

Abstract
The design of β-peptide foldamers targeting the transmembrane (TM) domains of complex natural membrane proteins has been a formidable challenge. A series of β-peptides was designed to stably insert in TM orientations in phospholipid bilayers. Their secondary structures and orientation in the phospholipid bilayer was characterized using biophysical methods. Computational methods were then devised to design a β-peptide that targeted a TM helix of the integrin αIIbβ3. The designed peptide (β-CHAMP) interacts with the isolated target TM domain of the protein, and activates the intact integrin in vitro.
The discovery of the β-peptide class of foldamers1–6 has resulted in the development of β- and mixed α–β peptides capable of binding specific biological targets.2,7,8 Considerable progress has been made in the design of β-peptides that compete for water-soluble protein-protein interactions, or that bind to the surface of membranes.2,7–11 However, the design of β-peptide foldamers that target the transmembrane (TM) domains of complex natural membrane proteins has been a formidable challenge, which is addressed in the present paper. Our first objective was to design β-peptides that stably insert in TM orientations in bilayers, and adapt biophysical methods to demonstrate their orientations. Next, computational methods were devised to design a β-peptide that targeted the αIIb TM helix of the integrin αIIbβ3. The designed peptide indeed interacts with the isolated target TM domain of the protein, and activates the intact integrin in vitro.
We have chosen the well characterized platelet integrin αIIbβ3 as a model system for our design.12 The αIIb and β3 TMs associate in the resting state of the integrin, but this interaction is disrupted when the integrin is activated to bind to its extracellular ligand, fibrinogen. Thus, disruption of the αIIb–β3 TM helical interaction – either by introducing disruptive mutations13–16 or by addition of α-peptides that compete for this interaction17–20 – leads to integrin activation. Our goal here was to design a β-peptide that binds specifically to the αIIb TM domain. This endeavor not only tests and extends our fundamental understanding of the mechanism of TM helix-helix interaction, but takes an important step towards the goal of designing non-natural molecules that specifically target TM regions of proteins in a sequence-specific manner.
Previously we developed the CHAMP method (computed helical anti-membrane protein) to enable computational design of α-peptides that target TM helices of natural proteins.18 The original CHAMP method uses the crystallographic database of membrane proteins to define interacting TM helical pairs, whose backbones are compatible with the target sequence and that serve as starting points for a sidechain repacking algorithm. As there are no experimental structures of TM β-peptides, let alone TM β-peptides interacting with α-peptides, it was necessary to discover an optimal pose of the β-peptide backbone onto the α-peptide, as well as to design a sequence of the β-peptide that stably interacts with the α-peptide in this pose. We began with the previous model of a complex of the αIIb with a successfully designed CHAMP peptide to guide the selection of the β-CHAMP sequence. In our previous work,18 the CHAMP peptide targeted a GXXXG motif in the αIIb sequence. The exposed backbone atoms of the Gly residues in this motif are recognized by a similar motif in the CHAMP peptide, resulting in inter-helical backbone-backbone interactions that help drive helix-association in membranes. Therefore, we positioned a β-peptide poly-homo-Gly (hGly) helix against the GXXXG of the αIIb helix, using a grid search and the CHARM force field to determine the optimal position of the β-helix against the α-helix.
There are two common helix types for β-amino acids: while the 14-helix is most stable for β3-substituted amino acids in polar solvents, preliminary studies showed the co-existence of a 12-helical conformation.21 Thus, both were considered in the design of the CHAMP peptide. In each case, multiple poses of the poly-hGly backbone were exhaustively sampled to discover the optimal backbone orientation. The sequence of the β-peptide was then outfitted with sidechains using a packing algorithm that evaluates various sidechains in low-energy rotamers to obtain combinations that optimize the geometric complementarity between the β-peptide and the α-helical target.22 The β-peptide positions that directly contacted the mainchain could not sterically accommodate a sidechain, and hence remained hGly. The design was completed by inclusion of Trp and Lys residues near the termini to properly orient the peptide in the bilayer. The initial design focused on the 14-helix and the backbone docking restraints induced an (hGly-X-X)3 motif with the hGly residues lined along one face of the helix. Somewhat surprisingly, this same motif shows a good fit for the 12-helix; the major difference is that the hGly residues line up along the helix in the 3-fold screw of the 14-helix, but spiral around the helix in the 12-helix. This difference leads to different predicted packing angles in the complex.
To test the designed binding mode, two control peptides were prepared. We disrupted the GXXGXXG motif by mutating the central β3-homo-glycine (hG14) to β3-homo-isoleucine (β-CHAMP G14I) and scrambled the β-CHAMP sequence (β-CHAMPscr). All β-peptides were synthesized using optimized microwave-assisted solid phase peptide synthesis techniques outlined in the Supporting Information, TM α-peptide was synthesized using a previously established protocol.23 The final sequences are shown in Table 1. Our protocols allow us to consistently obtain high purity products in high yield.
Table 1.
Sequences of the designed β-CHAMP peptides

|
One letter code refers to the corresponding β3-amino acid
The CD spectrum of β-CHAMP was measured in trifluoroethanol (TFE), dodecyl phosphatidylcholine (DPC) micelles, and in phospholipid vesicles composed of a mixture of palmitoyl oleoyl phosphatidylcholine (POPC) and palmitoyl oleoyl phosphatidylglycerol (POPG) (7:3) (Figure 2). In DPC micelles and phospholipid vesicles there were minima at 204 nm and 206 nm, respectively, and a maximum at ca. 220 nm, indicative of the 12-helical structure. In TFE, the β-CHAMP CD spectrum suggested an equilibrium between 12- and 14-helical structures, consistent with increasing destabilization of the 12-helix with increasing solvent polarity.21,24 Control β-CHAMP peptides followed the same pattern, showing higher 14-helical content in TFE.21 Also, when the β-CHAMP mixed with the αIIb TM peptide under conditions in which they formed a 1:1 complex (Figure 2), the spectrum was well described as a mixture of the TM α-helical and the 12-helical form of the β-peptide, strongly suggesting that the β-CHAMP binds its target in a 12-helical conformation.
Figure 2.

CD spectra of β-CHAMP in DPC micelles (blue circles, 40 μM peptide, 10 mM DPC, 10 mM phosphate buffer, pH 7.4), POPC/POPG (7:3) vesicles (green circles, 167 μM total lipid, 1:100 peptide:lipid), and in TFE (red squares, 40 μM peptide). CD spectrum of β-CHAMP in the presence of 1 equiv. of αIIb-TM (black diamonds, DPC micelles, 40 μM total peptide). The line represents linear combination of the 12-helical spectrum of β-CHAMP and the CD spectrum of αIIb-TM in equal proportions.
To determine the orientation of the β-CHAMP peptides in phospholipid bilayers, they were examined in hydrated multilayers by attenuated total internal reflectance IR. (Figure 3, Figure S2, Supporting Information). The order parameter of the amide I′ band (1646 cm−1) of β-CHAMP was 0.83, indicative of the helix being aligned close to the membrane normal.25–28
Figure 3.

FT-IR/ATR spectrum of β-CHAMP in POPC/POPG (7:3) lipid membranes (1:80 peptide:lipid ratio). The blue and red traces represent light polarized parallel and perpendicular, respectively, to the plane of incidence.
The binding of the β-CHAMP peptides to their target αIIb-TM was studied using analytical ultracentrifugation (AUC) in density matched DPC micelles. In order to exclude the contribution of αIIb TM absorbance we labeled the N-termini of the β-peptides with a 2,4-dinitrophenyl (DNP) group. Analysis of the sedimentation equilibrium profile at 356 nm allows one to monitor the radial distribution of the β-peptide, which is sensitive to its association with the intended target. In the absence of the αIIb TM peptide, the designed β-CHAMP and the control peptides sedimented with an apparent molecular weight close to that expected for the corresponding monomers. A small but significant increase in the molecular weight observed for the β-CHAMP suggests a small degree of homo-dimerization, which is likely due to the GXXGXXG motif, as both β-CHAMP G14I and β-CHAMPscr showed no evidence of self-association. In the presence of the αIIb TM, β-CHAMP sedimented with a molecular weight consistent with a 1:1 complex with the target, while β-CHAMP G14I and β-CHAMPscr displayed no association with the αIIb TM (Table 2, Figures S3–S8, Supporting Information).
Table 2.
Association of DNP-labeled β-peptides with αIIb-TM in DPC micelles as measured by AUC
| Peptide | Monomer MW, Da | MWobs in absence of αIIb TM peptide, Da | MWobs in presence of αIIb TM peptide, Da |
|---|---|---|---|
| β-CHAMP | 3420.56 | 4380 ± 61 | 6737 ± 71 |
| β-CHAMP G14I | 3476.67 | 3542 ± 172 | 3905 ± 106 |
| β-CHAMPscr | 3445.81 | 3446 ± 147 | 3542 ± 172 |
Having established interaction of the β-CHAMP with the αIIb TM domain, we explored whether β-CHAMP can disrupt interaction of the αIIb and β3 TM domains enabling the activated αIIbβ3 to bind fibrinogen. Activation of αIIbβ3 was monitored by optical trap-based single-molecule rupture force spectroscopy. Full-length αIIbβ3 was isolated from human platelets (as described in Supporting Information) and the rupture forces between the surface-bound αIIbβ3 and fibrinogen were measured after incubation of the integrin with β-CHAMP or β-CHAMPscr. The β–peptides were flanked with polyethylene-glycol groups on the N-terminus to enhance their solubility. The cumulative probabilities of αIIbβ3 binding to fibrinogen in the presence of β–peptides are shown in Figure 4. In order to assess non-specific background binding, the experiments were also performed in the presence of the αIIbβ3-specific Fab antibody fragment, abciximab, that prevents interaction of fibrinogen and the integrin. β-CHAMP almost completely activates αIIbβ3 in the absence of the antibody fragment and has essentially no activity in the presence of abciximab. The integrin-activating effect of the control peptide β-CHAMPscr is substantially lower. Mn2+, previously shown to activate αIIbβ3,29 was used as a positive control and solvent (TFE) was used as a negative control. The rupture force spectroscopy results were also confirmed by transmission electron microscopy data (Figure 5) clearly showing opening of the αIIbβ3 structure (a morphological equivalent of activation) as a result of an αIIb TM helix interacting with β-CHAMP. These results that show strong and selective interaction of the β-CHAMP with the αIIb TM domain are in good agreement with the AUC data.
Figure 4.

Rupture force spectroscopy data on interaction of the isolated surface-bound αIIbβ3 and fibrinogen in the presence of the β-CHAMP peptides (5 μM). Negative control represents trifluoroethanol, positive control - 2 mM Mn2+. The difference between the β-CHAMP data (column 1) and the β-CHAMPscr data (column 3) is statistically significant (p=0.008).
Figure 5.
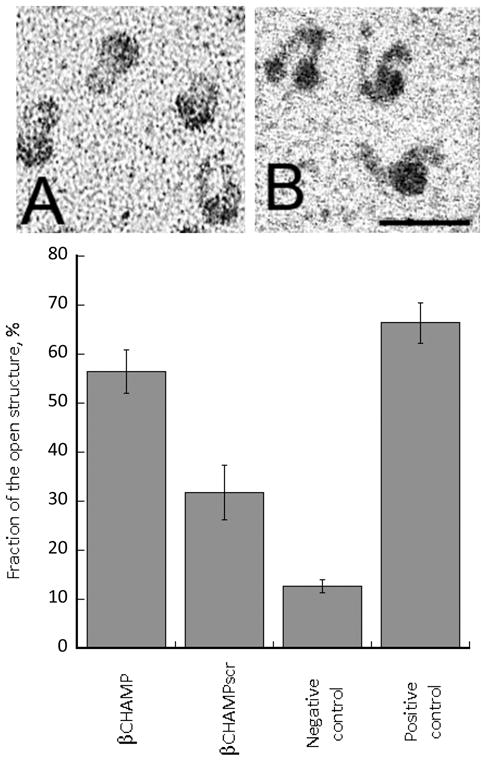
Transmission electron microscopy of purified αIIbβ3 in “closed” (A) and “open” (B) forms, corresponding to inactive and active conformations, respectively. Magnification bar = 30 nm. The bar graph shows fraction of the open structure in the presence of the β-CHAMP peptides (5 μM). Negative control represents trifluoroethanol, positive control - 2 mM Mn2+.
In conclusion, these studies indicate the feasibility of designing β-peptides to target TM helices of natural proteins. This required the design of a β-peptide that spanned the bilayer, which was accomplished by inclusion of a block of apolar residues sufficiently long to span the bilayer, as well as inclusion of Trp and Lys residues near the headgroup region of the bilayer. The TM orientation was established using polarized IR spectroscopy. Specific interaction with the αIIb TM was computationally designed by; 1) placing Gly residues at regular spacings along one face of the 12-helix; 2) optimizing the interaction of the 12-helix with αIIb TM; and 3) optimizing the placement of other sidechains in the β-CHAMP to allow favorable van der Waals contacts with the α-helical target. The interaction was shown to be specific using variants of the β-CHAMP sequence. These studies provide a well-defined and automated approach to design β-peptides that recognize membrane targets.
Supplementary Material
Figure 1.

Design of β-CHAMP. a) Docking of a CHAMP peptide on αIIb TM helix (cyan), the α-carbons of Gly residues are shown as red spheres.18 b) The position of the interacting motif on an α-helix (GX3GX3G ); c) a 14 helix (GX2GX2G); d) a 12-helix (GX2GX2G); e) final sequence of β-CHAMP modeled as a 12 helix, C3 atoms of the GX2GX2G are shown as red spheres.
Acknowledgments
The authors thank Chandrasekaran Nagaswami for technical assistance with transmission electron microscopy. This work has been supported by the NIH grants GM54616, GM60610, HL40387 and NSF MRSEC.
Footnotes
Supporting Information Available: Details of the computational design, preparation, and experimental (AUC, CD, FT-IR) characterization of peptides. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Horne WS, Gellman SH. Acc Chem Res. 2008;41:1399. doi: 10.1021/ar800009n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kritzer JA, Stephens OM, Guarracino DA, Reznik SK, Schepartz A. Bioorg Med Chem. 2005;13:11. doi: 10.1016/j.bmc.2004.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seebach D, Gardiner J. Acc Chem Res. 2008;41:1366. doi: 10.1021/ar700263g. [DOI] [PubMed] [Google Scholar]
- 4.Goodman CM, Choi S, Shandler S, DeGrado WF. Nat Chem Biol. 2007;3:252. doi: 10.1038/nchembio876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee EF, Sadowsky JD, Smith BJ, Czabotar PE, Peterson-Kaufman KJ, Colman PM, Gellman SH, Fairlie WD. Angew Chem Int Ed Engl. 2009;48:4318. doi: 10.1002/anie.200805761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Di Blasio B, Pavone V, Lombardi A, Pedone C, Benedetti E. Biopolymers. 1993;33:1037. doi: 10.1002/bip.360330706. [DOI] [PubMed] [Google Scholar]
- 7.Sadowsky JD, Fairlie WD, Hadley EB, Lee HS, Umezawa N, Nikolovska-Coleska Z, Wang SM, Huang DCS, Tomita Y, Gellman SH. J Am Chem Soc. 2007;129:139. doi: 10.1021/ja0662523. [DOI] [PubMed] [Google Scholar]
- 8.Stephens OM, Kim S, Welch BD, Hodsdon ME, Kay MS, Schepartz A. J Am Chem Soc. 2005;127:13126. doi: 10.1021/ja053444+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sadowsky JD, Schmitt MA, Lee HS, Umezawa N, Wang S, Tomita Y, Gellman SH. J Am Chem Soc. 2005;127:11966. doi: 10.1021/ja053678t. [DOI] [PubMed] [Google Scholar]
- 10.Bautista AD, Appelbaum JS, Craig CJ, Michel J, Schepartz A. J Am Chem Soc. 2010;132:2904. doi: 10.1021/ja910715u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Imamura Y, Watanabe N, Umezawa N, Iwatsubo T, Kato N, Tomita T, Higuchi T. J Am Chem Soc. 2009;131:7353. doi: 10.1021/ja9001458. [DOI] [PubMed] [Google Scholar]
- 12.Bennett JS. J Clin Invest. 2005;115:3363. doi: 10.1172/JCI26989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu H, Metcalf DG, Streu CN, Billings PC, DeGrado WF, Bennett JS. J Mol Biol. 2010;401:882. doi: 10.1016/j.jmb.2010.06.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luo BH, Takagi J, Springer TA. J Biol Chem. 2004;279:10215. doi: 10.1074/jbc.M312732200. [DOI] [PubMed] [Google Scholar]
- 15.Litvinov RI, Vilaire G, Li W, DeGrado WF, Weisel JW, Bennett JS. Biochemistry. 2006;45:4957. doi: 10.1021/bi0526581. [DOI] [PubMed] [Google Scholar]
- 16.Partridge AW, Liu S, Kim S, Bowie JU, Ginsberg MH. J Biol Chem. 2004 doi: 10.1074/jbc.M412701200. [DOI] [PubMed] [Google Scholar]
- 17.Yin H, Litvinov RI, Vilaire G, Zhu H, Li W, Caputo GA, Moore DT, Lear JD, Weisel JW, DeGrado WF, Bennett JS. J Biol Chem. 2006;281:36732. doi: 10.1074/jbc.M605877200. [DOI] [PubMed] [Google Scholar]
- 18.Yin H, Slusky JS, Berger BW, Walters RS, Vilaire G, Litvinov RI, Lear JD, Caputo GA, Bennett JS, DeGrado WF. Science. 2007;315:1817. doi: 10.1126/science.1136782. [DOI] [PubMed] [Google Scholar]
- 19.Caputo GA, Litvinov RI, Li W, Bennett JS, DeGrado WF, Yin H. Biochemistry. 2008;47:8600. doi: 10.1021/bi800687h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang J, Yin H, Qiu J, Tucker MJ, DeGrado WF, Gai F. J Am Chem Soc. 2009;131:3816. doi: 10.1021/ja809007f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Korendovych IV, Shandler SJ, Montalvo GL, DeGrado WF. Org Lett. 2011;13:3474. doi: 10.1021/ol201218y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Korendovych IV, Senes A, Kim YH, Lear JD, Fry HC, Therien MJ, Blasie JK, Walker FA, DeGrado WF. J Am Chem Soc. 2010;132:15516. doi: 10.1021/ja107487b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rathore N, Gellman SH, De Pablo JJ. Biophys J. 2006 doi: 10.1529/biophysj.106.084491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Korendovych IV, Kim YH, Ryan AH, Lear JD, DeGrado WF, Shandler SJ. Org Lett. 2010;12:5142. doi: 10.1021/ol102092r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tucker MJ, Getahun J, Nanda V, DeGrado WF, Gai F. J Am Chem Soc. 2004;126:5078. doi: 10.1021/ja032015d. [DOI] [PubMed] [Google Scholar]
- 26.Goormaghtigh E, Raussens V, Ruysschaert J. Biochim Biophys Acta. 1999;1422:105. doi: 10.1016/s0304-4157(99)00004-0. [DOI] [PubMed] [Google Scholar]
- 27.Axelsen PH, Braddock WD, Brockman HL, Jones CM, Dluhy RA, Kaufman BK, Puga FJ. Applied Spectroscopy. 1995;49:526. [Google Scholar]
- 28.Axelsen PH, Citra MJ. Prog Biophys Mol Biol. 1996;66:227. doi: 10.1016/s0079-6107(97)00007-2. [DOI] [PubMed] [Google Scholar]
- 29.Litvinov RI, Nagaswami C, Vilaire G, Shuman H, Bennett JS, Weisel JW. Blood. 2004;104:3979. doi: 10.1182/blood-2004-04-1411. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.