

Abstract
Hereditary sensory and autonomic neuropathy type II (HSANII) is a rare autosomal-recessive disorder characterized by peripheral nerve degeneration resulting in a severe distal sensory loss. Although mutations in FAM134B and the HSN2 exon of WNK1 were associated with HSANII, the etiology of a substantial number of cases remains unexplained. In addition, the functions of WNK1/HSN2 and FAM134B and their role in the peripheral nervous system remain poorly understood. Using a yeast two-hybrid screen, we found that KIF1A, an axonal transporter of synaptic vesicles, interacts with the domain encoded by the HSN2 exon. In parallel to this screen, we performed genome-wide homozygosity mapping in a consanguineous Afghan family affected by HSANII and identified a unique region of homozygosity located on chromosome 2q37.3 and spanning the KIF1A gene locus. Sequencing of KIF1A in this family revealed a truncating mutation segregating with the disease phenotype. Subsequent sequencing of KIF1A in a series of 112 unrelated patients with features belonging to the clinical spectrum of ulcero-mutilating sensory neuropathies revealed truncating mutations in three additional families, thus indicating that mutations in KIF1A are a rare cause of HSANII. Similarly to WNK1 mutations, pathogenic mutations in KIF1A were almost exclusively restricted to an alternatively spliced exon. This study provides additional insights into the molecular pathogenesis of HSANII and highlights the potential biological relevance of alternative splicing in the peripheral sensory nervous system.
Introduction
Hereditary sensory and autonomic neuropathies (HSANs) are a clinically and genetically heterogeneous group of neurological conditions characterized by peripheral nerve degeneration resulting in a loss of sensory perception accompanied by variable degrees of autonomic dysfunction.1,2 Mutations in several genes were identified for autosomal-recessive (WNK1 [MIM 605232], NTRK1 [MIM 191315], IKBKAP [MIM 603722], NGFB [MIM 162030], CCT5 [MIM 610150], and FAM134B [MIM 613114])3–9 and autosomal-dominant (SPTLC1 [MIM 605712], SPTLC2 [MIM 605713], RAB7 [MIM 602298], and ATL1 [MIM 606439]) forms 10–13 of HSANs. Hereditary sensory and autonomic neuropathy type II (HSANII [MIM 201300]) is characterized by a recessive inheritance, onset of symptoms in the first decade of life, and severe distal sensory loss with minimal autonomic dysfunction.14,15 We previously established that mutations in the nervous system-specific HSN2 exon of WNK1 cause HSANII3,4 and reported that two founder mutations in the HSN2 exon were responsible for the higher prevalence of HSANII in the French-Canadian population of Quebec.16 Although mutations in WNK1 and FAM134B were shown to cause HSANII in some families,3,4,9 the etiology of a substantial number of cases remains unexplained.17 In addition, the functions of WNK1/HSN2 and FAM134B and their role in the peripheral nervous system remain poorly understood.
To understand the disease pathophysiology and WNK1/HSN2 function, we used a yeast two-hybrid (Y2H) screen to identify proteins interacting with the domain encoded by the HSN2 exon. We also performed genome-wide homozygosity mapping in a consanguineous Afghan family affected by HSANII. These two parallel approaches converged to the identification of causative mutations for HSANII in KIF1A (MIM 601255), which is a gene encoding a motor protein involved in the anterograde transport of synaptic-vesicle precursors along axons.18
Material and Methods
Yeast Two-Hybrid Screen
A human cDNA encoding the HSN2 exon was subcloned in a pGBKT7 bait vector (Clontech, #630443) so that it could be expressed as a bait GAL4 DNA binding domain (BD) fusion protein when transformed in S. cerevisiae AH109 (MATa) (Clontech, #630444). We performed a Y2H screen by mating these cells with Y187 cells (MATα) pretransformed with an oligo(dT) human fetal brain cDNA library (Clontech, #638869). The prey proteins encoded in Y187 cells are GAL4 activation domain (AD) fusion proteins. As a positive control, AH109 pretransformed with pGBKT7-53 were mated with Y187 pretransformed with pGADT7-T (MatchMaker positive vectors, Clontech). These diploid cells are transcriptionally active for their reporter genes, and colonies grow on SD/-LW. As a negative control, these diploid cells were tested on SD/-LWHA medium. The stringency of diploid clones was controlled with a 3-amino-1,2,4-triazole concentration (1 mM 3-AT) on minimal medium lacking Leu, Trp, and His, and the clones were tested for B-galactosidase activity by overlay. Unique interactors were identified by PCR, digestion profiles (HaeIII), and sequencing. To define the protein segments of KIF1A interacting with the domain encoded by the HSN2 exon, we subcloned 17 fragments of KIF1A in pGADT7, transformed them in Y187, and mated the cells as described above with AH109 yeast cells expressing pGBKT7-HSN2.
Coimmunoprecipitation and Immunolotting
Cultured human embryonic kidney cells (HEK293) were transfected with an N-tagged truncated version of WNK1/HSN2 (domains encoded by exons 1 to the HSN2 exon and a Myc tag in the N terminus) for 48 hr with Lipofectamine Plus. After cell lysis and centrifugation, the supernatants were precleared and immunoprecipitated with Dynabeads Protein G (Invitrogen, #100-03D) complexed to mouse monoclonal anti-KIF1A (8 μg) (BD Transduction Laboratories, #612094) or species- and isotype-matched IgG1 (R&D Systems, #MAB002) overnight on a rotator at 4°C. After extensive washes with cold PBS, samples were eluted from the beads and analyzed, separated by SDS-PAGE, transferred to nitrocellulose, and immunoblotted. Monoclonal anti-HSN2 (Novus Biologicals, NBP1-32917) and anti-KIF1A (BD Transduction Laboratories, #612094) antibodies were used for the detection of WNK1/HSN2 and KIF1A, respectively.
Patients and DNA Samples
Informed written consent was obtained from each individual prior to enrollment in the study. All procedures were approved by the local ethics committees on human experimentation. The study included a consanguineous Afghan family affected by HSANII, 112 additional unrelated HSAN cases, and 665 unrelated European control subjects who were screened for KIF1A mutations. Standard methods were used for blood sampling and DNA extraction from peripheral blood.
Genome-wide SNP Genotyping and Homozygosity Mapping
DNA samples from the three affected siblings and their father were genotyped with the Illumina HumanHap300-Duov2 Genotyping BeadChip (HumanHap300v2_A) at the McGill University and Génome Québec Innovation Centre. Genotyping was performed according to the manufacturer's instructions. The call rate of the four samples was greater than 98%. The 318,237 genotyped single nucleotide polymorphisms (SNPs) were analyzed with the “runs of homozygosity” tool available in the PLINK software (v1.05)19 so that homozygous intervals across the 22 autosomes could be mapped. For identification of homozygous intervals, a fixed threshold of 1 Mb in length was used. Five missing calls and one heterozygous SNP within a segment were allowed.
KIF1A Mutation Screening and Haplotype Analysis
All the coding exons of KIF1A (GenBank accession number: AB290172.1) were amplified by PCR with the primers listed in Table S1, available online. Primers were designed to amplify fragments covering coding exons and their intronic flanking regions. PCR products were sequenced on a 3730XL DNA analyzer at the McGill University and Génome Québec Innovation Centre. Mutations were confirmed by reamplification of the fragment and resequencing of the proband and its available relatives. Mutation Surveyor (version 3.10, SoftGenetics) was used for mutation-detection analysis. For the purpose of haplotype analysis, 34 single nucleotide polymorphisms (SNPs) located within the KIF1A locus were genotyped by PCR amplification and sequencing in the affected individuals carrying the c.2840 delT mutation and in their parents.
Analysis of KIF1A Splicing Events by Reverse Transcriptase PCR
cDNA synthesis and PCR analysis were performed via standard methods. Total RNA (1 μg) from commercially available RNA samples (Clontech) was used as template for first-strand cDNA synthesis. Long-range PCR was performed with Long PCR Mix (Fermentas). Beta-actin (encoded by ACTB [MIM 102630]) was coamplified as an internal control to correct for loading variations. The amplified fragments were analyzed by agarose gel electrophoresis, and sequence analysis was performed. Oligonucleotide sequences are listed in Table S1.
Quantitative Real-Time PCR Experiments
Total RNA samples (1 μg) from adult human brain and dorsal root ganglia (DRG) (Clontech) were used as templates for first-strand cDNA synthesis. Real-time quantitative PCR experiments were performed on an ABI 7900HT Real-Time Instrument (Applied Biosystems) with QuantiTect SYBR green (QIAGEN). The KIF1A isoform lacking exon 25b was amplified with a forward primer flanking the splice junction of exons 25 and 26 and a reverse primer in exon 27. The KIF1A exon-25b-containing isoform was amplified with primers in exon 25b and 27. PCR primer pairs are listed in Table S1. POLR2A (MIM 180660) was selected as the endogenous reference on the basis of its constant expression in different tissues.20 Relative expression levels of KIF1A isoforms were calculated with the 2-ΔΔCT method.21 All data are expressed as means ± standard error of the mean (SEM) of three independent experiments performed in triplicate.
Immunodetections of Primary-Neuron Culture
Adult-mouse sensory neuronal cultures and immunodetections were performed essentially as described previously.4 Dorsal root ganglia were dissected from adult mouse C57BL/6 and incubated with 10 mg/ml of collagenase D (Roche) for 45 min at 37°C. Trypsin (0.25%) was then added for 30 min at 37°C. The tissues were washed once with cold Neurobasal medium (Invitrogen) plus 2% inactivated goat serum and then triturated in warm Neurobasal medium plus serum. One milliliter of the cell suspension was overlaid on 1 ml of 35% Percoll in saline (Pharmacia; Amersham Biosciences) and centrifuged at 10°C at 285 × g for 15 min. The cell pellet, which included sensory neurons, was washed in 5 ml of fresh medium, resuspended in fresh warm medium with 50 ng/ml of nerve growth factor, and plated on pol-d-lysine- and laminin-coated coverslips. Depending on whether or not they were to be infected with lentiviral particles, cells were cultured for 5 days or 48 hr, respectively, prior to their immunodetection. A mouse anti-KIF1A (1:200; BD Transduction Laboratories, #612094), a previously described rabbit anti-WNK1/HSN2,4 and beta III tubulin (1:2,000; Abcam, #ab41489) were used for triple immunolabeling of the neurons. Confocal microscopy analysis was performed with a Leica TCS SP5 broadband confocal microscope as previously described.4
Lentiviral shRNA-Mediated Knockdown of KIF1A
Lentiviruses were produced from pLKO.1 vector containing a short hairpin RNA (shRNA) sequence targeting the KIF1A gene. After immunoblot detections, two clones (Open Biosystems) that efficiently silenced the expression of KIF1A were chosen for neuronal infections (kif43 #TRCN0000091343 and kif46 #TRCN0000091346); control infections were made with another clone from Open Biosystems (shGFP #RHS4459). For lentiviruses production, 293FT packaging cells were cotransfected with the ViraPower Packaging Mix (Invitrogen) and the vector of interest. The supernatant was collected after 3 days, filtered, and either used directly on cells or concentrated via ultracentrifugation. Titer for each virus production was evaluated with serial dilution of the supernatant and infection of 293 cells. ShGFP at 3 × 106 TU/ml, kif43 at 3.9 × 106 TU/ml, and kif46 at 3.2 × 106 TU/ml were used (TU = transducing unit). Pregnant mice were sacrificed at embryonic day 15.5 (E15.5) so that dissociated primary cortical neurons could be prepared essentially as described previously.4 The freshly plated primary neurons were kept in a humidified incubator at 37°C for one night before they were infected overnight with the viral particles described above; growing cells for 4 days after infection allowed shRNA silencing. Whole-cell protein lysates were then prepared with SUB lysis buffer and resolved by SDS-PAGE. The anti-KIF1A antibody was used at 1:1,000, and an anti-actin (Sigma) was used at 1:5,000.
Results
KIF1A Interacts with the Domain Encoded by the HSN2 Exon of WNK1
To identify proteins interacting with the HSN2 exon of WNK1, we performed a Y2H screen by using a human fetal brain cDNA library (bait, pGBKT7-HSN2 [Figure 1A]; prey, pGADT7-human fetal brain cDNA library). One of the positive clones identified corresponded to KIF1A (Figure 1B). We validated this initial interaction by performing a direct Y2H assay with pGBKT7-HSN2 and pGADT7-KIF1A (cDNA clone from Kazusa DNA Research Institute) (Figure 1C, KIF1A lane). We then performed separate Y2H interaction assays between the domain encoded by the HSN2 exon and 17 overlapping fragments of KIF1A and identified three distinct KIF1A segments interacting with the domain encoded by the HSN2 exon (Figures 1C and 1D). Coimmunoprecipitation of KIF1A and WNK1/HSN2 from HEK293 cells expressing a truncated form of WNK1/HSN2 further validated the interaction between WNK1/HSN2 and KIF1A (Figure 2).
Figure 1.
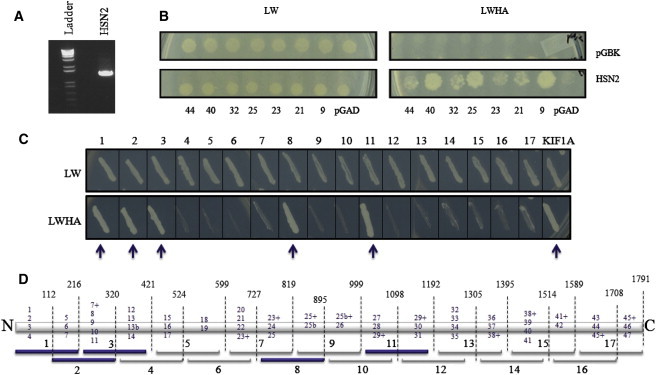
Yeast Two-Hybrid Screen for Interactors with the Domain Encoded by the HSN2 Exon
(A) PCR confirmation of the presence of the HSN2 exon in the pGBKT7 vector; the nature of the excised band was confirmed by direct sequencing.
(B) Y2H assays between domain encoded by the HSN2 exon and some of the identified interactors inserted in pGADT7 vector (KIF1A is clone #23). To test the interaction, we first grew mated yeasts on a plate with the mating medium (left panel. SD/-LW, lacking leucine and tryptophan), then transferred them onto a plate with the selection medium (right panel. SD/-LWHA, lacking leucine, tryptophan, histidine, and adenine). As a control, empty pGBK constructs were used against the interactors (top lane), and empty-pGAD constructs were used against the pGBKT7-HSN2 (bottom right corner).
(C) Separate Y2H interaction assays between the domain encoded by the HSN2 exon and 17 overlapping fragments of KIF1A, as shown in (D). The KIF1A lane corresponds to the full-length cDNA clone.
(D) Schematic representation of the 1791 amino acids of KIF1A (with regions encoded by exons 13b and 25b) showing breakpoints for the 17 fragments used in (C). Blue numbers indicate which exons were included so that each fragment could be generated. Exon numbers with a + sign indicate that the majority of the exon was included in that fragment. Fragments indicated by blue arrows (1, 2, 3, 8, and 11) showed the strongest affinity for the domain encoded by the HSN2 exon.
Figure 2.
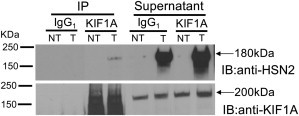
Transient Expression of WNK1/HSN2 and Detection of Its Interaction with Endogenous KIF1A
Whole-protein lysates prepared from HEK293 cells transfected (T) with truncated (180 KDa) WNK1/HSN2 and nontransfected cells (NT) were immunoprecipitated (IP) with commercial KIF1A antibody and control IgG1 isotype antibody before they were loaded and separated by SDS-PAGE. The gel was transferred to a nitrocellulose membrane and separately immunoblotted (IB) with anti-HSN2 or anti-KIF1A antibodies. Supernatants of the immunoprecipitations were also collected. Anti-KIF1A immunoprecipitation showed the presence of a protein with the appropriate size (180 KDa) when anti-HSN2 was used.
Clinical Description of the Afghan Family
Three Afghan brothers born from first-degree cousins presented at Shariati Hospital, Tehran, and were accompanied by their father. The index patient (individual IV:2, Figure 3) was 13 years of age and reported an age of onset of 10 years. He presented with numbness in his feet and hands and mutilation of the fingers and toes but did not complain of weakness. The problem had gradually worsened, and he had developed ulcerative lesions refractory to therapy; these lesions led to amputation. The patient's past medical history was unremarkable, and he had no other neurological disorders. On examination, his mental state and cranial nerves were normal. Muscle strength was normal in the proximal upper extremities. In the hands, the first dorsal interossei was 4/5, the abductor pollicis brevis was 3/5, and the finger extension was 4/5. Intrinsic hand muscles were slightly wasted. In the lower extremities, hip flexors were 5/5, foot dorsiflexors were 0/5, and plantar flexors were 4/5. He had wasting of the pretibial and calf muscles. Deep tendon reflexes were generally absent. All fingers were mutilated, and only one toe was intact. Pinprick and touch were normal, but vibration and position sense was absent in his feet and hands. Nerves were not hypertrophic. Nerve-conduction studies showed normal ulnar motor, reduced median compound muscle action potential (CMAP) amplitude, and slightly reduced motor velocities. Tibial and peroneal motor potentials were absent; sural and superficial peroneal sensory potentials were absent; and ulnar and median sensory potentials had a very low amplitude. Motor unit action potentials (MUAPs) were neurogenic in the first dorsal interosseus (FDI) and tibialis anterior (TA) muscles.
Figure 3.
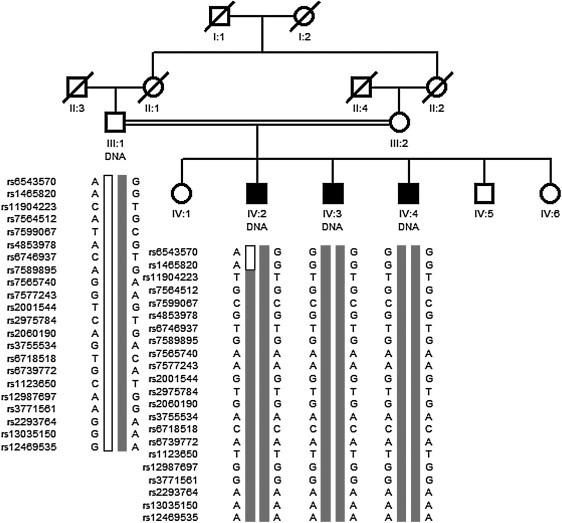
Family Tree of the Afghan HSANII Family and Haplotype Analysis of the 2q37.3-qter Chromosomal Region Identified by Genome-wide Homozygosity Mapping
The parents of the patients are first-degree cousins. Only DNA samples from the father and his three affected sons were available. Genotyping data from 20 informative SNPs among the 276 SNPs spanning the 2q37.3 locus found in a homozygous state in all three patients as well as two proximal recombinant markers (patient IV:2, rs6543570 and rs1465820) are shown.
Patient IV:3 was 9 years old. Clinical manifestations had started a year earlier and included mutilation of the digits, particularly the toes, which were all mutilated on examination. Muscle strength was normal in the upper extremities, where there was no wasting. Hip flexors, quadriceps, and gastrocnemius were 5/5; foot dorsiflexors were 4−/5; and leg and foot muscles were wasted. Deep tendon reflexes were 1+ in upper extremities, and knee and ankle jerks were absent. Pinprick and touch senses were normal, and vibration was impaired in the feet and hands. Median and ulnar motor nerve conduction velocities (NCVs) were normal. Tibial and peroneal CMAPs were absent. Sural, median, and ulnar sensory potentials were absent. Needle examination of tibialis anterior muscle showed neurogenic MUAPs.
Patient IV:4 was 7 years old at examination and presented with mutilation of the right foot. The left foot and fingers had only minor changes, and there was no muscle wasting. Muscle strength was normal except for foot dorsiflexors, which were 4+/5. Deep tendon reflexes in the biceps and the triceps were 1+; deep tendon reflexes in the knee, ankle, and brachioradialis were absent. Pinprick sense was normal; vibration and position senses were impaired in the hands and feet. There was no nerve hypertrophy. Median and ulnar motor NCVs were normal. Tibial and peroneal CMAPs were of low amplitude and had slightly reduced onduction velocities. Sural sensory potential was absent; median and ulnar sensory potentials were of very low amplitude and had mildly reduced conduction velocities. Needle examination of tibialis anterior muscle showed mildly neurogenic MUAPs.
Overall, the clinical picture was consistent with an ulcero-mutilating sensory neuropathy with prominent involvement of position and vibration senses and concomitant distal motor involvement, particularly in the feet.
Genome-wide Homozygosity Mapping
Genetic analysis of the Afghan family revealed neither mutation in WNK1 nor linkage to the WNK1 chromosomal locus. Given the parental consanguinity, we performed a homozygosity mapping to map the disease locus. A unique region of homozygosity larger than 1 Mb was identified on chromosome 2q37.3-qter spanning 2.68 Mb and comprising 38 known genes (Table 1), among which is KIF1A, which encodes the protein we identified in our Y2H screen. Marker rs1465820 was identified as the proximal recombinant breakpoint, and no distal recombination event occurred. Among the 276 consecutive SNPs homozygous for the same alleles in the three affected individuals, 105 were found in a heterozygous state in the father, as expected for a recessive mode of inheritance (20 of them are shown in Figure 3).
Table 1.
Shared Homozygous Regions Identified by Genome-wide Homozygosity Mapping in the Afghan Family
| Chromosome |
Homozygous SNPs |
Informative SNPs | Proximal Recombinant Breakpoint | Distal Recombinant Breakpoint | Interval (NCBI36/hg18) | Size (Mb) |
|---|---|---|---|---|---|---|
| 2 | 276 | 105 | rs1465820 | qter | 240,270,700–242,951,149 | 2,680,449 |
| 12 | 53 | 0 | rs149411 | rs590460 | 48,736,243–49,666,499 | 930,256 |
| 2 | 50 | 0 | rs707718 | rs11898850 | 72,209,957–73,053,127 | 843,107 |
| 5 | 70 | 29 | rs6867206 | rs4507452 | 50,688,451–51,468,426 | 779,975 |
| 4 | 69 | 3 | rs6841266 | rs2293131 | 190,271,285–190,979,685 | 708,400 |
| 18 | 63 | 0 | rs8087644 | rs1561234 | 49,683,496–50,225,187 | 541,691 |
| 6 | 64 | 0 | rs1418968 | rs1390459 | 18,881,077–19,411,155 | 530,078 |
| 13 | 50 | 0 | rs462954 | rs1929185 | 91,169,091–91,696,831 | 527,740 |
| 2 | 57 | 22 | rs2583526 | rs1401838 | 76,038,324–76,486,648 | 448,324 |
| 5 | 60 | 22 | rs4074670 | rs4701077 | 178,137,065–178,536,983 | 399,918 |
| 2 | 57 | 0 | rs4854306 | rs6757749 | 423,274–812,116 | 388,842 |
| 4 | 51 | 0 | rs1510716 | rs13128291 | 28,138,936–28,499,076 | 360,140 |
| 22 | 51 | 0 | rs8137243 | rs4044210 | 44,822,499–45,164,979 | 342,480 |
| 10 | 52 | 19 | rs17157052 | rs11251048 | 1,853,551–2,110,407 | 256,856 |
SNP: single-nucleotide polymorphism. Mb: megabase pairs. Informative SNPs: SNPs found in a heterozygous state in the father. Only regions larger than 250 kb are shown.
KIF1A Mutation Screening and Haplotype Analysis
Previously reported homozygous KIF1A-knockout mice showed severe motor and sensory deficits and a substantial extent of neuronal death.22 These observations led us to speculate that the interaction between the domain encoded by the HSN2 exon of WNK1 and KIF1A was relevant to the pathogenesis of HSANII. KIF1A comprises 47 exons (46 coding) plus three alternatively spliced coding exons that we here refer to as 13b, 25b, and 36b. Sequencing of the coding exons and flanking intronic sequence of the gene in the Afghan family revealed a 1 bp deletion (GenBank accession number AB290172.1: c.2840delT [p.Leu947Argfs∗4]) in the alternatively spliced exon 25b, which would be predicted to cause a frameshift. This mutation was homozygous in all three affected individuals, heterozygous in the father (family 1, Figure 4), and not present in 665 population controls of European ancestry. We then screened KIF1A in a cohort of 112 unrelated HSAN cases negative for mutations in the HSN2 exon; 23 of these cases were HSANII (Table 2). The c.2840delT mutation was again found in a homozygous state in four patients from two unrelated families from Turkey and Belgium (families 2 and 3, Figure 4) and in a heterozygous state in another Belgian patient. This last individual also carried in exon 46 a heterozygous 1 bp insertion predicted to cause a frameshift and a premature stop codon (GenBank accession number AB290172.1: c.5271dupC [p.Ser1758Glnfs∗7]). Segregation analysis confirmed the compound heterozygous state of this last patient; the c.2840delT and c.5271dupC mutations were transmitted by the mother and father, respectively (family 4, Figure 4). No truncating mutations in KIF1A, either in a heterozygous or in a homozygous state, were detected in the 665 control individuals (1330 chromosomes), further confirming that truncating mutations in KIF1A cause HSANII. Haplotype analysis by genotyping 34 SNPs located within the KIF1A locus in all four families identified a shared haplotype of approximately 45 kb flanking the c.2840delT mutations. However, given the high frequency of this haplotype block in the HapMap CEU population, it remains unclear whether these mutations originated from a common ancestor or arose independently (Table 3). Affected individuals from three of the four families (families 1, 2, and 4) with KIF1A mutations were initially diagnosed with HSANII. Patients from the fourth family (family 3) had a slightly later age of onset and, despite the lack of evidence for an autosomal-dominant pattern of inheritance, were diagnosed as having a severe form of HSANI (Table 4). Similarly to patients presenting with WNK1 mutations3 and unlike patients with FAM134B mutations,9 individuals with KIF1A mutations showed no clinical autonomic dysfunction.
Figure 4.
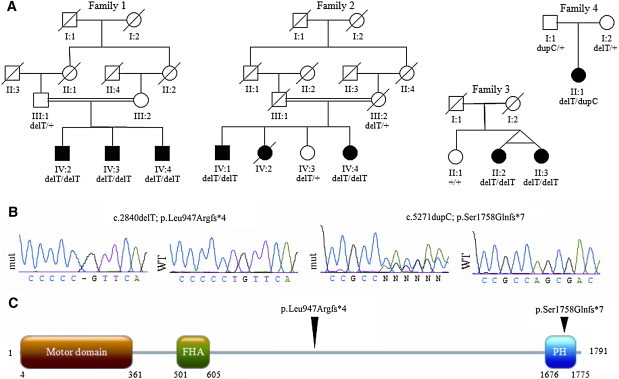
KIF1A Mutations in Individuals with HSANII
(A) Family trees and segregation analysis of the mutations.
(B) Sequencing traces of the mutations. Abbreviations are as follows: mut, mutated sequencing trace; WT, control sequencing trace.
(C) Linear protein structure of KIF1A indicating the kinesin motor domain, the Forkhead-associated domain (FHA), and the Pleckstrin homology domain (PH). Arrowheads above the protein indicate the position of the mutations.
Table 2.
HSAN Individuals Screened for KIF1A Mutations
| Type | Inheritance | Unrelated Individuals |
|---|---|---|
| HSANI/HSANIB/CMT2B | AD | 28 |
| HSANII | AR | 23 |
| HSANIII | AR | 2 |
| HSANIV | AR | 4 |
| HSANV | AR | 1 |
| HSAN with spastic paraplegia and/or pyramidal-tract signs | AR | 7 |
| Unspecified | unknown | 47 |
| Total | 112 |
Inclusion criteria: unrelated patients with clinical features belonging to the clinical spectrum of ulcero-mutilating sensory neuropathies. 62 patients were negative for mutations in the HSN2 exon of WNK1 and for mutations in SPTLC1, RAB7, WNK1, NTRK1, NGFB, and CCT5.17 Abbreviations are as follows: CMT2B, Charcot-Marie-Tooth neuropathy type 2B; AD, autosomal dominant; AR, autosomal recessive.
Table 3.
Haplotype Sharing in Patients with the c.2840delT Mutation
| SNP ID (rs) | Position (NCBI36) | Alleles | Family 1 | Family 2 | Family 3 | Family 4 | Shared Allele | Frequency | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rs755302 | 241,301,068 | G/A | G | G | G | G | G | G | A | G | G | G = 50% |
| rs4613 | 241,301,960 | A/G | G | G | A | A | A | A | G | A | A | A = 64% |
| rs3732341 | 241,304,217 | G/A | G | G | A | A | A | A | A | A | A | A = 46.7% |
| rs3772050 | 241,321,413 | T/C | C | C | C | C | C | C | C | C | C | C = 76.7% |
| rs11886658 | 241,324,642 | G/A | G | G | G | G | A | A | A | A | A | A = 50% |
| rs10174559 | 241,325,449 | G/A | G | G | G | G | G | G | G | G | G | G = 78.3% |
| rs7578279 | 241,325,937 | T/C | T | T | C | C | T | T | T | T | T | T = 39.2% |
| rs3772054 | 241,327,201 | A/G | A | A | A | A | A | A | A | A | A | A = 66.7% |
| rs4414678 | 241,329,306 | G/T | T | T | T | T | T | T | T | T | T | T = 35.1% |
| rs1013225 | 241,329,317 | C/G | G | G | G | G | G | G | G | G | G | G = 73.6% |
| rs56024577 | 241,329,475 | A/G | A | A | A | A | A | A | A | A | A | A = 26.9% |
| rs3755534 | 241,330,687 | A/G | A | A | G | G | G | G | G | G | G | G = 85% |
| rs3772056a | 241,332,581 | A/G | G | G | G | G | G | G | G | G | G | G = 71% |
| rs7576602a | 241,333,365 | C/T | C | C | C | C | C | C | C | C | C | C = 78.8% |
| rs10196604 | 241,334,748 | C/T | C | C | C | C | C | C | C | C | C | C = 72.6% |
| rs3755535a | 241,336,265 | A/C | A | A | A | A | A | A | A | A | A | A = 40% |
| rs11693670 | 241,340,129 | C/G | G | G | G | G | G | G | G | G | G | G = 47.5% |
| rs12624059a | 241,341,913 | A/G | G | G | G | G | G | G | G | G | G | G = 69.2% |
| rs4676447a | 241,343,769 | C/T | C | C | C | C | C | C | C | C | C | C = 40% |
| p.Leu947Argfs∗4 | 241,345,427 | c.2840 delT | delT | delT | delT | delT | delT | delT | T | delT | delT | |
| rs7598218a | 241,346,971 | A/G | G | G | G | G | G | G | G | G | G | G = 61.7% |
| rs11688298 | 241,351,234 | A/T | T | T | T | T | T | T | T | T | T | T = 45.4% |
| rs3821345a | 241,352,633 | G/T | G | G | G | G | G | G | G | G | G | G = 39.2% |
| rs11690282a | 241,353,054 | C/T | T | T | T | T | T | T | T | T | T | T = 77.6% |
| rs3755536 | 241,354,706 | A/G | G | G | G | G | G | G | G | G | G | G = 70.7% |
| rs11681427 | 241,354,900 | C/T | C | C | C | C | C | C | C | C | C | C = 77.5% |
| rs2288751a | 241,362,102 | T/C | T | T | T | T | T | T | T | T | T | T = 60.8% |
| rs1063353 | 241,362,319 | C/T | T | T | T | T | T | T | T | T | T | T = 64.9% |
| rs2288750 | 241,371,118 | C/T | C | C | C | C | C | C | T | C | C | C = 80.6% |
| rs2288746 | 241,375,319 | C/T | C | C | T | T | T | T | C | T | T | T = 58.5% |
| rs12989742 | 241,377,750 | C/T | T | T | C | C | T | T | C | T | T | T = 69.2% |
| rs6708456 | 241,378,666 | C/T | C | C | T | T | C | C | T | C | C | C = 69.2% |
| rs6732174 | 241,380,213 | C/T | C | C | T | T | C | C | T | C | C | C = 71.7% |
| rs734586 | 241,383,427 | A/G | A | A | G | G | A | A | G | A | A | A = 58.3% |
| rs7599943 | 241,390,650 | C/T | C | C | T | T | C | C | T | C | C | C = 66.7% |
Haplotype blocks shared by two or more families are indicated in bold. Alleles shared by all affected individuals are indicated in italics. Allele frequencies were either derived from the CEU HapMap database or from direct sequencing of the coding exons of KIF1A in our 665 population controls of European ancestry. To estimate the frequency of the 45 kb haplotype shared by all four families (dark gray shading), we used the phasing data of the HapMap3 project for the CEU population. Genotyping data were available for nine (indicated by a superscript “a”) of the 16 SNPs constituting this haplotype. The “GCAGCGGTT” haplotype identified in the HSANII families was found at a frequency of 0.37 in the CEU population (87/234 chromosomes). The probability of n independent mutational events for the c.2840delT mutation on a haplotype whose frequency is 0.37 was calculated as follows: p = 0.37n, giving p = 0.019, p = 0.05, and p = 0.14 for four, three, and two independent mutational events, respectively.
Table 4.
Clinical Features of Affected Individuals with KIF1A Mutations
| Family | PID | AOO | AE | Sensory Symptoms | Skin Changes | Amputations | Muscle Weakness | Nerve Conduction Study | Intellectual Disability | Additional Information |
|---|---|---|---|---|---|---|---|---|---|---|
| Family 1 Afghanistan HSANII |
IV:2 | 10 | 13 | distal loss of vibration sense and proprioception | ulcerations | yes, digits and toes | distal UL and LL | see detailed clinical description in the Results section | no | |
| IV:3 | 8 | 9 | ulcerations | yes, digits and toes | distal LL | no | ||||
| IV:4 | 6 | 7 | ulcerations | yes, toes | mild in distal LL | no | ||||
| Family 2 Turkey HSANII |
IV:1 | 7 | 22 | painless ulcerations | ulcerations, skin grafts | yes | distal LL, mild in distal UL | absent median SNAPs and muscle denervation | no | seizures at age 18; renal AA amyloidosis |
| IV:4 | 9 | 20 | painless ulcerations | ulcerations | no | distal LL, mild in distal UL | right radial SNAP 3uV, normal motor conduction. Muscle denervation | no | ||
| Family 3 (CMT-155) Belgium HSANI |
II:2 | 15 | 61 | panmodal distal sensory loss | ulcerations | right lower leg (age 23), all fingers except thumbs (spontaneous amputations) | complete paralysis (0/5) distal left LL, normal strength in wrists | no | spontaneous pains UL and LL, absent tendon reflexes. Tibial and fibular nerve biopsy in amputated limb: thickened peri- and epineurium with near complete loss of myelinated axons |
|
| II:3 | 13 | 61 | panmodal distal sensory loss | ulcerations | left lower leg, toes on the right foot, spontaneous amputation of parts of the fingers | complete paralysis (0/5) distal right LL, paresis fingers | normal ulnar motor NCV (60.0 m/s), EMG neurogenic | no | ||
| Family 4 (CMT-869) Belgium HSANII |
II:1 | CG | 8 | panmodal distal sensory loss | ulcerations, hyperkeratosis hand palms, nail dystrophy | terminal phalanx digit II right side | distal > proximal LL and UL. Severe dorsiflexor weakness in feet, weakness hipflexors | SNAPs reduced to absent in sural and median nerve; reduced CMAP amplitudes with borderline NCV for median and fibular nerve, EMG neurogenic | slow speech development with bad articulation; IQ of 80 normal MRI scan of the brain | Charcot joint deformities in the terminal phalanges; difficult gait, pronounced equinus deformities, wheelchair, small stature |
None of the affected individuals had autonomic dysfunction. Abbreviations are as follows: PID, individual identification from pedigree (see Figure 4A); AOO, age of onset;l AE, age at last exam; CG, congenital; UL, upper limbs; LL, lower limbs; SNAP, sensory nerve action potential; NCV, nerve conduction velocity; EMG, electromyogram; CMAP, compound muscle action potential; IQ, intelligence quotient; MRI, magnetic resonance imaging. Both Belgian families are of European ancestry; see Rotthier et al.17 for further discussion on family 3 (CMT-155).
Analysis of KIF1A Splicing Events
Exon 25b was originally identified in a brain-expressed human mRNA, and this isoform is predicted to encode a 1791 amino acid peptide (GenBank accession number BAG06726.1). A sequence cluster alignment based on multi-species alignment from the UCSC Genome Browser indicated that this exon and its splice junctions are conserved through evolution (Figure S1). To further confirm that exon 25b is an alternatively spliced exon of KIF1A, we performed RT-PCR reactions with human RNA samples and primers located within exon 25b and its neighboring exons (Figures 5A and 5B) and observed that the KIF1A exon-25b-containing isoform was strongly expressed in the nervous system. Real-time quantitative PCR experiments revealed that the KIF1A isoform lacking exon 25b was the most abundant form in both adult brain and DRG. The relative abundance of the exon-25b-containing isoform was nonetheless higher in the DRG (Figure 5C), which is consistent with the peripheral nervous system phenotype observed in patients lacking this isoform. Long-range RT-PCR reactions were initiated with sets of primers flanking the region between the first coding exon of KIF1A and exon 25b and the region between exon 25b and the last exon of KIF1A; sequencing PCR products confirmed the extent of the entire coding sequence of this specific isoform in the brain and DRG (Figures 5D and 5E).
Figure 5.
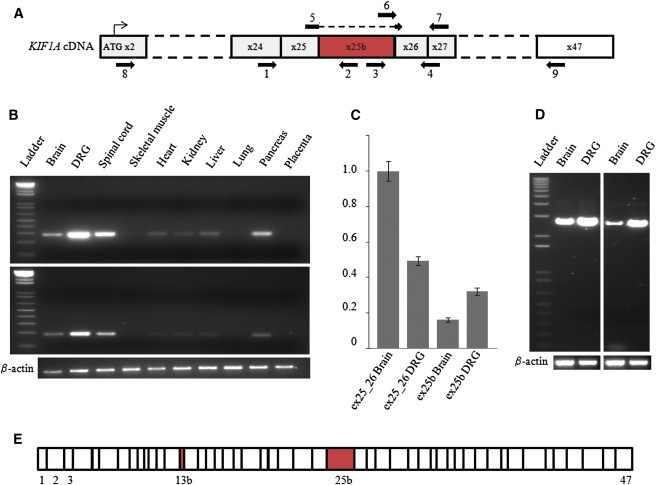
Analysis of Human-Tissue Expression of the KIF1A Messenger Containing Exon 25b
(A) Diagram of KIF1A cDNA from the first coding exon (x2) to the last exon (x47). Exon 25b is in red. The numbered arrows indicate the primers (listed in Table S1) used for PCR experiments on cDNA samples.
(B) PCR amplifications with cDNAs from different tissues. Top: amplifications of a 281 bp product between exon 24 (primer 1) and exon 25b (primer 2). Bottom: amplifications of a 185 bp product between exon 25b (primer 3) and exon 26_27 (primer 4). Beta-actin was coamplified as an internal control.
(C) Relative quantification of isoforms of KIF1A lacking exon 25b (ex25_26, primers 5 and 7) and KIF1A containing exon 25b (ex25b, primers 6 and 7) in adult human brain and DRG tissues by real-time quantitative PCR via the 2-ΔΔCT method. POLR2A was used as an endogenous reference. The brain KIF1A isoform lacking exon 25b was arbitrarily chosen as the calibrator. The bars show the mean ± SEM for each target.
(D) Long-range PCR amplifications from brain and DRG cDNAs. Left: amplifications between exon 2 (primer 8) and exon 25b (primer 2). Right: amplifications between exon 25b (primer 3) and exon 47 (primer 9). Beta-actin was coamplified as internal control.
(E) The linear structure of the KIF1A transcript is shown to contain exon 25bon the basis of the sequencing traces of the long-range PCR products. This isoform contains all the known exons of KIF1A plus exons 13b and 25b (in red).
Immunodetections of WNK1/HSN2 and KIF1A and shRNA-mediated Knockdown of KIF1A in Primary-Neuron Culture
To follow up on our observations that WNK1/HSN2 and KIF1A interact at the protein level, we used anti-HSN2 and anti-KIF1A antibodies to examine the localization of the two proteins in cultured primary sensory neurons prepared from adult mouse DRG. Both proteins were found to localize in cell bodies and along axons (Figures 6A–6E). Using identical cells, our group previously showed that WNK1 localized exclusively in the cell body, whereas WNK1/HSN2 localized in both the cell body and the axons.4 Given the localization of this second isoform and the well-established role of KIF1A in anterograde transport, we hypothesized that KIF1A could be responsible for the anterograde transport of WNK1/HSN2 along axons.18 To test this hypothesis, we used lentiviral delivery of shRNAs to silence KIF1A (Figures 6F–6J). Infection of cultured DRG sensory neurons substantially reduced KIF1A in cell bodies and rendered it undetectable in axons (Figure 6G) but did not reduce WNK1/HSN2 in axons (Figure 6H). This last observation suggests that KIF1A might not be the only transporter of WNK1/HSN2 along axons. However, we cannot rule out the possibility that the reduced remaining amount of KIF1A protein (Figures 5F and 5G) was still sufficient for the transport of WNK1/HSN2.
Figure 6.
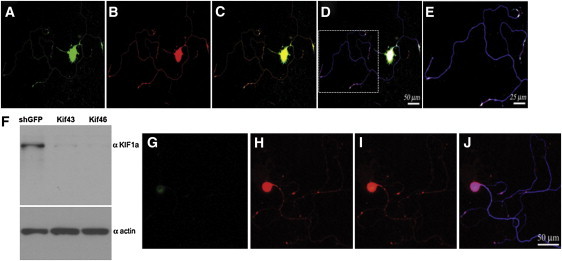
Localization of KIF1A and WNK1/HSN2 and Silencing of KIF1A in Adult Mouse DRG Neurons
(A–E) Sequential laser confocal scans to compare the localization of KIF1A and WNK1/HSN2. Neurons were cultured 48 hr prior to their immunodetection. KIF1A and WNK1/HSN2 colocalize well, as evidenced by the use of anti-KIF1A (A, green), anti-HSN2 (B, red) and anti-betaIII tubulin (blue).
(C) Merged image of (A) and (B); strong colocalization of green and red is yellow.
(D) Merged image of (C) and the matching betaIII tubulin detection; strong colocalization of red and blue is pink.
(E) Enlargement of the boxed image area in (D).
(F–J) Lentiviral shRNA-mediated knockdown of KIF1A. Twenty-four hours after their plating, primary neurons were infected overnight with lentiviral particles expressing shRNA (kif46) so that KIF1A would be silenced. (F) Immunoblot analysis confirmed the knockdown of KIF1A in neurons infected by lentivirus expressing shRNA against KIF1A (Kif43 and Kif46). Control lysates were prepared from cells infected with lentiviruses expressing shRNA against GFP, and an actin antibody showed equal loadings. Cells were grown for 4 days after infection before their immunodetection with anti-KIF1A (G, green), anti-HSN2 (H, red), and anti-betaIII tubulin. (I) Merged image of (G) and (H). (J) Merged image of (I) and the matching betaIII tubulin detection (blue).
Discussion
Our study used two parallel approaches, homozygosity mapping and identification of the binding partners of the domain encoded by the HSN2 exon. These approaches converged to allow us to identify HSANII-causative mutations in the KIF1A gene. Similarly to mutations in the HSN2 exon of WNK1,4,17 all the patients carry mutations in an alternatively spliced exon, at least in a heterozygous state. The HSANII link to the alternatively spliced exon 25b of KIF1A is further supported by Y2H data that showed that, in addition to other regions of KIF1A, the amino acids encoded by this exon participate in the interaction with the HSN2 portion of WNK1 (Figures 1C and 1D). Similarly to HSANII patients with WNK1 mutations, all individuals with KIF1A mutations presented with marked sensory loss and severe mutilations. However, they also showed distal muscle weakness, which is not a typical feature of HSANII patients (Table 4). A variable motor involvement is nonetheless well documented in other HSAN types, particularly in the autosomal-dominant forms.10–13
The only compound heterozygous patient with a mutation in exon 46 exhibits the most severe motor phenotype and is the only one presenting with developmental delay and short stature (Table 4); an interesting observation when one considers that homozygous KIF1A-knockout mice display severe motor and sensory deficits, die within a day of birth, and are significantly smaller than their littermates.22 We could thus speculate that as a result of the central role of KIF1A in neurons,23 homozygous loss-of-function mutations in exons that are always spliced-in may either be lethal or lead to a more severe phenotype than HSANII. However, further families and pathogenic KIF1A mutations will be needed if researchers are to determine whether such a genotype-phenotype correlation exists.
Two recent studies reported potential pathogenic mutations in KIF1A. Hamdan et al. identified a de novo missense mutation in a patient with nonsyndromic intellectual disability,24 and Erlich et al. found a homozygous missense change in a single consanguineous family with Hereditary Spastic Paraparesis.25 Both mutations were located in the kinesin motor domain of KIF1A. Although these mutations were reported in single families and hence should be interpreted with caution, it is noteworthy that all the HSANII patients carrying mutations in KIF1A present with variable degrees of muscle weakness and that one of them has a speech delay.
Although we showed that KIF1A interacts with WNK1/HSN2 and both proteins colocalize along axons of DRG sensory neurons, the functional role of this interaction remains unclear. KIF1A did not appear to be responsible for the presence of WNK1/HSN2 along axons despite its well-established role in anterograde transport and the differential localization between the WNK1 and WNK1/HSN2 isoforms in DRG sensory neurons. Interestingly though, WNK1 phosphorylation of synaptotagmin-2, which is transported with synaptic vesicle precursors by KIF1A,18 reduces its binding to phospholipid vesicles.26 In addition, the phosphorylation state of kinesins modulates both cargo insertion and removal27 and influences the binding of kinesins to microtubules.28 Therefore, it can be hypothesized that the kinase activity of WNK1/HSN2 regulates the unloading of KIF1A cargos at axonal tips of neurons, and such a hypothesis is consistent with the slightly increased colocalization of KIF1A and WNK1/HSN2 at the distal portion of axons (Figure 6C). Of note, the two frameshift mutations identified here are located downstream of the motor and the Forkhead-associated (FHA) domains of KIF1A, but both are predicted to affect the C-terminal lipid binding Pleckstrin homology (PH) domain (Figure 4C), which is necessary for vesicle transport.29
Although the functional relationship between KIF1A and WNK1/HSN2 and the role of the KIF1A exon-25b-containing isoform remain to be elucidated, this study identifies mutations of KIF1A as a rare cause of HSANII and provides clues to the pathogenesis of HSANII and the function of WNK1/HSN2 in addition to further emphasizing the essential role of KIF1A in the peripheral nervous system. Finally, for both WNK1 and KIF1A, the mutations were largely restricted to alternatively spliced exons, underscoring the importance of alternative splicing in the peripheral nervous system.
Acknowledgments
We are grateful to the patients for their contribution to this study. We thank Dr. R.M. King and Prof. Claus for their assistance with this project. J.-B.R. is the recipient of a Canadian Institutes of Health Research scholarship; V.L. and N.M. are the recipients of a Claude Laberge fellowship from the Réseau de Médecine Génétique Appliqué; B.B. was funded by the Association de la Neuropathie Sensorielle et Autonomique Hereditaire de Type 2; G.A.R. was funded by a Canadian Institutes of Health Research grant (#179251). J.B. is supported by a PhD fellowship from the Fund for Scientific Research (FWO-Flanders). Granting The work of the Antwerp-Belgian team was in part funded by the University of Antwerp, the Fund for Scientific Research (FWO-Flanders), the Medical Foundation Queen Elisabeth, the Association Belge contre les Maladies Neuromusculaires, and the Interuniversity Attraction Poles P6/43 program of the Belgian Federal Science Policy Office.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
GenBank, http://www.ncbi.nlm.nih.gov/genbank
HapMap, http://hapmap.ncbi.nlm.nih.gov
McGill University and Génome Québec Innovation Centre, http://gqinnovationcenter.com/index.aspx
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Axelrod F.B. Hereditary sensory and autonomic neuropathies. Familial dysautonomia and other HSANs. Clin. Auton. Res. 2002;12(Suppl 1):I2–I14. doi: 10.1007/s102860200014. [DOI] [PubMed] [Google Scholar]
- 2.Hilz M.J. Assessment and evaluation of hereditary sensory and autonomic neuropathies with autonomic and neurophysiological examinations. Clin. Auton. Res. 2002;12(Suppl 1):I33–I43. doi: 10.1007/s102860200017. [DOI] [PubMed] [Google Scholar]
- 3.Lafreniere R.G., MacDonald M.L., Dube M.P., MacFarlane J., O'Driscoll M., Brais B., Meilleur S., Brinkman R.R., Dadivas O., Pape T., Study of Canadian Genetic Isolates Identification of a novel gene (HSN2) causing hereditary sensory and autonomic neuropathy type II through the Study of Canadian Genetic Isolates. Am. J. Hum. Genet. 2004;74:1064–1073. doi: 10.1086/420795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shekarabi M., Girard N., Rivière J.B., Dion P., Houle M., Toulouse A., Lafrenière R.G., Vercauteren F., Hince P., Laganiere J. Mutations in the nervous system—Specific HSN2 exon of WNK1 cause hereditary sensory neuropathy type II. J. Clin. Invest. 2008;118:2496–2505. doi: 10.1172/JCI34088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Einarsdottir E., Carlsson A., Minde J., Toolanen G., Svensson O., Solders G., Holmgren G., Holmberg D., Holmberg M. A mutation in the nerve growth factor beta gene (NGFB) causes loss of pain perception. Hum. Mol. Genet. 2004;13:799–805. doi: 10.1093/hmg/ddh096. [DOI] [PubMed] [Google Scholar]
- 6.Indo Y., Tsuruta M., Hayashida Y., Karim M.A., Ohta K., Kawano T., Mitsubuchi H., Tonoki H., Awaya Y., Matsuda I. Mutations in the TRKA/NGF receptor gene in patients with congenital insensitivity to pain with anhidrosis. Nat. Genet. 1996;13:485–488. doi: 10.1038/ng0896-485. [DOI] [PubMed] [Google Scholar]
- 7.Bouhouche A., Benomar A., Bouslam N., Chkili T., Yahyaoui M. Mutation in the epsilon subunit of the cytosolic chaperonin-containing t-complex peptide-1 (Cct5) gene causes autosomal recessive mutilating sensory neuropathy with spastic paraplegia. J. Med. Genet. 2006;43:441–443. doi: 10.1136/jmg.2005.039230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Slaugenhaupt S.A., Blumenfeld A., Gill S.P., Leyne M., Mull J., Cuajungco M.P., Liebert C.B., Chadwick B., Idelson M., Reznik L. Tissue-specific expression of a splicing mutation in the IKBKAP gene causes familial dysautonomia. Am. J. Hum. Genet. 2001;68:598–605. doi: 10.1086/318810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kurth I., Pamminger T., Hennings J.C., Soehendra D., Huebner A.K., Rotthier A., Baets J., Senderek J., Topaloglu H., Farrell S.A. Mutations in FAM134B, encoding a newly identified Golgi protein, cause severe sensory and autonomic neuropathy. Nat. Genet. 2009;41:1179–1181. doi: 10.1038/ng.464. [DOI] [PubMed] [Google Scholar]
- 10.Dawkins J.L., Hulme D.J., Brahmbhatt S.B., Auer-Grumbach M., Nicholson G.A. Mutations in SPTLC1, encoding serine palmitoyltransferase, long chain base subunit-1, cause hereditary sensory neuropathy type I. Nat. Genet. 2001;27:309–312. doi: 10.1038/85879. [DOI] [PubMed] [Google Scholar]
- 11.Verhoeven K., De Jonghe P., Coen K., Verpoorten N., Auer-Grumbach M., Kwon J.M., FitzPatrick D., Schmedding E., De Vriendt E., Jacobs A. Mutations in the small GTP-ase late endosomal protein RAB7 cause Charcot-Marie-Tooth type 2B neuropathy. Am. J. Hum. Genet. 2003;72:722–727. doi: 10.1086/367847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rotthier A., Auer-Grumbach M., Janssens K., Baets J., Penno A., Almeida-Souza L., Van Hoof K., Jacobs A., De Vriendt E., Schlotter-Weigel B. Mutations in the SPTLC2 subunit of serine palmitoyltransferase cause hereditary sensory and autonomic neuropathy type I. Am. J. Hum. Genet. 2010;87:513–522. doi: 10.1016/j.ajhg.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guelly C., Zhu P.P., Leonardis L., Papić L., Zidar J., Schabhüttl M., Strohmaier H., Weis J., Strom T.M., Baets J. Targeted high-throughput sequencing identifies mutations in atlastin-1 as a cause of hereditary sensory neuropathy type I. Am. J. Hum. Genet. 2011;88:99–105. doi: 10.1016/j.ajhg.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murray T.J. Congenital sensory neuropathy. Brain. 1973;96:387–394. doi: 10.1093/brain/96.2.387. [DOI] [PubMed] [Google Scholar]
- 15.Ota M., Ellefson R.D., Lambert E.H., Dyck P.J. Hereditary sensory neuropathy, type II. Clinical, electrophysiologic, histologic, and biochemical studies of a Quebec kinship. Arch. Neurol. 1973;29:23–37. doi: 10.1001/archneur.1973.00490250041005. [DOI] [PubMed] [Google Scholar]
- 16.Roddier K., Thomas T., Marleau G., Gagnon A.M., Dicaire M.J., St-Denis A., Gosselin I., Sarrazin A.M., Larbrisseau A., Lambert M. Two mutations in the HSN2 gene explain the high prevalence of HSAN2 in French Canadians. Neurology. 2005;64:1762–1767. doi: 10.1212/01.WNL.0000161849.29944.43. [DOI] [PubMed] [Google Scholar]
- 17.Rotthier A., Baets J., De Vriendt E., Jacobs A., Auer-Grumbach M., Lévy N., Bonello-Palot N., Kilic S.S., Weis J., Nascimento A. Genes for hereditary sensory and autonomic neuropathies: A genotype-phenotype correlation. Brain. 2009;132:2699–2711. doi: 10.1093/brain/awp198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okada Y., Yamazaki H., Sekine-Aizawa Y., Hirokawa N. The neuron-specific kinesin superfamily protein KIF1A is a unique monomeric motor for anterograde axonal transport of synaptic vesicle precursors. Cell. 1995;81:769–780. doi: 10.1016/0092-8674(95)90538-3. [DOI] [PubMed] [Google Scholar]
- 19.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Radonić A., Thulke S., Mackay I.M., Landt O., Siegert W., Nitsche A. Guideline to reference gene selection for quantitative real-time PCR. Biochem. Biophys. Res. Commun. 2004;313:856–862. doi: 10.1016/j.bbrc.2003.11.177. [DOI] [PubMed] [Google Scholar]
- 21.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 22.Yonekawa Y., Harada A., Okada Y., Funakoshi T., Kanai Y., Takei Y., Terada S., Noda T., Hirokawa N. Defect in synaptic vesicle precursor transport and neuronal cell death in KIF1A motor protein-deficient mice. J. Cell Biol. 1998;141:431–441. doi: 10.1083/jcb.141.2.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Otsuka A.J., Jeyaprakash A., García-Añoveros J., Tang L.Z., Fisk G., Hartshorne T., Franco R., Born T. The C. elegans unc-104 gene encodes a putative kinesin heavy chain-like protein. Neuron. 1991;6:113–122. doi: 10.1016/0896-6273(91)90126-k. [DOI] [PubMed] [Google Scholar]
- 24.Hamdan F.F., Gauthier J., Araki Y., Lin D.T., Yoshizawa Y., Higashi K., Park A.R., Spiegelman D., Dobrzeniecka S., Piton A., S2D Group Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. Am. J. Hum. Genet. 2011;88:306–316. doi: 10.1016/j.ajhg.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Erlich Y., Edvardson S., Hodges E., Zenvirt S., Thekkat P., Shaag A., Dor T., Hannon G.J., Elpeleg O. Exome sequencing and disease-network analysis of a single family implicate a mutation in KIF1A in hereditary spastic paraparesis. Genome Res. 2011;21:658–664. doi: 10.1101/gr.117143.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee B.H., Min X., Heise C.J., Xu B.E., Chen S., Shu H., Luby-Phelps K., Goldsmith E.J., Cobb M.H. WNK1 phosphorylates synaptotagmin 2 and modulates its membrane binding. Mol. Cell. 2004;15:741–751. doi: 10.1016/j.molcel.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 27.Sato-Yoshitake R., Yorifuji H., Inagaki M., Hirokawa N. The phosphorylation of kinesin regulates its binding to synaptic vesicles. J. Biol. Chem. 1992;267:23930–23936. [PubMed] [Google Scholar]
- 28.Morfini G., Pigino G., Szebenyi G., You Y., Pollema S., Brady S.T. JNK mediates pathogenic effects of polyglutamine-expanded androgen receptor on fast axonal transport. Nat. Neurosci. 2006;9:907–916. doi: 10.1038/nn1717. [DOI] [PubMed] [Google Scholar]
- 29.Klopfenstein D.R., Vale R.D. The lipid binding pleckstrin homology domain in UNC-104 kinesin is necessary for synaptic vesicle transport in Caenorhabditis elegans. Mol. Biol. Cell. 2004;15:3729–3739. doi: 10.1091/mbc.E04-04-0326. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.