long-term refrigerated platelets [12].
Regarding hyperthermia, there is less reliable information about the impact of higher temperatures on the function of platelets. The available studies have focused on optimization of storage conditions for transfusional platelet products and on refining aggregation techniques, rather than on the biological consequences of this temperature sensitivity. The generally accepted conclusion from these studies is that storage of platelets at temperatures < 37°C induces platelet activation whereas temperatures > 37°C exert the opposite effect [14, 15]. Yet, the in vivo responses of platelets to changes in temperature may be of far reaching consequence. A recent study focusing on the thermo-mechanical behavior of thoracic aortas in New Zealand White rabbits with different degrees of atherosclerosis, demonstrated that increases of corporal temperature, both locally or systemically, plays an important role in increasing the risk of acute coronary syndromes [16]. The thermographic assessment of an atherosclerotic plaque (a surrogate indicator of the plaque metabolic state) has become an accepted predictive marker of vulnerable plaque at risk of rupture [17].
Similarly, heat stroke, a life-threatening state characterized by the rise of core body temperature above 40°C, is associated with a systemic inflammatory response leading to multiorgan dysfunction including abnormal coagulation [18]. Epidemiologic studies indicate that from 1979 to 1997, more than 7,000 deaths in United States were attributable to excessive heat, especially among the vulnerable population of the very young and elderly [19]. In Saudi Arabia, the incidence of heat stroke increases 10 times at summer season and the mortality rate is estimated at 50 percent [18]. While the haemorrhagic manifestations and microvessel thrombosis are widespread and common in patients with severe heat stroke [20], the pathogenesis of this phenomenon is not fully understood.
Local changes in temperature are also typical during acute inflammatory processes of hypermetabolic states such as tumor growth. To improve the understanding about the local and systemic effect of increased temperature on platelet physiology, we have exposed human platelets to varying temperatures. We found that exposure of platelets to heat stress resulted in an impairment of platelet spreading, activation of αIIbβ3, aggregation, ATP release and thromboxane A2 (TXA2) formation. In addition, contrary to the present dogma that platelets “degranulate” and release their contents en masse, the content of platelet alpha-granules was altered very selectively, and was dependent on inhibition of tyrosine kinases, p38 and nuclear factor-kappa B (NF-kappaB) signaling pathways.
Materials and methods
Cell preparation
Blood samples were obtained in plastic tubes containing sodium citrate (3.8%) from healthy donors who had not taken nonsteroidal anti-inflammatory drugs for at least 10 days prior to sampling. The study was performed according to institutional guidelines (National Academy of Medicine, Buenos Aires, Argentina) and received the approval of the institutional ethics committee. A written consent was obtained from all subjects. PRP was obtained by centrifugation at 180 x g for 10 minutes. Washed platelet suspensions (WPs) prepared as previously described [21], were resuspended in Tyrode’s buffer at a concentration of 4×108/ml, unless otherwise stated. CaCl2 (1 mM) was added before platelet stimulation.
Flow cytometry
To evaluate P-selectin and CD63 expression, platelets pre-incubated at different temperatures were stimulated, fixed and stained with a FITC-conjugated CD62P (anti P-selectin antibody), anti CD63 antibody or matched IgG isotype as a negative control (BD Biosciences, San Jose, CA, USA). Flow cytometry analysis was performed on a FACSCalibur flow cytometer using FCS Express V3 software. A similar technique was employed to evaluate CD61 expression (BD Biosciences) and αIIbβ3 integrin activation by using FITC-conjugated PAC1 (BD Biosciences) or Alexa488-conjugated fibrinogen (Invitrogen, Eugene, OR, USA). Results are expressed as percentage of positive cells.
Platelet adhesion and spreading on fibrinogen
WPs (5×107/ml) were plated on fibrinogen (100μg/ml)-coated slides (Sigma) for 20 minutes. Adhered platelets were fixed, permeabilized, stained with TRITC-Phalloidin, mounted, and visualized using fluorescent microscopy [22].
Platelet aggregation and ATP release
Aggregation and ATP release were measured simultaneously in a Lumi-aggregometer (Chrono-Log, Havertown, PA, USA) as previously described [22]. Aggregation was induced by thrombin, arachidonic acid (AA), ADP (Sigma), or collagen (Nycomed Pharma, Unterschleibheim, Germany). ATP levels were calculated at the end of the assay by adding a known amount of ATP (Sigma). Results are expressed as the percentage of maximal light transmission.
Measurement of Thromboxane B2 (TXB2) release
WPs were incubated with ADP for 5 minutes in an aggregometer chamber stirring at 1000 rpm. The addition of ice-cold PBS containing EDTA (2 mM) and aspirin (500 μM) was used to stop the reaction. The samples were centrifuged and TXB2 was measured in the supernatants using an ELISA kit from Cayman Chemical (Ann Arbor, MI, USA).
Measurement of von Willebrand factor (vWF), vascular endothelial growth factor (VEGF), and endostatin levels
WPs were stimulated with thrombin for 5, 15 or 30 minutes. The reaction was stopped by the addition of PBS containing prostacyclin (75 nM) (Cayman). Samples were centrifuged twice at 1100 x g for 5 minutes and then at 9300 x g for 5 minutes and supernatants stored at −80°C until assayed for vWF (Dako, Glostrup, Denmark), VEGF, and endostatin (RayBiotech Inc, Norcross, GA, USA) levels by ELISA.
Immunoblotting
WPs were lysed (1–2×109/ml) in loading buffer in the presence of a protease inhibitor cocktail (Sigma). Equivalent amounts of proteins were subjected to electrophoresis on a 12% SDS-PAGE and electrotransferred to nitrocellulose membranes (GE Healthcare, Buckinghamshire, UK). After blocking, the membranes were incubated overnight at 4°C with primary antibodies (pSrc-Tyr416, pSyk-Tyr525/526, pp38-Thr180/Tyr182 from Cell Signaling, Danvers, MA, USA), pERK E-4 and pAkt1/2/3-Ser473 from Santa Cruz CA, USA, followed by an HRP-linked secondary antibody (Santa Cruz Biotechnology) for 1 hour at 22°C. Protein bands were visualized by using the ECL reaction. Immunoblotting results were semi-quantitated using Gel-Pro analyzer 3.1 software and values from blot reprobes were used for normalization of data for protein loads.
Statistical analysis
Results were expressed as means ± SEM and analyzed by one-way analysis of variance followed by Newman-Keuls multiple comparison test to determine significant differences between groups. P values lower than 0.05 were considered statistically significant.
Results
Inside-out platelet signaling is decreased at high temperature
In order to improve the understanding of the effect of hyperthermia on platelet physiology, several responses resulting from platelet activation were evaluated at different temperatures ranging from the physiological 37°C to 42°C. The interaction of a soluble platelet agonist with its receptor leads to a cascade of intracellular events, broadly classified as inside-out signaling. The subsequent induction of conformational changes of the αIIbβ3 integrin, leads to exposure of neoepitopes permissible to fibrinogen binding [23]. We found that thrombin-induced αIIbβ3 activation, as reflected by lower PAC1 and fibrinogen binding, is down-regulated in a temperature-dependent fashion (Fig. 1). To rule out that the decreased fibrinogen binding was due to a temperature dependent alteration of either the fibrinogen molecule or the fluorophore, Alexa 488-conjugated fibrinogen alone was preincubated at 37°, 40° or 42°C for 10 minutes, before adding it to the platelets stimulated with thrombin at room temperature. The percentage of fibrinogen binding obtained after staining with the pre-warmed fibrinogen molecule was similar to the values obtained with a non pre-warmed one (Fig. S1A). The same control experiment was performed with the FITC-conjugated PAC1 antibody, and, again, neither the FITC fluorochrome nor the antibody was affected by the temperature increases (data not shown). In addition, no changes in fibrinogen binding were observed when platelets at RT were stimulated with pre-warmed (37°C–42°C) thrombin (data not shown), indicating that the decreased activation of αIIbβ3 integrin is due to a defect on platelet function rather than a reagent alteration/degradation.
Fig. 1.
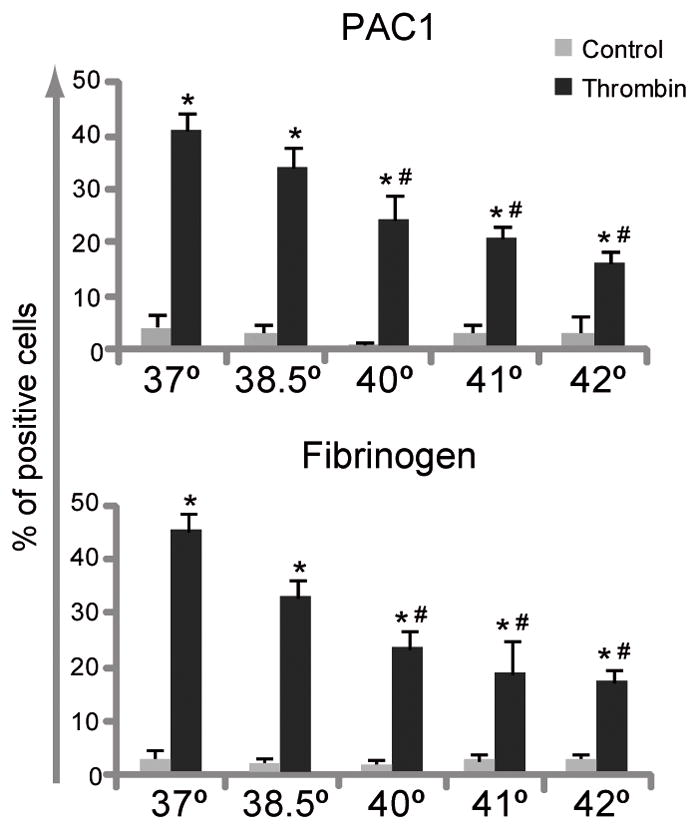
Heat exposure negatively regulated thrombin-induced αIIbβ3 activation. WPs were incubated at 37°, 38.5°, 40°, 41° or 42°C for 10 minutes and then stimulated with thrombin (0.05 U/ml) for 5 minutes. Analysis of PAC-1 and fibrinogen binding were performed by flow cytometry (n=6). *P < 0.05 vs. unstimulated control; #P < 0.05 vs. thrombin at 37°C.
Effect of temperature on platelet adhesion and spreading
The development of lamellipodia and filopodia was used as a surrogate of the interaction between glycoprotein αIIbβ3 and its binding partners. Platelets attachment and spreading is understood to be a direct consequence of the outside-in signaling triggered by αIIbβ3 interaction with its ligand. It represents one of the essential steps for the initiation of haemostasis and thrombosis [23]. Even though higher temperatures did not dramatically affect the adhesion of platelets to fibrinogen-coated surfaces, temperatures of 40°C and 42°C completely inhibited platelet spreading and lamellipodia formation seen at the physiological temperature of 37°C (Fig. 2).
Fig. 2.

High temperature inhibits platelet spreading. WPs were incubated at 37°, 40° or 42°C for 10 minutes and then plated on BSA- or fibrinogen-coated slides and stained with TRITC-Phalloidin. Platelet spreading was visualized under fluorescent microscopy (original magnification 600×). Platelet perimeter and number per field were measured using Image-Pro software (n=4). #P < 0.01 vs. 37°C.
Inhibition of platelet aggregation, ATP release and TXA2 formation by high temperatures
Having shown that hyperthermia decreased fibrinogen and PAC1 binding, we went on to examine the effect of temperature on platelet aggregation. Figure 3 reveals a temperature and agonist sensitive pattern of platelet aggregation. At 40°C, the low concentrations of arachidonic acid (AA) and ADP have close to a full effect on platelet aggregation (Fig. 3B and C), whereas thrombin, collagen and higher AA concentrations reach this level of inhibition only at temperature of 42°C (Fig. 3A and B). The degree of inhibition of platelet aggregation was in all cases associated to the ATP release. As it is shown in Fig. 3C, the marked reduction in platelet aggregation by ADP (2 μM) was accompanied with a complete inhibition of ATP release. In the same line, the moderate inhibition of the aggregation response induced by thrombin, collagen or AA (0.5 mM) at 40°C was parallel to a moderate decrease in ATP release (data not shown).
Fig. 3.

Inhibition of platelet aggregation, ATP release and TXB2 formation at high temperatures. (A) WPs or (B) PRP were incubated at 37°, 40° or 42°C for 10 minutes and then aggregation was induced by the indicated agonists. Graphics are representative of 4 independent experiments. (C) PRP was incubated at 37°, 40° or 42°C for 10 minutes and then stimulated with ADP. Aggregation and ATP release were determined simultaneously in a Lumi-aggregometer (n=4). After 5 minutes stimulation, supernatants were collected to measure TXB2 levels by ELISA. *P < 0.05 vs. unstimulated control. #P < 0.05 vs. thrombin at 37°C (n = 3).
Although the inhibitory effect of the 40°C temperature on platelet aggregation could be overcome by higher concentrations of ADP (5μM),even in the absence of TXA2 and ATP (data not shown), this gain achieved by higher dose of ADP was lost at 42°C (Fig. 3C). The more potent agonists of platelet aggregation, such as thrombin and high collagen concentrations were less affected by temperature. Thus, there was a very graded reaction to temperature for each of the individual agonists, as well as a very typical and distinct pattern of response by the various tested agonists.
Differential regulation of platelet alpha-granule release by temperature
The activation of various signaling cascades during platelet activation ultimately leads to release of the contents of dense and alpha-granules. The generally accepted paradigm that “degranulation” of platelet alpha granules leads to secretion of most substances en mass is presently being revised to reflect a more sequential and selective release of granular content in tissues [24–26]. Platelet alpha granule release at the site of vascular injury is now thought to be regulated by many biological processes including inflammation [27]. Our experiments were in agreement with this new understanding. While a remarkable release of all of these proteins normally stored in platelet granules can be induced by thrombin at 37°C, higher temperatures of 40°C and 42°C temper this dramatic release (Fig. 4). There was a significant variability in the grade of the response. For example, exposure of P-selectin on the platelet surface and the release of VEGF was dramatically inhibited at 40°C and almost completely suppressed at 42°C. In contrast, the inhibition of thrombin-induced vWF or endostatin release was less dramatic and more gradual (Fig. 4A). In fact, while P-selectin and VEGF levels triggered by thrombin at 42°C are almost not different from those found in unstimulated platelets, vWF and endostatin levels are still significantly raised by thrombin.
Fig. 4.

Effect of hyperthermia on platelet alpha-granule secretion induced by thrombin. WPs were preincubated at 37° (dark line), 40° (grey line) or 42°C (dashed line) for 10 minutes and then stimulated with thrombin (0.05 U/ml) for 5 minutes (A) or at the indicated time (B). P-selectin was detected by flow cytometry (n=10). vWF, VEGF, and endostatin levels in the supernatants were quantified by ELISA (n=5). Since the basal levels of each protein release in unstimulated controls were not affected by the temperature results are expressed as fold increase vs. unstimulated controls at the same temperature. *P < 0.05 vs. unstimulated control; #P < 0.05 vs. thrombin at 37°C.
Since during the later secretory stages platelets undergo contraction and increased granule release into the medium, we next examined the effect of temperature after 15 and 30 minutes thrombin activation, respectively. Among the different proteins evaluated under these conditions, only the release of VEGF was slightly increased after 30 minutes compared to 5 minutes stimulation. While the secretion of P-selectin and VEGF was markedly decreased with higher temperatures, the inhibition of vWF and endostatin was less dramatic regardless platelet stimulation by thrombin was performed during 5, 15 or 30 minutes (Fig. 4B). Since P-selectin is found not only in alpha but also in dense granules [28], we analyzed the expression of CD63, an ubiquitous protein found mainly in the membrane of lysosomal, dense but also in alpha granules to some extent [29]. We found that like P-selectin, hyperthermia negatively regulates CD63 expression (Fig. S1B).
Effect of temperature on platelet signaling
To explore the molecular changes that accompany the temperature-induced changes in platelet function, we measured the degree of phosphorylation of proteins participating in the activation of different signaling pathways. We found that thrombin caused a robust phosphorylation of all kinases tested (ERK1/2, p38, AKT, Syk and Src) in a concentration-dependent manner. While the degree of thrombin-induced phosphorylation was not modified by temperature in the case of pERK1/2 and pAKT, p38, Syk and Src phosphorylation levels were significantly reduced at 40°C and completely abolished at 42°C (Fig. 5A and B). It has been shown that heat shock proteins are present in platelets and several members of this family mediate platelet function such as adhesion, aggregation, and granule release [30]. Considering that phosphorylation of HSP27 is downstream to and catalyzed by p38 MAPK [31], we examined the effect of temperature on pHSP27. The heat-mediated negative regulation of p38 phosphorylation, thrombin-induced phosphorylation of HSP27 was decreased in platelets exposed to high temperature (Fig. 5A).
Fig. 5.

Effect of temperature on platelet signaling. WPs were preincubated at 37°, 40° or 42°C for 10 minutes. (A) WPs were stimulated with the indicated concentration of thrombin for 5 minutes and lysates were immunoblotted with anti-pp38, pERK1/2, pHSP27 or pAKT antibody. (B) WPs were stimulated with thrombin for 2 minutes and lysates were immunoblotted with anti-pSyk and pSrc antibody. Each membrane was reprobed with anti-actin antibody to calculate the relative IOD using Gel-Pro software (n=3–5). *P < 0.05 vs. thrombin at 37°C.
We have recently shown that platelets express the NF-kappaB machinery and that the activation of this transcription factor is involved in platelet activation [21]. We now observed that similarly to p38, I-kappaB degradation, a measure of NF-kappaB activation, was dampened as temperature increased (Fig. 5A).
In view of these findings, we choose to analyze the role of p38 and the recently described NF-kappaB pathway in the temperature-mediated differential release of alpha-granule contents by using specific inhibitors of p38 [SB203580] and NF-kappaB [RO1069920] [21] at concentrations that have been confirmed to completely inhibit their phosphorylation (Fig. S1C). We found that the individual inhibitors failed to produce a statistically significant inhibition of the thrombin-induced release of alpha-granule proteins (Fig. 6), but the combination of p38 and NF-kappaB inhibition led to a statistically significant inhibition of P-selectin expression, VEGF release, vWF and endostatin (Fig. 6). Conversely, inhibition of pERK1/2 or PI3K/pAKT (by U0126 or LY294002 and Wortmannin respectively) failed to regulate the release/expression of alpha-granule proteins (data not shown) although they efficiently suppressed phosphorylation of ERK1/2 or AKT (Fig. S1C).
Fig. 6.

Inhibition of p38 and NF-kappaB pathways mimics the effect of heat exposure on platelet alpha-granule secretion. WPs were preincubated with SB203580 (25 μM), RO1069920 (6 μM) or the combination of both for 10 minutes and then stimulated with thrombin (0.05 U/ml) for 5 minutes. P-selectin was detected by flow cytometry (n=5). vWF, VEGF, and endostatin levels in the supernatants were quantified by ELISA (n=4). Results are expressed as fold increase vs. unstimulated controls. *P < 0.05 vs. unstimulated control; #P < 0.05 vs. thrombin alone.
Discussion
Platelet homeostasis and function is maintained within a very narrow physiological range of temperature. The understanding, however, suggests that this “sensitivity” of platelets to temperature has more to do with platelet fragility than with a regulation of their function. We would like to propose that the sensitivity of platelets to external stimuli such as changes in temperature may be an evolutionally beneficial mechanism of facilitating a selective and sequential release of factors needed for inflammatory and angiogenic processes during development, tissue regeneration, wound healing or tumor growth. Our studies provide early evidence that platelet exposure to temperatures higher than the physiological temperature of 37°C but within the pathological range of in vivo events, inhibit aggregation in a dose-dependent fashion. The pattern of platelet aggregation response to temperature is specific to individual agonists (Fig. 3B). Smaller rises in temperature (at 40°C) and low doses of arachidonic acid and ADP result in close to maximal inhibitory effect on platelet aggregation, suggesting that these acute inflammatory responders are responsible for early platelet response. In contrast, thrombin and collagen reach this higher level of aggregation inhibition only at temperatures of 42°C. Of note is that thrombin and collagen are deposited in tissues in late stages of tissue organization and in chronic wounds and this differential response to different agonists at different temperatures provides means to fine-tune a tissue response. The dose dependent response to hyperthermia and the agonist-dependent platelet response on platelet aggregation may provide clues as to the meticulous orchestration of early and late stages of wound healing.
Several other platelet responses including spreading, PAC1 and fibrinogen binding, and ATP release were similarly regulated in resting and thrombin-activated platelets exposed to varying physiologically relevant temperatures. Interestingly, while preincubation of thrombin or fibrinogen at 40°C or 42°C failed to modify the platelet ability to bind fibrinogen, other thrombin-induced parameters such as phosphorylation of ERKs and AKT were not modified by temperature variations (see Fig. 5) pointing out that hyperthermia is affecting platelet function rather than agonists or reagent quality/integrity.
The fact that hyperthermia dampened the hemostatic function of platelets would suggest that high fever should lead to hemorrhage. Although the mechanisms of bleeding have been extensively explored in the hemorrhagic fever related diseases, no deep analysis regarding the linkage between platelet function and body temperature has been performed in those patients. However, there are evidences suggesting that an abnormal platelet function is found on patients with fever since it has been reported that while the threshold for prophylactic transfusions in cancer patients can safely be set at 5 × 109/l in the absence of fever, the threshold should be at least 20 × 109/l in those patients with fever > 38° C or under hyperthermia therapy to avoid bleeding [32]
The newly revised paradigms of platelet selective “alpha granule degranulation” [25, 33], and the concept that a sequential platelet granule release at the site of vascular injury [26, 27], contributes to the up- or down-regulation of processes such as inflammation and angiogenesis [34] was supported by our findings. We showed that exposure of P-selectin on the platelet surface and the release of VEGF were dramatically inhibited at 40°C and almost completely suppressed at 42°C (Fig. 4), suggesting that these acute phase reactants are early and transient responders. In contrast, the inhibition of thrombin–induced vWF or endostatin release at 40°C and 42°C was much less dramatic suggesting a lingering pattern of their release. A possible explanation for this differential response is that higher temperatures such as those occurring in high metabolic states (burns, wounds or tumors), may require ongoing activity of stromal (vWF) and inhibitory proteins (endostatin), whereas chemoatractants (P-selectin) and inducers of angiogenesis (VEGF) are required for the initial induction of inflammatory chemotaxis and angiogenesis. In addition, it might be possible that released granule components have (transient) interaction with binding partners on the surface of the open canalicular system.
Interestingly, it has been shown previously that vWF colocalizes with endostatin but not with VEGF, and that a distinct and separate organization exists for the different proteins within the alpha-granules [25]. Our findings indicate that the differential granule release is regulated not only by tissue platelet agonists but also by changes in microenvironmental factors such as temperature. The different regulatory effect of temperature on P-selectin expression and vWF release is intriguing since both proteins appeared to be stored in similar alpha granules. Using 3D analysis and combined immune-electronmicroscopy characterization, van Nispen tot Pannerden H et al [29] suggested that besides the existence of morphologically different alpha-granule subtypes (tubular and spherical), a complex compartmentalization and a high spatial segregation of cargo exists within individual α-granules. This leads us to speculate that soluble and membrane-bound proteins appear highly compartmentalized within individual alpha granules and its release may be governed by different mechanisms. In fact, we found that CD63, another granule membrane bound protein followed a temperature-inhibition pattern similar to P-selectin (Fig. S1C). To what extent differences in granule morphology and spatial protein segregation within individual alpha granules contribute to the observed temperature-dependent differential secretory activity of platelet alpha granules contents remains to be determined.
Local hyperthermia is used to enhance the effectiveness of cancer treatments such as ionizing radiation [35]. Platelets are crucial regulators of tumor vascular homeostasis by delivering angiogenesis-modulating factors to the tumor site [36]. Our results showed that high temperatures tempered the release of VEGF while sparing the secretion of the anti-angiogenic endostatin. The control of platelet activation might therefore be one of the physiological mechanisms involved in local hyperthermia effect during radiation therapy.
A deeper analysis of the mechanisms involved in the hyperthermia-mediated inhibition of platelet aggregation revealed that lack of aggregation after ADP stimulation was associated with a robust decrease in TXA2 generation and in p38 phosphorylation. It is well known that pp38 activates cyclooxygenase-1, -2 and thromboxane synthase [37], suggesting that the cyclo-oxygenase-1, and/or thromboxane synthase may be the main targets of the hyperthermia–mediated effects on AA metabolism.
In addition to the extensively studied PI3K/AKT pathway in platelets [38, 39], the multiple MAPK and Src family kinases (SFKs) involving pathways have now become the focus of growing research. Evidence now exists that ERK1/2, p38 and JNK1 are present in platelets and become phosphorylated in response to platelet stimulation with thrombin, ADP, and collagen among others [40]. Platelets express at least six SFKs, among which Fyn and Lyn are involved in the activation of platelets following binding of collagen or laminin to the platelet-specific GPVI/Fc receptor (FcR) c-chain complex. Another important platelet activation pathway that involves SFKs, specifically Src itself and Fyn, is that of outside-in signaling by the platelet-specific integrin, αIIbβ3 [41]. To study the molecular changes associated with physiological hyperthermia effect on platelet biology, we analyzed the aforementioned pathways in platelets incubated at 37°C, 40°C and 42°C. We have found that even though thrombin-induced phosphorylation of ERK1/2 and AKT were not modified by higher temperatures, pSrc, pSyk and p38 activation was completely abolished. Similar results were reported in synoviocyte, where short-term hyperthermia prevented IL-1β-induced p38 phosphorylation but not pERK1/2 or pAKT [42]. Moreover, heat-mediated inhibition of the small HSP27 phosphorylation, one of the main targets of active p38 in human platelets, further confirmed the negative regulation of high temperature on the p38 signaling pathway [30].
Although platelets are anucleate cellular structures, they express several transcription factors that exert non-genomic functions, including the positive and negative regulation of platelet activation. We [21] have along with others [43, 44] previously demonstrated that NF-kappaB was expressed in platelets and its pharmacological inhibition by RO1069920 suppressed different platelet activation responses including P-selectin expression and vWF release. A combined pharmacological blockade of pp38 and NF-kappaB by SB203580 and RO1069920 respectively, mimicked the effect of temperature on alpha-granule release. The thrombin-induced P-selectin expression and VEGF release were completely suppressed by the synergistic action of the dual pathway inhibition, whereas vWF and endostatin levels remained elevated, suggesting that the differential effect of temperature on alpha-granule release may be linked to a selective inhibition of p38 and NF-kappaB signaling pathways.
In conclusion the sum of effects of physiologically relevant hyperthermia on platelets presented above suggests that the sensitivity of platelets to temperature may be of pathophysiological relevance. We demonstrated that exposure to higher temperatures had a negative effect on platelet haemostatic function, and that the release of alpha-granule contents varied with increasing temperatures. This graded response may be reflective of a carefully orchestrated tissue-controlled response by platelets rather than platelet fragility. The recognition of this phenomenon may aid future development of therapeutic agents, which are presently tested at ambient temperatures.
Supplementary Material
Acknowledgments
The authors are grateful to the CEMIC technicians for collecting the blood samples. GLK is supported by NIH/NCI R01 GM093050, ASA/NSCOR NNJ06HA28G and DOE DE-SC0002606 grants, SN by PIP 1142009010016301 and MS by ANPCYT PICTs 1990-06 and 230-08 grants.
References
- 1.Lindemann S, Kramer B, Seizer P, Gawaz M. Platelets, inflammation and atherosclerosis. J Thromb Haemost. 2007;5 (Suppl 1):203–11. doi: 10.1111/j.1538-7836.2007.02517.x. [DOI] [PubMed] [Google Scholar]
- 2.Wang Y, Zhang H. Platelet-induced inhibition of tumor cell growth. Thromb Res. 2008;123:324–30. doi: 10.1016/j.thromres.2008.06.021. [DOI] [PubMed] [Google Scholar]
- 3.McNicol A, Israels SJ. Beyond hemostasis: the role of platelets in inflammation, malignancy and infection. Cardiovasc Hematol Disord Drug Targets. 2008;8:99–117. doi: 10.2174/187152908784533739. [DOI] [PubMed] [Google Scholar]
- 4.Yeaman MR. Bacterial-platelet interactions: virulence meets host defense. Future Microbiol. 5:471–506. doi: 10.2217/fmb.09.112. [DOI] [PubMed] [Google Scholar]
- 5.Fujimi S, MacConmara MP, Maung AA, Zang Y, Mannick JA, Lederer JA, Lapchak PH. Platelet depletion in mice increases mortality after thermal injury. Blood. 2006;107:4399–406. doi: 10.1182/blood-2005-09-3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wagner DD, Frenette PS. The vessel wall and its interactions. Blood. 2008;111:5271–81. doi: 10.1182/blood-2008-01-078204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wagner DD. New links between inflammation and thrombosis. Arterioscler Thromb Vasc Biol. 2005;25:1321–4. doi: 10.1161/01.ATV.0000166521.90532.44. [DOI] [PubMed] [Google Scholar]
- 8.Wolberg AS, Meng ZH, Monroe DM, 3rd, Hoffman M. A systematic evaluation of the effect of temperature on coagulation enzyme activity and platelet function. J Trauma. 2004;56:1221–8. doi: 10.1097/01.ta.0000064328.97941.fc. [DOI] [PubMed] [Google Scholar]
- 9.Michelson AD, MacGregor H, Barnard MR, Kestin AS, Rohrer MJ, Valeri CR. Reversible inhibition of human platelet activation by hypothermia in vivo and in vitro. Thromb Haemost. 1994;71:633–40. [PubMed] [Google Scholar]
- 10.Xavier RG, White AE, Fox SC, Wilcox RG, Heptinstall S. Enhanced platelet aggregation and activation under conditions of hypothermia. Thromb Haemost. 2007;98:1266–75. doi: 10.1160/th07-03-0189. [DOI] [PubMed] [Google Scholar]
- 11.Zhang JN, Wood J, Bergeron AL, McBride L, Ball C, Yu Q, Pusiteri AE, Holcomb JB, Dong JF. Effects of low temperature on shear-induced platelet aggregation and activation. J Trauma. 2004;57:216–23. doi: 10.1097/01.ta.0000093366.98819.fe. [DOI] [PubMed] [Google Scholar]
- 12.Rumjantseva V, Grewal PK, Wandall HH, Josefsson EC, Sorensen AL, Larson G, Marth JD, Hartwig JH, Hoffmeister KM. Dual roles for hepatic lectin receptors in the clearance of chilled platelets. Nat Med. 2009;15:1273–80. doi: 10.1038/nm.2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoffmeister KM, Josefsson EC, Isaac NA, Clausen H, Hartwig JH, Stossel TP. Glycosylation restores survival of chilled blood platelets. Science. 2003;301:1531–4. doi: 10.1126/science.1085322. [DOI] [PubMed] [Google Scholar]
- 14.Han P, Ardlie NG. The influence of pH, temperature, and calcium on platelet aggregation: maintenance of environmental pH and platelet function for in vitro studies in plasma stored at 37 degrees C. Br J Haematol. 1974;26:373–89. doi: 10.1111/j.1365-2141.1974.tb00479.x. [DOI] [PubMed] [Google Scholar]
- 15.Scharbert G, Kalb M, Marschalek C, Kozek-Langenecker SA. The effects of test temperature and storage temperature on platelet aggregation: a whole blood in vitro study. Anesth Analg. 2006;102:1280–4. doi: 10.1213/01.ane.0000199399.04496.6d. [DOI] [PubMed] [Google Scholar]
- 16.Guinea GV, Atienza JM, Fantidis P, Rojo FJ, Ortega A, Torres M, Gonzalez P, Elices ML, Hayashi K, Elices M. Effect of atherosclerosis on thermo-mechanical properties of arterial wall and its repercussion on plaque instability. Int J Cardiol. 2009;132:444–6. doi: 10.1016/j.ijcard.2007.08.087. [DOI] [PubMed] [Google Scholar]
- 17.Schaar JA, Mastik F, Regar E, den Uil CA, Gijsen FJ, Wentzel JJ, Serruys PW, van der Stehen AF. Current diagnostic modalities for vulnerable plaque detection. Curr Pharm Des. 2007;13:995–1001. doi: 10.2174/138161207780487511. [DOI] [PubMed] [Google Scholar]
- 18.Bouchama A, Knochel JP. Heat stroke. N Engl J Med. 2002;346:1978–88. doi: 10.1056/NEJMra011089. [DOI] [PubMed] [Google Scholar]
- 19.Heat-related illnesses, deaths, and risk factors--Cincinnati and Dayton, Ohio, 1999, and United States, 1979-1997. MMWR Morb Mortal Wkly Rep. 2000;49:470–3. [PubMed] [Google Scholar]
- 20.Roberts GT, Ghebeh H, Chishti MA, Al-Mohanna F, El-Sayed R, Al-Mohanna F, Bouchama A. Microvascular injury, thrombosis, inflammation, and apoptosis in the pathogenesis of heatstroke: a study in baboon model. Arterioscler Thromb Vasc Biol. 2008;28:1130–6. doi: 10.1161/ATVBAHA.107.158709. [DOI] [PubMed] [Google Scholar]
- 21.Malaver E, Romaniuk MA, D’Atri LP, Pozner RG, Negrotto S, Benzadon R, Schattner M. NF-kappaB inhibitors impair platelet activation responses. J Thromb Haemost. 2009;7:1333–43. doi: 10.1111/j.1538-7836.2009.03492.x. [DOI] [PubMed] [Google Scholar]
- 22.Romaniuk MA, Tribulatti MV, Cattaneo V, Lapponi MJ, Molinas FC, Campetella O, Schattner M. Human platelets express and are activated by galectin-8. Biochem J. 2010;432:535–47. doi: 10.1042/BJ20100538. [DOI] [PubMed] [Google Scholar]
- 23.Shattil SJ, Newman PJ. Integrins: dynamic scaffolds for adhesion and signaling in platelets. Blood. 2004;104:1606–15. doi: 10.1182/blood-2004-04-1257. Epub 2004 Jun 17. [DOI] [PubMed] [Google Scholar]
- 24.Sehgal S, Storrie B. Evidence that differential packaging of the major platelet granule proteins von Willebrand factor and fibrinogen can support their differential release. J Thromb Haemost. 2007;5:2009–16. doi: 10.1111/j.1538-7836.2007.02698.x. [DOI] [PubMed] [Google Scholar]
- 25.Italiano JE, Jr, Richardson JL, Patel-Hett S, Battinelli E, Zaslavsky A, Short S, Ryeom S, Folkman J, Klement GL. Angiogenesis is regulated by a novel mechanism: pro- and antiangiogenic proteins are organized into separate platelet alpha granules and differentially released. Blood. 2008;111:1227–33. doi: 10.1182/blood-2007-09-113837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma L, Perini R, McKnight W, Dicay M, Klein A, Hollenberg MD, Wallace JL. Proteinase-activated receptors 1 and 4 counter-regulate endostatin and VEGF release from human platelets. Proc Natl Acad Sci U S A. 2005;102:216–20. doi: 10.1073/pnas.0406682102. 0406682102 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma L, Elliott SN, Cirino G, Buret A, Ignarro LJ, Wallace JL. Platelets modulate gastric ulcer healing: role of endostatin and vascular endothelial growth factor release. Proc Natl Acad Sci U S A. 2001;98:6470–5. doi: 10.1073/pnas.111150798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Israels SJ, Gerrard JM, Jacques YV, McNicol A, Cham B, Nishibori M, Bainton DF. Platelet dense granule membranes contain both granulophysin and P-selectin (GMP-140) Blood. 1992;80:143–52. [PubMed] [Google Scholar]
- 29.van Nispen tot Pannerden H, de Haas F, Geerts W, Posthuma G, van Dijk S, Heijnen HF. The platelet interior revisited: electron tomography reveals tubular alpha-granule subtypes. Blood. 2010;116:1147–56. doi: 10.1182/blood-2010-02-268680. [DOI] [PubMed] [Google Scholar]
- 30.Polanowska-Grabowska R, Gear AR. Heat-shock proteins and platelet function. Platelets. 2000;11:6–22. doi: 10.1080/09537100075742. [DOI] [PubMed] [Google Scholar]
- 31.Benjamin IJ, McMillan DR. Stress (heat shock) proteins: molecular chaperones in cardiovascular biology and disease. Circ Res. 1998;83:117–32. doi: 10.1161/01.res.83.2.117. [DOI] [PubMed] [Google Scholar]
- 32.Gmur J, Burger J, Schanz U, Fehr J, Schaffner A. Safety of stringent prophylactic platelet transfusion policy for patients with acute leukaemia. Lancet. 1991;338:1223–6. doi: 10.1016/0140-6736(91)92098-m. [DOI] [PubMed] [Google Scholar]
- 33.Klement GL, Yip TT, Cassiola F, Kikuchi L, Cervi D, Podust V, Italiano JE, Wheatley E, Abou-Slaybi A, Bender E, Almog N, Kieran M, Folkman J. Platelets actively sequester angiogenesis regulators. Blood. 2009;113:2835–42. doi: 10.1182/blood-2008-06-159541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McGregor L, Martin J, McGregor JL. Platelet-leukocyte aggregates and derived microparticles in inflammation, vascular remodelling and thrombosis. Front Biosci. 2006;11:830–7. doi: 10.2741/1840. [DOI] [PubMed] [Google Scholar]
- 35.Horsman MR, Overgaard J. Hyperthermia: a potent enhancer of radiotherapy. Clin Oncol (R Coll Radiol) 2007;19:418–26. doi: 10.1016/j.clon.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 36.Ho-Tin-Noe B, Goerge T, Wagner DD. Platelets: guardians of tumor vasculature. Cancer Res. 2009;69:5623–6. doi: 10.1158/0008-5472.CAN-09-1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Borsch-Haubold AG, Pasquet S, Watson SP. Direct inhibition of cyclooxygenase-1 and -2 by the kinase inhibitors SB 203580 and PD 98059. SB 203580 also inhibits thromboxane synthase. J Biol Chem. 1998;273:28766–72. doi: 10.1074/jbc.273.44.28766. [DOI] [PubMed] [Google Scholar]
- 38.Jackson SP, Yap CL, Anderson KE. Phosphoinositide 3-kinases and the regulation of platelet function. Biochem Soc Trans. 2004;32:387–92. doi: 10.1042/bst0320387. [DOI] [PubMed] [Google Scholar]
- 39.Woulfe DS. Akt signaling in platelets and thrombosis. Expert Rev Hematol. 2010;3:81–91. doi: 10.1586/ehm.09.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adam F, Kauskot A, Rosa JP, Bryckaert M. Mitogen-activated protein kinases in hemostasis and thrombosis. J Thromb Haemost. 2008;6:2007–16. doi: 10.1111/j.1538-7836.2008.03169.x. [DOI] [PubMed] [Google Scholar]
- 41.Newman DK. The Y’s that bind: negative regulators of Src family kinase activity in platelets. J Thromb Haemost. 2009;7 (Suppl 1):195–9. doi: 10.1111/j.1538-7836.2009.03369.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stuhlmeier KM. Short term hyperthermia prevents the activation of mitogen-activated protein kinase p38. Exp Gerontol. 2009;44:406–12. doi: 10.1016/j.exger.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 43.Gambaryan S, Kobsar A, Rukoyatkina N, Herterich S, Geiger J, Smolenski A, Lohmann SM, Walter U. Thrombin and collagen induce a feedback inhibitory signaling pathway in platelets involving dissociation of the catalytic subunit of protein kinase A from an NFkappaB-IkappaB complex. J Biol Chem. 2010;285:18352–63. doi: 10.1074/jbc.M109.077602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Spinelli SL, Casey AE, Pollock SJ, Gertz JM, McMillan DH, Narasipura SD, Mody NA, King MR, Maggirwar SB, Francis CW, Taubman MB, Blumberg N, Phipps RP. Platelets and megakaryocytes contain functional nuclear factor-kappaB. Arterioscler Thromb Vasc Biol. 30:591–8. doi: 10.1161/ATVBAHA.109.197343. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.