

Abstract
Monogenic disorders offer unique opportunities for researchers to shed light upon fundamental physiological processes in humans. We investigated a large family affected with autosomal-dominant adermatoglyphia (absence of fingerprints) also known as the “immigration delay disease.” Using linkage and haplotype analyses, we mapped the disease phenotype to 4q22. One of the genes located in this interval is SMARCAD1, a member of the SNF subfamily of the helicase protein superfamily. We demonstrated the existence of a short isoform of SMARCAD1 exclusively expressed in the skin. Sequencing of all SMARCAD1 coding and noncoding exons revealed a heterozygous transversion predicted to disrupt a conserved donor splice site adjacent to the 3′ end of a noncoding exon uniquely present in the skin-specific short isoform of the gene. This mutation segregated with the disease phenotype throughout the entire family. Using a minigene system, we found that this mutation causes aberrant splicing, resulting in decreased stability of the short RNA isoform as predicted by computational analysis and shown by RT-PCR. Taken together, the present findings implicate a skin-specific isoform of SMARCAD1 in the regulation of dermatoglyph development.
Main Text
Epidermal ridges are characteristic features of the human skin1 and in wide use in the modern era as almost unsurpassed identification tools. The physiological role of epidermal ridges remains controversial. Recent data have dismissed the theory that fingerprints might improve the grip by ramping up friction levels.2 Instead, epidermal ridges might amplify vibratory signals to deeply embedded nerves involved in fine texture perception.3
The factors underlying the formation of epidermal ridges during embryonic development and their pattern remain unknown but are likely to include both genetically determined traits4 as well as environmental elements5 and to involve some form of interactions between the mesenchymal and the dermal and the epidermal elements. At 24 weeks postfertilization, the epidermal-ridge system displays an adult morphology6 that remains permanent without any modification throughout life. The congenital absence of epidermal ridges is a rare condition known as adermatoglyphia (ADG). To date only four families with congenital absence of fingerprints have been described.7–10 In three of these families,7–9 additional features such as congenital facial milia, skin blisters, and fissures associated with heat or trauma were reported. A number of more complex syndromes such as Naegeli-Franceschetti-Jadassohn syndrome (MIM 161000) and dyskeratosis congenita (MIM 305000) also feature abnormal development of epidermal ridges,11,12 as detailed in a recent review of the topic.13
In the present study we investigated a large Swiss kindred presenting with autosomal-dominant adermatoglyphia recently coined as the “immigration delay disease”13 because affected individuals report significant difficulties entering countries that require fingerprint recording. All affected members of this family displayed since birth an absence of fingerprints (Figure 1A); histological analysis13 revealed that this absence was associated with a reduced number of sweat glands and a sweat test showed a reduced ability for hand transpiration (Figure 1B).
Figure 1.
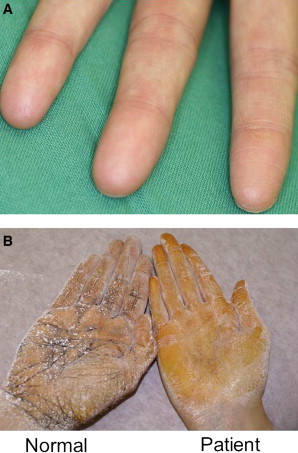
Clinical Features
(A and B) Absence of fingerprints (A) and reduced hand perspiration demonstrated by sweat test (B) in a patient with adermatoglyphia.
All affected (n = 9) and healthy (n = 7) family members or their legal guardian provided written and informed consent according to a protocol approved by the institutional review board of University Hospital Basel in adherence with the principles of the declaration of Helsinki. DNA was extracted from peripheral blood lymphocytes. We initially genotyped all family members by using the Illumina Human Linkage-12 chip comprising 6000 tagged SNPs distributed across the genome. Two hundred ng of DNA were hybridized according to the Infinium II assay (Illumina, San Diego, CA) and scanned with an Illumina BeadArray reader. The scanned images were imported into BeadStudio 3.1.3.0 (Illumina) for extraction and quality control, with an average call rate of 99.9%.
Multipoint linkage analysis with the Superlink software14 generated a LOD score of 2.85 at marker rs1509948 (Figure 2). Fine mapping of the disease interval was performed with polymorphic microsatellite markers that were selected from the National Center for Biotechnology Infromation (NCBI) database. Genotypes were established with fluorescently labeled primer pairs (Research Genetics, Invitrogen, Carlsbad, CA) according to the manufacturer's recommendations. PCR products were separated by PAGE on an automated sequencer (ABI PRISM 3100 Genetic Analyzer; Applied Biosystems, Foster City, CA), and allele sizes were determined with Gene Mapper v4.0 software. Haplotype analysis refined the disease locus to a 5.1 Mb interval between markers D4S423 and D4S1560 (Figure 2).
Figure 2.
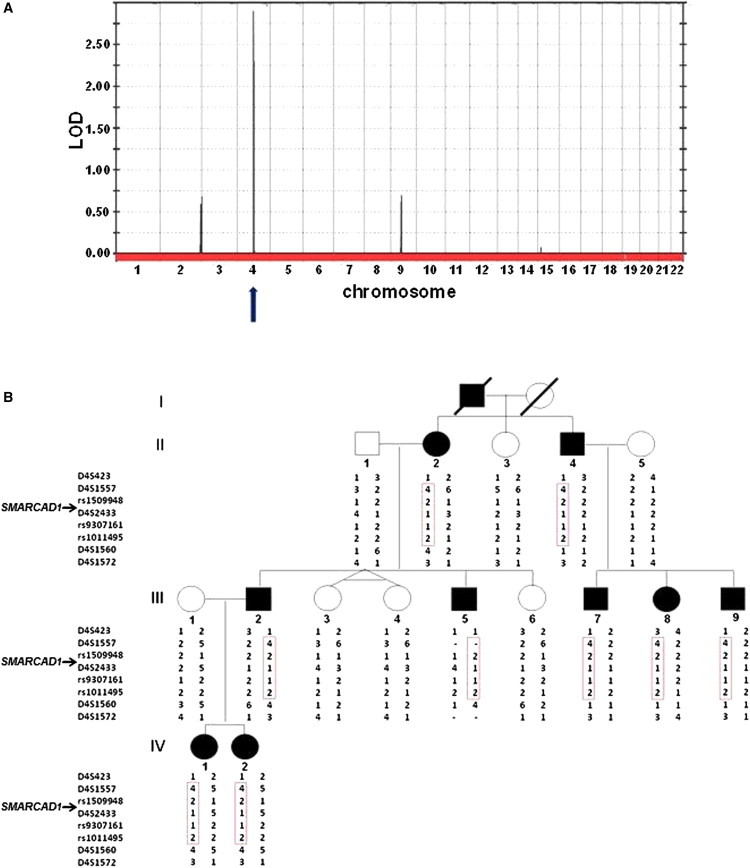
Genetic Mapping of ADG
(A) Multipoint LOD score analysis was performed with the SuperLink software. LOD scores are plotted against all SNP markers distributed across the genome.
(B) Haplotype analysis with polymorphic markers on chromosomal region 4q22 reveals a heterozygous 5.1 Mb interval between markers D4S423 and D4S1560 uniquely shared by all patients (boxed in red).
We found the disease interval contained 17 genes. All coding and noncoding exons of the disease interval genes were fully sequenced. Initially, no mutation was identified. We therefore carefully scrutinized all currently available databases for rare transcripts. We identified one minor transcript (ENST00000509418, NM_001128430.1), sharing a common nucleotide sequence with the 3′-end of SMARCAD1 (MIM 612761). SMARCAD1 encodes a protein that is structurally related to the SWI2/SNF2 superfamily of DNA-dependent ATPases, which function as catalytic subunits of chromatin-remodeling complexes and are consequently considered to be major regulators of transcriptional activity.15 The two SMARCAD1 isoforms differ in lengths and sites of transcription initiation. The shortest SMARCAD1 isoform is predicted to contain a unique 5′-nontranslated exon (Figure 3A). It is of interest that, in contrast with the major large isoform, which was found to be expressed ubiquitously as previously shown,16 the SMARCAD1 short isoform was mainly identifiable by RT-PCR in skin fibroblasts and to a lesser extent in keratinocytes and esophageal tissue (Figure 4), suggesting that it might represent an attractive candidate gene for a skin condition such as ADG.
Figure 3.
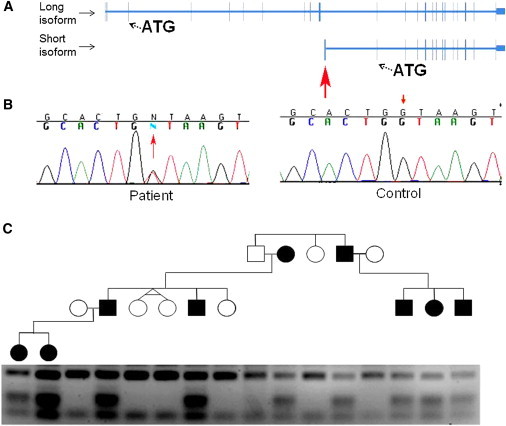
Mutation Analysis
(A) Bioinformatics analysis indicated the existence of two SMARCAD1 isoforms differing both in lengths and sites of transcription start site. The short SMARCAD1 isoform contains a unique nontranslated exon (red arrow).
(B) Sequence analysis revealed a heterozygous transversion, c.378+1G>T, in the short SMARCAD1 isoform (red arrow, left panel). The wild-type sequence is given for comparison (right panel).
(C) PCR-RFLP analysis confirmed segregation of the mutation in the family. Mutation c.378+1G>T creates a recognition site for MseI endonuclease; thus, healthy individuals display fragments of 163 bp and 46 bp, whereas affected heterozygous patients show in addition fragments of 73 bp and 90 bp.
Figure 4.
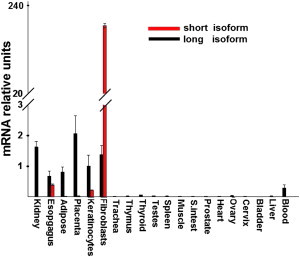
Tissue Expression of SMARCAD1 Isoforms
SMARCAD1 isoform expression was assessed with Clontech tissue blot cDNA array. Quantitative RT-PCR analysis showed that the long SMARCAD1 isoform is expressed ubiquitously at low level. In contrast, the short SMARCAD1 isoform was found to be expressed mainly in skin fibroblasts, keratinocytes, and the esophagus. Expression of SMARCAD1 was normalized to that of ACTB. Results are provided as the fold change of expression of SMARCAD1 long isoform expression in keratinocytes ± standard deviation.
To assess the possible involvement of SMARCAD1 in ADG, genomic DNA was amplified by PCR with primer pairs spanning the entire coding sequence of both SMARCAD1 isoforms (Table S1, available online) and Taq polymerase (QIAGEN, Valencia, CA). Cycling conditions were 94°C for 2 min followed by three cycles at 94°C for 40 s, 61°C for 40 s, and 72°C for 40 s; three cycles at 94°C for 40 s, 59°C for 40 s, and 72°C for 40 s; three cycles at 94°C for 40 s, 57°C for 40 s, and 72°C for 40 s; 33 cycles at 94°C for 40 s, 55°C for 40 s, and 72°C for 40 s; and a final extension step at 72°C for 10 min. DNA was extracted from gel and purified with QIAquick Gel Extraction kit (QIAGEN). Direct sequencing of the resulting PCR products with the BigDye terminator system on an automated sequencer (Applied Biosystems) revealed a heterozygous G>T transversion in the first intron of the skin-specific SMARCAD1 short isoform. The mutation, c.378+1G>T, was predicted to abolish the donor splice site adjacent to the 3′-end of the first unique exon of the short SMARCAD1 isoform. To confirm the existence of the mutation, we used a PCR-RFLP assay. A 537 bp long DNA fragment was amplified with the forward primer 5′-AGCTGATTGGCTGGGAATAC-3′ and reverse primer 5′-GGCATTCATAAAACTCAAAATGC-3′ (Figure 3B). The mutation creates a recognition site for MseI endonuclease (New England Biolabs, Ipswich, MA). Using this assay, we confirmed segregation of the mutation with the disease phenotype throughout the entire family and also excluded the mutation from a panel of 100 healthy Swiss individuals and 100 healthy Jewish individuals (data not shown); this suggests that the mutation does not represent a common neutral polymorphism but rather is a disease-causing mutation.
To assess the consequences of the mutation on the SMARCAD1-splicing pattern, we initially used RT-PCR to amplify cDNA derived from the RNA extracted from the fibroblast cell cultures that were established from a patient and a healthy individual. Total RNA was extracted with RNeasy Extraction Kit (QIAGEN). cDNA was synthesized (Thermo Scientific Verso cDNA Synthesis Kit, ABgene, Surrey, UK) and amplified by PCR with exon-crossing primers, 5′-GAAAGCAAGAATGTGGCAG-3′; 5′-GGGCTTGAGTGACAAACT-3′, located in exons 1 and 3 of the short SMARCAD1 isoform, respectively. DNA was extracted from gel, purified with QIAquick Gel Extraction kit (QIAGEN), and directly sequenced as described above. Only the wild-type splice product was identified, suggesting that aberrant splice variants might undergo degradation. To obtain further support for this possibility, we generated a minigene construct17 by subcloning exon 1, parts of intron 1 (because the first intron is very large [∼10.5 kb], we trimmed the intronic sequence) and exon 2 of the SMARCAD1 short isoform into the pEGFP-C3 vector (Figure 5A). More specifically, a 1.7 kb genomic DNA fragment comprising exon 1 and the first 1358 bp of intron 1 was cloned into the EcoR1 and Kpn1 restriction sites of the pEGFP-C3 vector with primers 5′-AAAAAGAATTCAAGAAATTAGAGCTTACATTTAG-3′ and 5′-AAAAAGGTACCTCACTGATTAACAGGGAAAAAG-3′, respectively. Then, a 0.7 kb genomic fragment comprising the last 500 bp of intron 1 followed by exon 2 was cloned into the Kpn1 and BamHI sites of the first construct with primers 5′-AAAAAGGTACCTATACTTTGATGATAGATGTGG-3′ and 5′-AAAAGGATCCCTTTGGTTTAGAATGGAAGG-3′, respectively. We sequenced the entire insert to verify the authenticity of the construct. Next, we introduced the c.378+1G>T mutation into the minigene by using the Quick Change Site-Directed Mutagenesis kit (Stratagene, Santa Clara, CA). Both the wild-type and the mutant minigene constructs were transiently transfected into HeLa cells with Lipofectamine 2000 (Invitrogen). Cells were harvested 48 hr after transfection; total RNA was extracted and subjected to RT-PCR and direct sequencing. Transfection of the wild-type minigene resulted as expected in the formation of one single and abundant spliced variant containing exons 1 and 2 of the short SMARCAD1 isoform; this was confirmed by sequencing analysis. In contrast, transfection of the mutation-carrying minigene was found to lead to the generation of two aberrant splice variants: the first one was found to contain an extra 51 bp from intron 1, and the second one was found to miss one G at the end of exon 1 because of the utilization of cryptic donor splice sites. Of interest, the abnormal splice products were only marginally detectable as compared with the wild-type RNA, both in HeLa cells (Figure 5B) and in primary human fibroblasts (data not shown). These results are in line with the fact that aberrant splice variants were not detectable in patient fibroblasts (see above).
Figure 5.
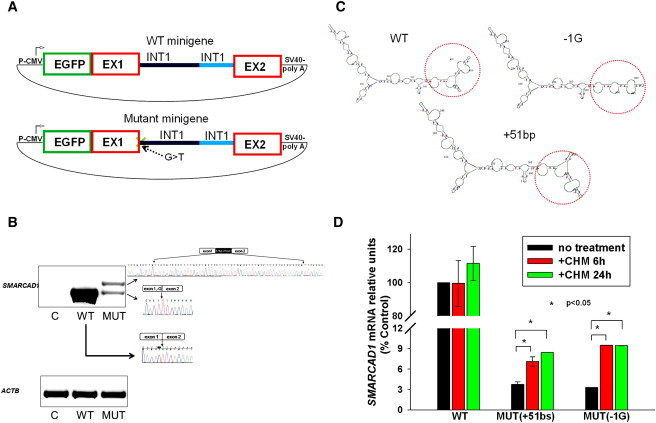
Consequences of Mutation c.378+1G>T
To assess the consequences of mutation c.378+1G>T on SMARCAD1 splicing, we used a minigene system. (A) Schematic representation of the SMARCAD1 short isoform wild-type and mutation-carrying minigenes.
(B) Sequence analysis of RT-PCR products generated from HeLa cells transfected with wild-type and mutant minigene constructs. Transfection of wild-type minigene resulted in the formation of one spliced variant containing exons 1 and 2 of the SMARCAD1 short isoform. In contrast, transfection of the mutant minigene resulted in two aberrant splice variants, containing an extra 51 bp from intron 1 or missing one G at the end of exon 1. A marked decrease in the level of expression of the spliced variants was also observed.
(C) Computational modeling predicts an altered mRNA secondary structure of both aberrant splice variants.
(D) Treatment with cycloheximide (at a concentration of 50 μg/ml for 24 hr), known to inhibit mRNA decapping, resulted in significantly increased levels of aberrant (but not wild-type) splice variants.
Two main mechanisms, alone or in combination, might explain this observation. First, authentic splicing is typically more efficient than splicing activated at cryptic sites.18 Therefore, it is possible that the significantly reduced level of aberrant splice variants is due to a decrease in splicing efficiency. Another possibility is that the abnormal 5′UTR variants affect RNA stability. Indeed, alteration in the secondary structure of an RNA molecule has been shown to inhibit translation initiation directly, by preventing the 40S subunit binding or scanning, or indirectly, by preventing the action of regulatory RNA-binding proteins. This in turn has been shown to foster mRNA degradation by increasing decapping and the deadenylation rate.19 To assess this possibility, we initially compared via computational analysis the secondary structure of wild-type and aberrant splice RNA variants by using the GeneBee RNA secondary-structure prediction software. As shown in Figure 5C, computational analysis predicts that both aberrant splice variations are likely to significantly affect RNA secondary configuration; this prediction is in agreement with the fact the 5′UTR region of the gene affected by the abnormal splicing is highly conserved across species at the nucleotide level (data not shown). To obtain experimental support for the possibility that aberrantly spliced variants of the SMARCAD1 short isoform undergo degradation, we treated cells transfected with both the wild-type and mutation-carrying constructs with cycloheximide at a concentration of 50 μg/ml for 24 hr, which is known to inhibit decapping of mRNA.20 As a result, we observed a significant increase in the aberrant splice variant levels but not in the wild-type splice variant (Figure 5D).
In conclusion, we have identified in a large family with ADG a splice site mutation causing aberrant splicing of a skin-specific isoform of SMARCAD1, implicating this gene in dermatoglyph ontogenesis. The mutation is likely to exert a loss-of-function effect.
Little is known about the function of the full-length SMARCAD1, and virtually nothing is known regarding the physiological role of the skin-specific isoform of this gene. Clearly, the tissue-specific pattern of expression of the short isoform is likely to underlie the very limited phenotype displayed by our patients, as attested by the severe phenotype observed in mice knocked out for the ubiquitous SMARCAD1 large isoform of the gene;21 those mice feature retarded growth, perinatal mortality, decreased fertility, and various skeletal defects.
The full-length SMARCAD1 seems to control the expression of a large spectrum of target genes encoding transcriptional factors and histone modifiers as well as regulators of the cell cycle and development.16 It is tempting to speculate that the skin-specific isoform of SMARCAD1 might target genes involved in dermatoglyph and sweat gland development, two structures jointly affected in the present family and in additional disorders such as Naegeli-Franceschetti-Jadassohn and Rapp-Hodgkin (MIM 129400) syndromes.11,22 Regardless of the exact mechanisms mediating the activity of the skin-specific isoform of SMARCAD1 in the skin, the present results once again underscore the fact that rare monogenic traits represent an invaluable tool for the investigation of concealed aspects of our biology.
Acknowledgments
We would like to acknowledge the participation of all family members in this study. We would like to thank Sylvia Kiese for her help. We wish to thank Gil Ast, Hadas Keren, and Mordechai Choder for helpful discussions.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows
Ensembl, http://www.ensembl.org/
GenBank, http://www.ncbi.nlm.nih.gov/Genbank/
GeneBee, http://www.genebee.msu.su/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Superlink, http://bioinfo.cs.technion.ac.il/superlink-online-twoloci/makeped/TwoLociMultiPoint.html
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Verbov J. Clinical significance and genetics of epidermal ridges—a review of dermatoglyphics. J. Invest. Dermatol. 1970;54:261–271. doi: 10.1111/1523-1747.ep12258550. [DOI] [PubMed] [Google Scholar]
- 2.Warman P.H., Ennos A.R. Fingerprints are unlikely to increase the friction of primate fingerpads. J. Exp. Biol. 2009;212:2016–2022. doi: 10.1242/jeb.028977. [DOI] [PubMed] [Google Scholar]
- 3.Scheibert J., Leurent S., Prevost A., Debrégeas G. The role of fingerprints in the coding of tactile information probed with a biomimetic sensor. Science. 2009;323:1503–1506. doi: 10.1126/science.1166467. [DOI] [PubMed] [Google Scholar]
- 4.Reed T., Viken R.J., Rinehart S.A. High heritability of fingertip arch patterns in twin-pairs. Am. J. Med. Genet. A. 2006;140:263–271. doi: 10.1002/ajmg.a.31086. [DOI] [PubMed] [Google Scholar]
- 5.Bokhari A., Coull B.A., Holmes L.B. Effect of prenatal exposure to anticonvulsant drugs on dermal ridge patterns of fingers. Teratology. 2002;66:19–23. doi: 10.1002/tera.10044. [DOI] [PubMed] [Google Scholar]
- 6.Babler W.J. Embryologic development of epidermal ridges and their configurations. Birth Defects Orig. Artic. Ser. 1991;27:95–112. [PubMed] [Google Scholar]
- 7.Baird H.W. Absence of fingerprints in four generations. Lancet. 1968;2:1250. doi: 10.1016/s0140-6736(68)91745-5. [DOI] [PubMed] [Google Scholar]
- 8.Basan M. Ectodermal dysplasia. Missing papillary pattern, nail disorders and furrows on 4 fingers. Arch. Klin. Exp. Dermatol. 1965;222:546–557. [PubMed] [Google Scholar]
- 9.Reed T., Schreiner R.L. Absence of dermal ridge patterns: Genetic heterogeneity. Am. J. Med. Genet. 1983;16:81–88. doi: 10.1002/ajmg.1320160113. [DOI] [PubMed] [Google Scholar]
- 10.Límová M., Blacker K.L., LeBoit P.E. Congenital absence of dermatoglyphs. J. Am. Acad. Dermatol. 1993;29:355–358. doi: 10.1016/0190-9622(93)70195-y. [DOI] [PubMed] [Google Scholar]
- 11.Lugassy J., Itin P., Ishida-Yamamoto A., Holland K., Huson S., Geiger D., Hennies H.C., Indelman M., Bercovich D., Uitto J. Naegeli-Franceschetti-Jadassohn syndrome and dermatopathia pigmentosa reticularis: Two allelic ectodermal dysplasias caused by dominant mutations in KRT14. Am. J. Hum. Genet. 2006;79:724–730. doi: 10.1086/507792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sirinavin C., Trowbridge A.A. Dyskeratosis congenita: Clinical features and genetic aspects. Report of a family and review of the literature. J. Med. Genet. 1975;12:339–354. doi: 10.1136/jmg.12.4.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burger B., Fuchs D., Sprecher E., Itin P. The immigration delay disease: Adermatoglyphia-inherited absence of epidermal ridges. J. Am. Acad. Dermatol. 2011;64:974–980. doi: 10.1016/j.jaad.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 14.Fishelson M., Geiger D. Exact genetic linkage computations for general pedigrees. Bioinformatics. 2002;18(Suppl 1):S189–S198. doi: 10.1093/bioinformatics/18.suppl_1.s189. [DOI] [PubMed] [Google Scholar]
- 15.Adra C.N., Donato J.L., Badovinac R., Syed F., Kheraj R., Cai H., Moran C., Kolker M.T., Turner H., Weremowicz S. SMARCAD1, a novel human helicase family-defining member associated with genetic instability: Cloning, expression, and mapping to 4q22-q23, a band rich in breakpoints and deletion mutants involved in several human diseases. Genomics. 2000;69:162–173. doi: 10.1006/geno.2000.6281. [DOI] [PubMed] [Google Scholar]
- 16.Okazaki N., Ikeda S., Ohara R., Shimada K., Yanagawa T., Nagase T., Ohara O., Koga H. The novel protein complex with SMARCAD1/KIAA1122 binds to the vicinity of TSS. J. Mol. Biol. 2008;382:257–265. doi: 10.1016/j.jmb.2008.07.031. [DOI] [PubMed] [Google Scholar]
- 17.Singh G., Cooper T.A. Minigene reporter for identification and analysis of cis elements and trans factors affecting pre-mRNA splicing. Biotechniques. 2006;41:177–181. doi: 10.2144/000112208. [DOI] [PubMed] [Google Scholar]
- 18.Roca X., Sachidanandam R., Krainer A.R. Intrinsic differences between authentic and cryptic 5′ splice sites. Nucleic Acids Res. 2003;31:6321–6333. doi: 10.1093/nar/gkg830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Day D.A., Tuite M.F. Post-transcriptional gene regulatory mechanisms in eukaryotes: An overview. J. Endocrinol. 1998;157:361–371. doi: 10.1677/joe.0.1570361. [DOI] [PubMed] [Google Scholar]
- 20.Schwartz D.C., Parker R. Mutations in translation initiation factors lead to increased rates of deadenylation and decapping of mRNAs in Saccharomyces cerevisiae. Mol. Cell. Biol. 1999;19:5247–5256. doi: 10.1128/mcb.19.8.5247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schoor M., Schuster-Gossler K., Roopenian D., Gossler A. Skeletal dysplasias, growth retardation, reduced postnatal survival, and impaired fertility in mice lacking the SNF2/SWI2 family member ETL1. Mech. Dev. 1999;85:73–83. doi: 10.1016/s0925-4773(99)00090-8. [DOI] [PubMed] [Google Scholar]
- 22.Atasu M., Akesi S., Elçioglu N., Yatmaz P.I., Ertas E.B. A Rapp-Hodgkin like syndrome in three sibs: Clinical, dental and dermatoglyphic study. Clin. Dysmorphol. 1999;8:101–110. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.