

Abstract
Charcot-Marie-Tooth disease is characterized by length-dependent axonal degeneration with distal sensory loss and weakness, deep-tendon-reflex abnormalities, and skeletal deformities. It is caused by mutations in more than 40 genes. We investigated a four-generation family with 23 members affected by the axonal form (type 2), for which the common causes had been excluded by Sanger sequencing. Exome sequencing of three affected individuals separated by eight meioses identified a single shared novel heterozygous variant, c.917A>G, in DYNC1H1, which encodes the cytoplasmic dynein heavy chain 1 (here, novel refers to a variant that has not been seen in dbSNP131or the August 2010 release of the 1000 Genomes project). Testing of six additional affected family members showed cosegregation and a maximum LOD score of 3.6. The shared DYNC1H1 gene variant is a missense substitution, p.His306Arg, at a highly conserved residue within the homodimerization domain. Three mouse models with different mutations within this domain have previously been reported with age-related progressive loss of muscle bulk and locomotor ability. Cytoplasmic dynein is a large multisubunit motor protein complex and has a key role in retrograde axonal transport in neurons. Our results highlight the importance of dynein and retrograde axonal transport in neuronal function in humans.
Main Text
Charcot-Marie-Tooth (CMT) disease is the most common inherited neuromuscular disorder and has an estimated prevalence of 1 in 2500. It is a chronic motor and sensory polyneuropathy characterized by distal muscle weakness and atrophy that might be associated with sensory loss, depressed tendon reflexes, and pes cavus. There are two main forms: the demyelinating type 1 that affects myelin (CMT1) and the axonal type 2 affecting the nerve axon (CMT2). Autosomal-dominant inheritance is most common, but autosomal-recessive and X-linked forms are also seen. There is considerable genetic heterogeneity, and more than 40 genes or loci have been identified.1 We studied a four-generation family with 23 members affected (Figure 1) with CMT2, characterized by delayed motor milestones and/or an abnormal gait. Sanger sequencing excluded the genes commonly associated with CMT2; MPZ (MIM 159440), NEFL (MIM 162280), MFN2 (MIM 608507), and LMNA (MIM 150330). Male to male transmission excluded GJB1 (MIM 304040), and after sequencing of PMP22 (MIM 601097) failed to identify a pathogenic mutation, we employed a whole-exome sequencing strategy.
Figure 1.
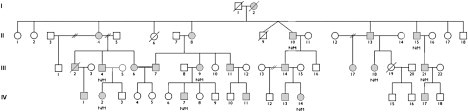
Partial Pedigree Showing Mutation Status
Open symbols represent unaffected individuals; filled squares represent affected males and filled circles affected females. Individual generations are numbered with roman numerals on the left. Individuals heterozygous for the p.His306Arg mutation are indicated as N/M. For the LOD-score calculation we assumed a rare autosomal-dominant model with a disease-allele frequency of 0.0001 and phenocopy rate of 0. The mutant DYNC1H1 allele is rare (frequency <0.001); because we only tested affected individuals to avoid issues of predictive testing and variable penetrance and because there are 12 meioses separating the nine sequenced individuals, the LOD score for the variant can be approximated as −log10(0.512) = 3.6.
Exonic sequences from three affected individuals (IV-2, -7, and -14) separated by eight meioses were enriched from genomic DNA with Agilent's SureSelect whole-exome kit (version 1) as recommended by the manufacturers except that gDNA fragmentation was carried out with a Diagenode Bioruptor. After hybridization reactions, captured DNA was amplified with 12 cycles of PCR then sequenced on an Illumina GAII sequencer with 76 bp paired-end reads. We used Novoalign (Novocraft Technologies) to align sequence reads to the hg19 reference genome and removed any duplicate reads from subsequent analyses. Seventy-six percent of targeted Consensus Coding Sequence project (CCDS) exonic bases were covered with at least 20 reads with an average coverage of 70× (Table S1, available online). We used Samtools2 to call SNPs and indels. Annovar3 was used for annotation of variants, and we filtered variants by using dbSNP131, 1000 Genomes (August 2010 release), and our in-house (exome data from 11 individuals of European descent) databases. Informed consent was obtained from all participants.
We assumed a rare autosomal-dominant model of inheritance, and after filtering, we found the three individuals had 177, 192, and 199 novel heterozygous variants (here, novel refers to a variant that has not been seen in dbSNP131or the August 2010 release of the 1000 Genomes project). annotated as missense, nonsense, frameshift, or splice site (Table 1 and Table S2). Four to six variants were shared by each pair, but only one variant in DYNC1H1 (NM_001376.4 [MIM 600112]) was shared by all three (the number expected on the basis of the number of meioses separating these individuals). Testing of six additional affected family members by Sanger sequencing showed cosegregation with a maximum LOD score of 3.6 (see Figure 1). The variant was not present in 322 ethnically matched control chromosomes. The shared DYNC1H1 variant is a missense change, p.His306Arg (c.917A>G), at a highly conserved residue (Figure S1) within the homodimerization domain of cytoplasmic dynein heavy chain 1.
Table 1.
Novel Heterozygous Missense, Nonsense, Frameshift, or Splice-Site Variants
|
Individuals |
Shared |
|||||
|---|---|---|---|---|---|---|
| IV-2 | IV-7 | IV-14 | IV-2 and IV-7 | IV-2 and IV-14 | IV-7 and IV-14 | All |
| 177 | 192 | 199 | 6 | 5 | 4 | 1 |
Number of novel heterozygous variants in the exome of each sequenced individual and the number shared across the individuals. Here, “novel” refers to a variant that has not been seen in dbSNP131, the August 2010 release of the 1000 Genomes project, or 11 in-house control exomes processed with the same laboratory and analysis pipeline.
Cytoplasmic dynein is a large multisubunit motor protein complex that has a range of cellular functions. In particular, it is the primary motor protein responsible for retrograde axonal transport in neurons—the movement of cargo such as organelles from the cell periphery to the cell body along cytoskeletal microtubules. A homodimer of the 532 kDa cytoplasmic dynein heavy chain 1 forms the core of dynein and is responsible for the protein complex binding to and moving along microtubules.4 Other subunits of dynein include the intermediate, light-intermediate, and light chains, thought to be responsible for interacting with cargo and maintaining the stability of the complex.4
Our results are corroborated by previous animal studies that have implicated disruption of Dync1h1 in neuropathic disease. Legs at odd angles (Loa)5, Cramping 1 (Cra1)5 and Sprawling (Swl)6 are mouse phenotypes that arose from N-ethyl-N-nitrosourea (ENU) or radiation-induced mutagenesis. Heterozygous mice have age-related progressive loss of muscle bulk and locomotor ability without a major reduction in life span.5,6 Although there are differences between the mouse models, and indeed different pathogenesis suggested for the same mouse model by different researchers,7,8 the mouse phenotype is broadly similar to that observed in our family. Affected patients typically have delayed motor milestones and/or an abnormal gait along with early-onset slowly progressive distal lower limb weakness and wasting with pes cavus deformity (see Figure S2). Clinical features of the best characterized family members are shown in Table 2. Upper limb involvement is less common, and ambulation is usually maintained through adulthood. Nerve conduction studies are within the normal range, sural nerve biopsies demonstrated minimal changes suggestive of increased axon degeneration, and muscle biopsies show findings consistent with secondary muscle involvement due to denervation (see Table 3). Transient paresthesia and neuropathic lower limb pains are also reported by several family members, typically those more severely affected. Although the core features consistent with CMT2 are seen across the family, there is significant variability in other findings: reflexes might be lost or preserved; fine touch, vibration sense, and proprioception are retained in many family members but lost in some; atypical features, such as predominant proximal muscle involvement, periscapular wasting and weakness, spinal and hip problems, and a broad-based waddling gait, are also found in a few.
Table 2.
Clinical Features of Selected Family Members
| Patient | Presentation | Features |
|---|---|---|
| II 10 | delayed motor milestones; recurrent surgery to feet and ankles in childhood | reduced power and wasting in distal upper and lower limbs; pes cavus; reduced proprioception, normal vibration sense; reflexes normal |
| II 13 | pes cavus at birth; recurrent surgery to feet and ankles in childhood | distal lower limb weakness and wasting; pes cavus; reduced proprioception, pin-prick, fine touch and vibration sense; reflexes normal; significant neuropathic pain; depression and paraphrenia; extrapyramidal features probably due to antipsychotics |
| III 4 | delayed motor milestones and recurrent falls | mild distal lower limb weakness and wasting; pes cavus; reduced fine touch, other sensory modalities normal; reflexes normal |
| III 7 | presented at age 11 with difficulties running | mild distal lower limb weakness and paresthesia; pes cavus; all sensory modalities reduced; reduced reflexes |
| III 9 | presented at age 14 with frequent falls | distal upper and lower limb weakness and wasting (LL > UL); pes cavus; all sensory modalities reduced; reduced ankle reflexes |
| III 11 | talipes at birth; delayed motor milestones | distal lower limb weakness and wasting; pes cavus; retained reflexes and sensation; retinoblastoma |
| III 14 | delayed motor milestones | broad-based gait; distal lower limb weakness and wasting; reduced reflexes; sensory modalities retained; mild intention tremor; strabismus |
| III 17 | abnormal gait in early childhood | minimally reduced power and wasting in distal lower limbs; normal reflexes and sensation; no pes cavus |
| III 18 | delayed motor milestones; speech delay | proximal and distal lower limbs weakness and wasting and scapula wasting; pes cavus and bilateral foot drop; reflexes reduced; paraesthesia but objectively normal sensation; significant neuropathic pain and back pain |
| III 21 | delayed motor milestones and learning difficulties | proximal and distal lower limb weakness and wasting; reduced reflexes; normal sensory modality testing; lumbar lordosis and reduced hip movements; behavioral problems including school exclusions |
| IV 2 | abnormal gait and falls in early childhood; global developmental delay | distal lower limb weakness; no pes cavus; normal reflexes and sensation |
| IV 7 | delayed motor milestones; speech delay and learning difficulties | broad-based gait; distal lower limb weakness and wasting; no pes cavus; reduced reflexes; reduced proprioception, other sensory modalities normal; lumbar lordosis and reduced hip movements; Achilles-tendon-lengthening operations |
| IV 14 | delayed motor milestones | Waddling gait; proximal lower limb weakness more dominant than distal lower limb weakness; no pes cavus; reduced knee reflexes; sensory modalities normal; lumbar lordosis |
Table 3.
Clinical Investigation Results
| Patient | Age at Study |
Motor |
Sensory |
Electromyography | Sural Nerve Biopsy | Muscle Biopsy | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Right Peroneal |
Right Tibial |
Right Sural |
Right Sup Peroneal |
|||||||||
| Amplitude (mV) | Velocity (m/s) | Amplitude (mV) | Velocity (m/s) | Pk-Pk (μV) | Velocity (m/s) | Pk-Pk (μV) | Velocity (m/s) | |||||
| II 10 | 29 | – | – | 3.85 | 46 | 20 | 39 | – | – | chronic denervation | – | – |
| II 13 | 31 | – | – | 4.3 | 51 | 12 | 41 | – | – | central neuronal atrophy | – | severe connective tissue replacement |
| II 15 | 36 | normal | normal | normal | normal | normal | normal | normal | normal | – | indolent axon degenerative process with some evidence of fiber regeneration consistent with a diagnosis of HMSN type 2 | – |
| III 4 | 25 | 2.0 | 56.6 | 8.9 | – | 20.3 | 42.9 | 13.3 | 45.5 | normal | – | – |
| III 14 | 28 | 5.4 | 44.2 | 7.1 | 39.6 | 8.6 | 37.7 | 7.8 | 40 | vastus medialis MUPs enlarged otherwise normal | – | – |
| III 17 | 20 | normal | normal | normal | normal | normal | normal | normal | normal | – | – | – |
| III 18 | 22 | 2.9 | 44 | 11.5 | – | 7 | 28 | 15 | 35 | mild degree of large fiber axonal sensory motor peripheral neuropathy | – | – |
| III 21 | 2 | normal | normal | normal | normal | normal | normal | normal | normal | interference pattern nonspecifically abnormal, occasional units up to 6 mV | – | – |
| IV 7 | 5 | not performed | not performed | not performed | not performed | not performed | not performed | not performed | not performed | – | – | atrophic fibers consistent with chronic partial denervation |
The mouse mutations of Dync1h1 (NM_030238.2; c.1739T>A [p.Phe580Tyr] in Loa; c.3164A>G [p.Tyr1055Cys] in Cra; and a 9 bp deletion, c.3119_3127delGCATAGTGA [p.(Gly1040_Thr1043delinsAla)], in Swl) also affect residues within the homodimerization region of the dynein stem domain (Figure 2). The embryonic lethality of the homozygous Dync1h1 null mouse but normal phenotype of heterozygous null mice9 raises the possibility of a dominant-negative effect for the neuropathic mutations. Given the multiple functions of dynein, the exact mechanism by which these mutations cause disease is still unclear. One recent study of the Cra mouse suggested an impact on synapse structure at neuromuscular junctions,7 whereas another proposed an effect on striatal neurons.8 However, a recent report with single-molecule and live-imaging techniques in the Loa mouse strongly suggested that defects in dynein motor processivity play a key role in disease causation.10 Measurement of retrograde run-lengths (periods of uninterrupted motion) showed a marked reduction in the ability of mutant dynein to move along microtubules both in vitro and in vivo.10 There was also evidence of altered interaction between the dynein motor and stem domains, and this interaction raises the possibility of motor domain miscoordination.10 This reduction in processivity could explain the observed neuropathy phenotype because the neurons most affected will be those with long axons such as the motor and sensory neurons.
Figure 2.
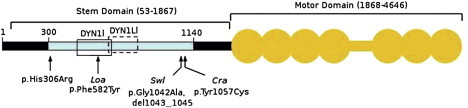
Schematic Representation of Human DYNC1H1
The N-terminal region of DYNC1H1 is represented by a horizontal black bar, and the stem domain (amino acids 53–1867) is indicated by a bracket above. Residues involved in DYNC1H1 dimerization (300–1140) are shown by a light blue bar; open boxes represent binding regions for intermediate (DYN1I; residues 448–703, solid line) and light-intermediate (DYN1LI; residues 651–802, broken line) chains, respectively. The motor domain (amino acids 1868–4646) is indicated by the orange shaded area; the seven ATPase domains are represented by circles, whereas the horizontal bar indicates the stalk region. The equivalent positions of mutations in three mouse models, Loa, Swl, and Cra1, are shown below the figure along with the p.His306Arg mutation identified in the family reported here. Note that the human protein contains two additional glycine residues at position 7 relative to mouse Dync1h1, that is numbering of equivalent residues in human DYNC1H1 is 2 higher than in mouse models. Phe582 is within a highly conserved domain responsible for binding the dynein intermediate chains as well as homodimerization.4 The 9 bp Swl deletion and Cra p.Tyr1057Cys mutation are outside the dynein intermediate-chain binding region but within the putative homodimerization domain.4 Numbering of amino acids is taken from Tynan et al.16 and the UniProtKB entry for human DYNC1H1 (identifier Q14204).
Mutations in other axonal transport genes cause related neuronal diseases. Autosomal-dominant mutations in the p150 subunit of dynactin (also known as dynein activator) cause a form of motor neuron disease (MIM 601143)11 and a heterozygous KIF1B (MIM 605995) mutation results in CMT2A (MIM 118210).12 KIF1B is a member of the kinesin superfamily of motor proteins that are involved in anterograde axonal transport (taking cargo from the cell body to the periphery of the neurons). Mutations in other kinesin genes have been shown to cause neuronal disease, for example KIF5A (MIM 602821) mutations cause hereditary spastic paraplegia (MIM 604187).13 Our findings provide further evidence of the importance of motor proteins and in particular retrograde axonal transport in the normal functioning of neurons.
Our work has used exome sequencing in selected individuals from a large pedigree to identify a disease-causing mutation. Charcot-Marie-Tooth disease can be caused by mutations in over 40 genes, and screening with Sanger sequencing is costly and time consuming. DYNC1H1 (14q32.31) is not within a known region of linkage for CMT. An alternative approach,14,15 linkage analysis followed by candidate gene Sanger sequencing, utilized in other studies would still have required the sequencing of at least the 78 exons of DYNC1H1.
In summary, we have identified a mutation in DYNC1H1 that causes Charcot-Marie-Tooth disease by using exome sequencing in a large dominant pedigree. The mouse models also harboring mutations in the homodimerization domain of cytoplasmic dynein heavy chain 1 support the pathogenicity of the human DYNC1H1 mutation and give valuable insights to pathophysiology. These results highlight the importance of dynein and retrograde axonal transport in neuronal function in humans.
Acknowledgments
We would like to express our gratitude to the participating family and to Alex Moorhouse and Andy Brash for technical assistance. Sequence analysis of MFN2 was performed by the Institute of Neurology (London, UK).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://www.1000genomes.org/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
References
- 1.Patzkó A., Shy M.E. Update on Charcot-Marie-Tooth disease. Curr. Neurol. Neurosci. Rep. 2011;11:78–88. doi: 10.1007/s11910-010-0158-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Banks G.T., Fisher E.M. Cytoplasmic dynein could be key to understanding neurodegeneration. Genome Biol. 2008;9:214. doi: 10.1186/gb-2008-9-3-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hafezparast M., Klocke R., Ruhrberg C., Marquardt A., Ahmad-Annuar A., Bowen S., Lalli G., Witherden A.S., Hummerich H., Nicholson S. Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science. 2003;300:808–812. doi: 10.1126/science.1083129. [DOI] [PubMed] [Google Scholar]
- 6.Chen X.J., Levedakou E.N., Millen K.J., Wollmann R.L., Soliven B., Popko B. Proprioceptive sensory neuropathy in mice with a mutation in the cytoplasmic Dynein heavy chain 1 gene. J. Neurosci. 2007;27:14515–14524. doi: 10.1523/JNEUROSCI.4338-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Courchesne S.L., Pazyra-Murphy M.F., Lee D.J., Segal R.A. Neuromuscular junction defects in mice with mutation of dynein heavy chain 1. PLoS ONE. 2011;6:e16753. doi: 10.1371/journal.pone.0016753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Braunstein K.E., Eschbach J., Ròna-Vörös K., Soylu R., Mikrouli E., Larmet Y., René F., De Aguilar J.L., Loeffler J.P., Müller H.P. A point mutation in the dynein heavy chain gene leads to striatal atrophy and compromises neurite outgrowth of striatal neurons. Hum. Mol. Genet. 2010;19:4385–4398. doi: 10.1093/hmg/ddq361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harada A., Takei Y., Kanai Y., Tanaka Y., Nonaka S., Hirokawa N. Golgi vesiculation and lysosome dispersion in cells lacking cytoplasmic dynein. J. Cell Biol. 1998;141:51–59. doi: 10.1083/jcb.141.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ori-McKenney K.M., Xu J., Gross S.P., Vallee R.B. A cytoplasmic dynein tail mutation impairs motor processivity. Nat. Cell Biol. 2010;12:1228–1234. doi: 10.1038/ncb2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Puls I., Jonnakuty C., LaMonte B.H., Holzbaur E.L., Tokito M., Mann E., Floeter M.K., Bidus K., Drayna D., Oh S.J. Mutant dynactin in motor neuron disease. Nat. Genet. 2003;33:455–456. doi: 10.1038/ng1123. [DOI] [PubMed] [Google Scholar]
- 12.Zhao C., Takita J., Tanaka Y., Setou M., Nakagawa T., Takeda S., Yang H.W., Terada S., Nakata T., Takei Y. Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bbeta. Cell. 2001;105:587–597. doi: 10.1016/s0092-8674(01)00363-4. [DOI] [PubMed] [Google Scholar]
- 13.Reid E., Kloos M., Ashley-Koch A., Hughes L., Bevan S., Svenson I.K., Graham F.L., Gaskell P.C., Dearlove A., Pericak-Vance M.A. A kinesin heavy chain (KIF5A) mutation in hereditary spastic paraplegia (SPG10) Am. J. Hum. Genet. 2002;71:1189–1194. doi: 10.1086/344210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ostergaard P., Simpson M.A., Brice G., Mansour S., Connell F.C., Onoufriadis A., Child A.H., Hwang J., Kalidas K., Mortimer P.S. Rapid identification of mutations in GJC2 in primary lymphoedema using whole exome sequencing combined with linkage analysis with delineation of the phenotype. J. Med. Genet. 2011;48:251–255. doi: 10.1136/jmg.2010.085563. [DOI] [PubMed] [Google Scholar]
- 15.Wang J.L., Yang X., Xia K., Hu Z.M., Weng L., Jin X., Jiang H., Zhang P., Shen L., Guo J.F. TGM6 identified as a novel causative gene of spinocerebellar ataxias using exome sequencing. Brain. 2010;133:3510–3518. doi: 10.1093/brain/awq323. [DOI] [PubMed] [Google Scholar]
- 16.Tynan S.H., Gee M.A., Vallee R.B. Distinct but overlapping sites within the cytoplasmic dynein heavy chain for dimerization and for intermediate chain and light intermediate chain binding. J. Biol. Chem. 2000;275:32769–32774. doi: 10.1074/jbc.M001537200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.