

Abstract
Adams-Oliver syndrome (AOS) is defined by the combination of aplasia cutis congenita (ACC) and terminal transverse limb defects (TTLD). It is usually inherited as an autosomal-dominant trait, but autosomal-recessive inheritance has also been documented. In an individual with autosomal-recessive AOS, we combined autozygome analysis with exome sequencing to identify a homozygous truncating mutation in dedicator of cytokinesis 6 gene (DOCK6) which encodes an atypical guanidine exchange factor (GEF) known to activate two members of the Rho GTPase family: Cdc42 and Rac1. Another homozygous truncating mutation was identified upon targeted sequencing of DOCK6 in an unrelated individual with AOS. Consistent with the established role of Cdc42 and Rac1 in the organization of the actin cytoskeleton, we demonstrate a cellular phenotype typical of a defective actin cytoskeleton in patient cells. These findings, combined with a Dock6 expression profile that is consistent with an AOS phenotype as well as the very recent demonstration of dominant mutations of ARHGAP31 in AOS, establish Cdc42 and Rac1 as key molecules in the pathogenesis of AOS and suggest that other regulators of these Rho GTPase proteins might be good candidates in the quest to define the genetic spectrum of this genetically heterogeneous condition.
Main Text
Adams-Oliver syndrome (AOS [MIM 100300]) is a multiple congenital anomaly syndrome that is characterized by aplasia cutis congenita (ACC) as well as terminal transverse limb defects (TTLD) in addition to variable involvement of the brain, eyes, and cardiovascular system.1–3 Original epidemiological data were consistent with a dominant mode of inheritance due to presumed de novo (in simplex cases) or familial (in multiplex families) mutations. Horizontal transmission was initially suspected as possible germline mosaicism, but multiple reports of recurrence in consanguineous unions made it likely that AOS can also occur as an autosomal-recessive trait.3
The classic combination of ACC and TTLD and their known association with vascular anomalies fueled speculation that vascular disruption is a major pathogenic mechanism in AOS.2,4 However, recently dominant mutations were found to cause AOS by virtue of Rac1 (MIM 602048) and Cdc42 (MIM 116952) inactivation, which leads to impaired actin cytoskeletal homeostasis.5 ARHGAP31 is a GTPase-activating protein (GAP) that stimulates the intrinsic GTPase activity of Rac1 and Cdc42 and thus replaces their active GTP-bound form with the inactive GDP-bound form.6 The two reported mutations in ARHGAP31 (MIM 610911) were hypermorphic in nature, causing sustained inactivation of Rac1 and Cdc42, which are known to be of critical importance in regulating the actin cytoskeleton.7 As a result, AOS appears to be a member of a growing list of actin cytoskeletopathies that includes such disparate disorders as Lowe Oculocerebrorenalsyndrome (OCRL [MIM 309000]), Nemaline Myopathy (NEM [MIM 609284, 256030, 161800, 609285, and 605355]) and Wiskott-Aldrich syndrome (WASF [MIM 605035, 605875, and 605068]).8–10
In our effort to molecularly characterize AOS in our inbred population, where autosomal-recessive AOS is seen more commonly,11 we have studied two unrelated individuals who have this syndrome and were born to consanguineous parents. We successfully combined autozygome analysis with exome data in one of these two individuals to identify a loss-of-function mutation in DOCK6, another modulator of Cdc42 and Rac1, and we then identified a second mutation in the other individual. Our data on Dock6 expression and the cellular phenotype of fibroblasts of individual 1 further confirm the role of DOCK6 in AOS pathogenesis, which appears to converge with that reported for ARHGAP31 in perturbation of the actin cytoskeleton through inactivation of Cdc42 and Rac1.
Individual 1 is an 11-month-old girl born to first-cousin Arab parents and who was referred for clinical genetics evaluation. She has four normal siblings and a cousin who is said to be similarly affected but who was unavailable for evaluation (Figure 1A). Pregnancy was uncomplicated. Abnormal hands and feet were noted at birth. At 11 months of age, she had severe and global developmental delay, recurrent seizures, and poor vision. Physical examination revealed microcephaly, large cutis aplasia of the scalp, optic atrophy, and axial hypotonia with appendicular hypertonia. In addition, there was distal reduction of the fingers and toes bilaterally and an absence of distal phalanges and nails (Figure 1B). Echocardiography was normal. Brain CT demonstrated hydrocephalus with dilatation of the lateral ventricles and multiple small periventricular and subependymal calcifications (Figure 1B). X-rays of the hands and feet revealed absence of the distal phalanges.
Figure 1.
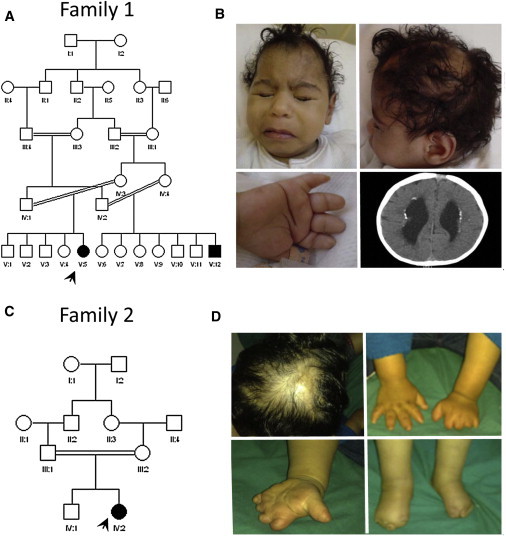
Pedigrees of the Two AOS Families Included in the Study and Clinical Photographs of Individuals 1 and 2
(A) Family 1 pedigree.
(B) Clinical photographs of individual 1.
(C) Family 2 pedigree.
(D) Clinical photographs of individual 2.
Note the typical ACC and TTLD in both individuals.
Individual 2 is a 3.5-year-old girl, the second child to healthy first-cousin Arab parents (Figure 1C). She was referred for evaluation of congenital deformity of the hands and feet. Family history is negative. There was history of oligohydramnios and decreased fetal movement during pregnancy. At birth, she was noted to have terminal-reduction defects of her hands and feet and cutis aplasia of the scalp. Her development appeared appropriate except for speech delay. Her physical examination at 3.5 years revealed microcephaly but normal height and weight. There was an area of alopecia with an underlying scar in the scalp. Finger and toe nails were either absent or severely hypoplastic, as were the distal phalanges, and her hands appeared stubby with distorted creases (Figure 1D). X-rays of the hands and feet revealed hypoplastic middle phalanges and absent distal phalanges. Echocardiography, EEG, and eye examination were all within normal limits.
The two individuals with AOS and their parents were enrolled in the study with an IRB-approved written informed consent (KFSHRC RAC#2080006). Blood was collected in EDTA tubes and Na-heparin tubes, and a small-punch skin biopsy was obtained whenever possible. Autozygome analysis was performed on individual 1 via the Axiom SNP Platform (Affymetrix) followed by autoSNPa genomewide determination of runs of homozygosity essentially as described before.12 Full-exome capture was performed with the TruSeq Exome Enrichment kit (Illumina) according to the manufacturer's protocol. Samples were prepared as an Illumina sequencing library, and in the second step, the sequencing libraries were enriched for the desired target via the Illumina Exome Enrichment protocol. The captured libraries were sequenced with Illumina HiSeq 2000 Sequencer. The reads were mapped against UCSC hg19 by BWA. The SNPs and indels were detected by SAMTOOLS.
We have previously proposed the effectiveness of combining autozygome data with next-generation sequencing.13 Indeed, using DNA from individual 1 alone, we were able to apply his autozygome as a filter of the resulting exome data; i.e., we only considered novel coding-sequence variants that were detected within runs of homozygosity (ten runs were identified for a total of 1303 genes). By applying this filter, we identified three variants (two missense and one indel mutation; Table S1, available online). We prioritized the 4 bp deletion (c.1362_1365delAACT, p.Thr455Serfs∗24; RefSeq accession number NM_020812.2) in DOCK6 because it was the only truncating mutation identified. Indeed, Sanger sequencing confirmed this homozygous mutation in individual 1, and her parents were found to be carriers (Figure 2). We then fully sequenced DOCK6 in individual 2 and identified a 1 bp duplication creating a stop codon (c.1245dupT, p.Asp416∗; RefSeq accession number NM_020812.2), and segregation was confirmed within the family. This second mutation represents an independent confirmation of the involvement of DOCK6 disruption in the pathogenesis of AOS.
Figure 2.
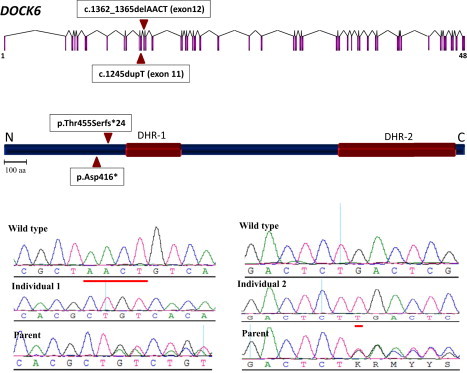
Identification of Two DOCK6 Mutations in AOS
Upper panel: Diagram of DOCK6 (triangles indicate the sites of the mutations). Middle panel: Diagram of DOCK6 (note that both truncating mutations are upstream of DHR-1 and DHR-2 domains). Bottom panel: Sequence chromatogram of the two mutations with the control tracing for comparison (sequence differences are underlined in red).
In order to determine the developmental expression pattern of Dock6, we performed WISH (whole-mount in situ hybridization) on mouse embryos of various stages of development. Expression of Dock6 at E9.5 was observed in the growing edge of the limb buds and in the developing heart (Figure 3A). At E10.5 the strongest expression was at the edge of the limb buds, but the heart expression was also maintained (Figure 3B). At E11.5 Dock6 mRNA was enriched in the apical ectodermal ridge of all four limbs (Figure 3C). By E12.5 and E13.5, the expression of Dock6 assumed a more diffused pattern in the four limbs, although the hind-limb, as in previous stages, was always stronger than the fore-limb (Figures 3D and 3E), and at E13.5 expression was clearly observed in the developing digits (Figures 3F and 3G). Lack of comparable staining with the corresponding sense probes confirmed specificity of the observed signals (see Figures 3H and 3I for representative examples). We also carried out (q) RT-PCR on various mouse adult tissues and found that although Dock6 is expressed in all tissues tested, there was significant Dock6 expression in the heart (Figure S1).
Figure 3.
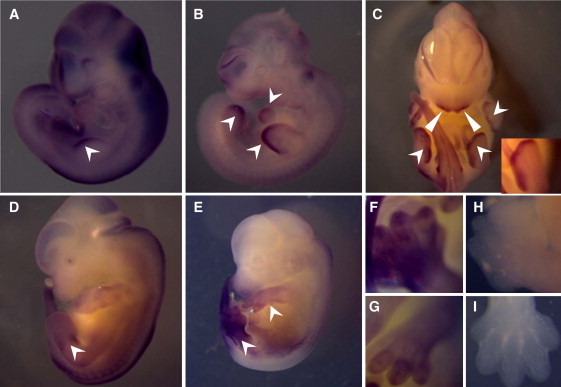
WISH of Dock6 during Mouse Embryonic Development
(A) E9.5 mouse embryo showing expression in the growing edge of the limb bud.
(B) E10.5 mouse embryo showing expression in the growing edge of limb buds and heart.
(C) E11.5 mouse embryo showing expression in the apical ectodermal ridge of all four limbs (arrowheads and inset) as well as the first-pharyngeal-arch-derived facial mesenchyme (triangles).
(D and E) E12.5 and 13.5 embryos showing the diffuse expression of Dock6 mRNA in the fore- and hind-limbs.
(F and G) Close-up view showing the expression in the limbs of an E13.5 embryo.
(H and I) Sense control for comparison with (F) and (G).
Because of the established role of Cdc42 and Rac1 in actin polymerization,14 we tried to gain insight into the cellular phenotype of DOCK6 mutation by staining patient fibroblasts homozygous for the p.Thr455Serfs∗24 mutation and assessing cytoskeletal organization. A small percentage of cells from individual 1 assumed a rounded phenotype (three to four cells per high-power field [HPF], or 16%) with “blebbing,” which we did not observe in control cells (Figures 4A–4C). Compared to control cells, cells from individual 1 also assumed an unusual elongated morphology and lacked lamellopodia and lateral ruffles (Figures 4D and 4E). The characteristics observed in cells from individual 1 were similar to observations of F-Actin distribution of Rac1-null fibroblast cells,15 and the rounded phenotype was also similar to that recently observed for HeLa cells after suppression of Cdc42 activity.16
Figure 4.
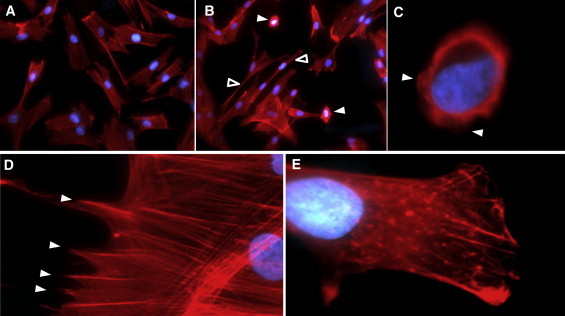
Patient and Control Fibroblast Cells Were Visualized by Fluorescent Microscopy for Phalloidin Staining of F-actin- and DAPI-Stained Nuclei
F-actin staining is in red, and DAPI staining is in blue.
(A and B) Control fibroblasts (A) show the typical spindle appearance, whereas patient fibroblasts (B) show relatively high frequency of rounded (solid arrowheads) and elongated (empty arrows heads) cells.
(C) A close-up view of one patient fibroblast with a rounded phenotype clearly indicates blebbing (arrow heads).
(D and E) The normal appearance of lamellopodia in control fibroblasts (D) is in sharp contrast to the “blunt” edges due to severe deficiency of lamellopodia formation in patient fibroblasts (E).
Cdc42 and Rac1 are Rho GTPases, a family of signaling proteins that, through induced polymerization of actin filaments, play key roles in a number of basic cellular processes, such as proliferation, polarization, migration, adhesion, secretion, maintenance of cell morphology, cytokinesis, apoptosis, and phagocytosis.17,18 They function as molecular switches that alternate between an active GTP-bound form and inactive GDP-bound form.19 This cycling in activity level is tightly regulated largely through the opposing action of two classes of proteins: GEFs, which stimulate the replacement of GDP with GTP; and GAPs, which stimulate the intrinsic GTPase activity of Rho GTPase and the consequent formation of the inactive GDP-bound form.19
GEFs are classically defined by their possession of Dbl homology (DH) and pleckstrin homology (PH) domains, which are required for exchange activity and membranous localization, respectively.20,21 More recently, an atypical family of GEFs, known as DOCKs, was identified in which the protein lacks DH and PH but contains two Dock homology region (DHR) domains, Dock homology region 1 (DHR1) and Dock homology region 2 (DHR2) which carry out membranous localization and exchange activity, respectively.22–24 DOCK proteins are further divided into four groups on the basis of substrate specificity and sequence alignment. DOCK6, along with DOCK7 and DOCK8, belongs to group C, which increases the availability of the active GTP-bound form of both Cdc42 and Rac1.25 Most available literature on DOCK-C proteins concerns their action on the cytoskeletal reorganization that is required for neurite outgrowth and dendrite formation in tissue culture experiments.26–28 However, available data on germline mutations in DOCK genes suggest a more widespread physiological role. For example, mice with inactivating Dock7 mutations have a generalized pigment deficiency, and humans with inactivating DOCK8 mutations have immunodeficiency.29,30
Similarly, DOCK6 was studied in the context of dendrite and axon formation, but data on the phenotypic consequences of germline mutation are lacking.25 In this study, we show that recessive germline mutations of this gene do indeed affect the actin cytoskeleton but primarily cause ACC and TTLD rather than the expected neuronal phenotype, although we note here that both individuals displayed microcephaly and that individual 1 had evidence of more severe brain involvement. Therefore, caution is required in extrapolating data on the likely phenotype of germline mutations from tissue culture experiments.
The two mutations we report are probably null in nature. Even though NMD is excluded for at least one of the two mutations (mutant transcript was readily identifiable on RT-PCR in fibroblasts from individual 1; data not shown), they are both predicted to encode mutant DOCK6 that lacks its two fundamental domains. DHR2 was clearly shown to be both necessary and sufficient for the GDP-to-GTP exchange activity, and DHR1 was shown to be responsible for the interaction with phosphatidylinositol-3, 4, 5-triphosphate and hence the membranous localization of DOCK proteins, but it was also shown to be necessary for Rac-dependent cell elongation and cell migration despite adequate Rac GTP loading.24,31 The latter observation suggests a model in which DHR1 is necessary for Rac GTP loading, whereas DHR2 is necessary for the actual Rac signaling.32
Consistent with the proposed null mechanism, our data on the cellular phenotype are virtually identical to those reported in the context of inactivation of Cdc42 and Rac1 as a result of ARHGAP31 mutations; specifically, this previous study reported a rounded cell appearance and lack of lamellopodia formation.5 Therefore, a model emerges in which impaired actin-cytoskeleton organization by inactivation of Cdc42 and Rac1 is a final common pathway in the pathogenesis of AOS, which can be caused by either null mutations of the activating DOCK6 or hypermorphic mutations of the inactivating ARHGAP31.
Speculation on the link between Cdc42 and Rac1 inactivation and the two key features of AOS can be informed by previously published work. For example, Rac1 inactivation in the developing limb buds in mice results in a TTLD remarkably similar to those observed in humans.33,34 This is thought to be a combination of impaired apoptosis of the presumptive interdigital spaces as well as improperly oriented cellular migration. Correctly oriented cellular migration was recently shown to be of critical importance in proper limb bud formation.35 The explanation of the ACC phenotype is less straightforward because it remains unclear why it preferentially affects the vortex area of the scalp. However, evidence shows that targeted inactivation of TGF-β in mouse skin results in an ACC phenotype with a location almost identical to that observed in AOS patients.36 When combined with the recent revelation that Cdc42 activation is necessary for the transduction of TGF-β-induced mobilization of the actin cytoskeleton,37 a potential mechanistic insight into ACC pathogenesis in AOS can be inferred.
In conclusion, our study shows the power of combining exome and autozygome data in unraveling Mendelian genetics by using simplex cases, a clear departure from the classical requirement of large pedigrees. We also demonstrate that recessive and dominant AOS can be caused by mutations in two modulators of the Cdc42 and Rac1 GTPase activity, which makes it possible that other modulators of their signaling might be potential candidate genes in this genetically heterogeneous condition.
Acknowledgments
We would like to express our deep appreciation to the family members for their enthusiastic and generous participation. We thank our Sequencing and Genomic Core Facilities at the King Faisal Specialist Hospital and Research Center. This study was supported in part by King Abdulaziz City for Science and Technology grant 09-MED941-20 (F.S.A.) as well as a Collaborative Research Grant from Dubai Harvard Foundation for Medical Research (F.S.A.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Burrows-Wheeler Aligner, http://bio-bwa.sourceforge.net/
Ensembl Genome Browser, http://www.ensembl.org/index.html
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
SAMtools, http://samtools.sourceforge.net/
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Adams F.H., Oliver C.P. Hereditary deformities in man due to arrested development. J. Hered. 1945;36:3–7. [Google Scholar]
- 2.Whitley C.B., Gorlin R.J. Adams-Oliver syndrome revisited. Am. J. Med. Genet. 1991;40:319–326. doi: 10.1002/ajmg.1320400315. [DOI] [PubMed] [Google Scholar]
- 3.Snape K.M., Ruddy D., Zenker M., Wuyts W., Whiteford M., Johnson D., Lam W., Trembath R.C. The spectra of clinical phenotypes in aplasia cutis congenita and terminal transverse limb defects. Am. J. Med. Genet. A. 2009;149A:1860–1881. doi: 10.1002/ajmg.a.32708. [DOI] [PubMed] [Google Scholar]
- 4.Papadopoulou E., Sifakis S., Raissaki M., Germanakis I., Kalmanti M. Antenatal and postnatal evidence of periventricular leukomalacia as a further indication of vascular disruption in Adams-Oliver syndrome. Am. J. Med. Genet. A. 2008;146A:2545–2550. doi: 10.1002/ajmg.a.32410. [DOI] [PubMed] [Google Scholar]
- 5.Southgate L., Machado R.D., Snape K.M., Primeau M., Dafou D., Ruddy D.M., Branney P.A., Fisher M., Lee G.J., Simpson M.A. Gain-of-function mutations of ARHGAP31, a Cdc42/Rac1 GTPase regulator, cause syndromic cutis aplasia and limb anomalies. Am. J. Hum. Genet. 2011;88:574–585. doi: 10.1016/j.ajhg.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tcherkezian J., Triki I., Stenne R., Danek E.I., Lamarche-Vane N. The human orthologue of CdGAP is a phosphoprotein and a GTPase-activating protein for Cdc42 and Rac1 but not RhoA. Biol. Cell. 2006;98:445–456. doi: 10.1042/BC20050101. [DOI] [PubMed] [Google Scholar]
- 7.Etienne-Manneville S., Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 8.Kolluri R., Tolias K.F., Carpenter C.L., Rosen F.S., Kirchhausen T. Direct interaction of the Wiskott-Aldrich syndrome protein with the GTPase Cdc42. Proc. Natl. Acad. Sci. USA. 1996;93:5615–5618. doi: 10.1073/pnas.93.11.5615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suchy S.F., Nussbaum R.L. The deficiency of PIP2 5-phosphatase in Lowe syndrome affects actin polymerization. Am. J. Hum. Genet. 2002;71:1420–1427. doi: 10.1086/344517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ilkovski B., Cooper S.T., Nowak K., Ryan M.M., Yang N., Schnell C., Durling H.J., Roddick L.G., Wilkinson I., Kornberg A.J. Nemaline myopathy caused by mutations in the muscle alpha-skeletal-actin gene. Am. J. Hum. Genet. 2001;68:1333–1343. doi: 10.1086/320605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Temtamy S.A., Aglan M.S., Ashour A.M., Zaki M.S. Adams-Oliver syndrome: Further evidence of an autosomal recessive variant. Clin. Dysmorphol. 2007;16:141–149. doi: 10.1097/MCD.0b013e3280f9df22. [DOI] [PubMed] [Google Scholar]
- 12.Shaheen R., Faqeih E., Seidahmed M.Z., Sunker A., Alali F.E., Khadijah A., Alkuraya F.S. A TCTN2 mutation defines a novel Meckel Gruber syndrome locus. Hum. Mutat. 2011;32:573–578. doi: 10.1002/humu.21507. [DOI] [PubMed] [Google Scholar]
- 13.Alkuraya F.S. Autozygome decoded. Genet. Med. 2010;12:765–771. doi: 10.1097/GIM.0b013e3181fbfcc4. [DOI] [PubMed] [Google Scholar]
- 14.Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 15.Vidali L., Chen F., Cicchetti G., Ohta Y., Kwiatkowski D.J. Rac1-null mouse embryonic fibroblasts are motile and respond to platelet-derived growth factor. Mol. Biol. Cell. 2006;17:2377–2390. doi: 10.1091/mbc.E05-10-0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yarbrough M.L., Li Y., Kinch L.N., Grishin N.V., Ball H.L., Orth K. AMPylation of Rho GTPases by Vibrio VopS disrupts effector binding and downstream signaling. Science. 2009;323:269–272. doi: 10.1126/science.1166382. [DOI] [PubMed] [Google Scholar]
- 17.Ridley A.J., Paterson H.F., Johnston C.L., Diekmann D., Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- 18.Tapon N., Hall A. Rho, Rac and Cdc42 GTPases regulate the organization of the actin cytoskeleton. Curr. Opin. Cell Biol. 1997;9:86–92. doi: 10.1016/s0955-0674(97)80156-1. [DOI] [PubMed] [Google Scholar]
- 19.Jaffe A.B., Hall A. Rho GTPases: Biochemistry and biology. Annu. Rev. Cell Dev. Biol. 2005;21:247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- 20.Hart M.J., Eva A., Evans T., Aaronson S.A., Cerione R.A. Catalysis of guanine nucleotide exchange on the CDC42Hs protein by the dbl oncogene product. Nature. 1991;354:311–314. doi: 10.1038/354311a0. [DOI] [PubMed] [Google Scholar]
- 21.Feng Q., Baird D., Cerione R.A. Novel regulatory mechanisms for the Dbl family guanine nucleotide exchange factor Cool-2/alpha-Pix. EMBO J. 2004;23:3492–3504. doi: 10.1038/sj.emboj.7600331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brugnera E., Haney L., Grimsley C., Lu M., Walk S.F., Tosello-Trampont A.C., Macara I.G., Madhani H., Fink G.R., Ravichandran K.S. Unconventional Rac-GEF activity is mediated through the Dock180-ELMO complex. Nat. Cell Biol. 2002;4:574–582. doi: 10.1038/ncb824. [DOI] [PubMed] [Google Scholar]
- 23.Côté J.F., Vuori K. Identification of an evolutionarily conserved superfamily of DOCK180-related proteins with guanine nucleotide exchange activity. J. Cell Sci. 2002;115:4901–4913. doi: 10.1242/jcs.00219. [DOI] [PubMed] [Google Scholar]
- 24.Yang J., Zhang Z., Roe S.M., Marshall C.J., Barford D. Activation of Rho GTPases by DOCK exchange factors is mediated by a nucleotide sensor. Science. 2009;325:1398–1402. doi: 10.1126/science.1174468. [DOI] [PubMed] [Google Scholar]
- 25.Miyamoto Y., Yamauchi J., Sanbe A., Tanoue A. Dock6, a Dock-C subfamily guanine nucleotide exchanger, has the dual specificity for Rac1 and Cdc42 and regulates neurite outgrowth. Exp. Cell Res. 2007;313:791–804. doi: 10.1016/j.yexcr.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 26.Miyamoto Y., Yamauchi J. Cellular signaling of Dock family proteins in neural function. Cell. Signal. 2010;22:175–182. doi: 10.1016/j.cellsig.2009.09.036. [DOI] [PubMed] [Google Scholar]
- 27.Watabe-Uchida M., John K.A., Janas J.A., Newey S.E., Van Aelst L. The Rac activator DOCK7 regulates neuronal polarity through local phosphorylation of stathmin/Op18. Neuron. 2006;51:727–739. doi: 10.1016/j.neuron.2006.07.020. [DOI] [PubMed] [Google Scholar]
- 28.Namekata K., Harada C., Taya C., Guo X., Kimura H., Parada L.F., Harada T. Dock3 induces axonal outgrowth by stimulating membrane recruitment of the WAVE complex. Proc. Natl. Acad. Sci. USA. 2010;107:7586–7591. doi: 10.1073/pnas.0914514107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blasius A.L., Brandl K., Crozat K., Xia Y., Khovananth K., Krebs P., Smart N.G., Zampolli A., Ruggeri Z.M., Beutler B.A. Mice with mutations of Dock7 have generalized hypopigmentation and white-spotting but show normal neurological function. Proc. Natl. Acad. Sci. USA. 2009;106:2706–2711. doi: 10.1073/pnas.0813208106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Q., Davis J.C., Lamborn I.T., Freeman A.F., Jing H., Favreau A.J., Matthews H.F., Davis J., Turner M.L., Uzel G. Combined immunodeficiency associated with DOCK8 mutations. N. Engl. J. Med. 2009;361:2046–2055. doi: 10.1056/NEJMoa0905506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Côté J.F., Motoyama A.B., Bush J.A., Vuori K. A novel and evolutionarily conserved PtdIns(3,4,5)P3-binding domain is necessary for DOCK180 signalling. Nat. Cell Biol. 2005;7:797–807. doi: 10.1038/ncb1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Côté J.F., Vuori K. GEF what? Dock180 and related proteins help Rac to polarize cells in new ways. Trends Cell Biol. 2007;17:383–393. doi: 10.1016/j.tcb.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suzuki D., Yamada A., Amano T., Yasuhara R., Kimura A., Sakahara M., Tsumaki N., Takeda S., Tamura M., Nakamura M. Essential mesenchymal role of small GTPase Rac1 in interdigital programmed cell death during limb development. Dev. Biol. 2009;335:396–406. doi: 10.1016/j.ydbio.2009.09.014. [DOI] [PubMed] [Google Scholar]
- 34.Wu X., Tu X., Joeng K.S., Hilton M.J., Williams D.A., Long F. Rac1 activation controls nuclear localization of beta-catenin during canonical Wnt signaling. Cell. 2008;133:340–353. doi: 10.1016/j.cell.2008.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hopyan S., Sharpe J., Yang Y. Budding behaviors: Growth of the limb as a model of morphogenesis. Dev. Dyn. 2011;240:1054–1062. doi: 10.1002/dvdy.22601. [DOI] [PubMed] [Google Scholar]
- 36.Zehnaly A., Hosokawa R., Urata M., Chai Y. TGF-beta signaling and aplasia cutis congenita: proposed animal model. J. Calif. Dent. Assoc. 2007;35:865–869. [PubMed] [Google Scholar]
- 37.Edlund S., Landström M., Heldin C.H., Aspenström P. Transforming growth factor-beta-induced mobilization of actin cytoskeleton requires signaling by small GTPases Cdc42 and RhoA. Mol. Biol. Cell. 2002;13:902–914. doi: 10.1091/mbc.01-08-0398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.