

Abstract
Autosomal-dominant adult-onset neuronal ceroid lipofuscinosis (ANCL) is characterized by accumulation of autofluorescent storage material in neural tissues and neurodegeneration and has an age of onset in the third decade of life or later. The genetic and molecular basis of the disease has remained unknown for many years. We carried out linkage mapping, gene-expression analysis, exome sequencing, and candidate-gene sequencing in affected individuals from 20 families and/or individuals with simplex cases; we identified in five individuals one of two disease-causing mutations, c.346_348delCTC and c.344T>G, in DNAJC5 encoding cysteine-string protein alpha (CSPα). These mutations—causing a deletion, p.Leu116del, and an amino acid exchange, p.Leu115Arg, respectively—are located within the cysteine-string domain of the protein and affect both palmitoylation-dependent sorting and the amount of CSPα in neuronal cells. The resulting depletion of functional CSPα might cause in parallel the presynaptic dysfunction and the progressive neurodegeneration observed in affected individuals and lysosomal accumulation of misfolded and proteolysis-resistant proteins in the form of characteristic ceroid deposits in neurons. Our work represents an important step in the genetic dissection of a genetically heterogeneous group of ANCLs. It also confirms a neuroprotective role for CSPα in humans and demonstrates the need for detailed investigation of CSPα in the neuronal ceroid lipofuscinoses and other neurodegenerative diseases presenting with neuronal protein aggregation.
Introduction
The neuronal ceroid lipofuscinoses (NCLs) are a heterogeneous group of inherited neurodegenerative disorders with an incidence of between 1 and 30 per 100,000. Common findings in the NCLs are an accumulation of autofluorescent storage material in neural and peripheral tissues and neurodegeneration. Although mutations in eight genes—CLN1 (PPT1 [MIM 256730]), CLN2 (TPP1 [MIM 204500]), CLN3 (MIM 204200), CLN5 (MIM 256731), CLN6 (MIM 601780), CLN7 (MFSD8 [MIM 610951]), CLN8 (MIM 600143), and CLN10 (CTSD [MIM 610127])—have been identified in autosomal-recessive childhood and juvenile NCLs1 and recently also in autosomal-recessive adult-onset NCL (Kufs disease [MIM 204300])2, the genetic and molecular basis of adult-onset NCL with dominant inheritance (Parry type [MIM 162350]) remains unknown.
Autosomal-dominant adult-onset neuronal ceroid lipofuscinosis (ANCL) was first described in a family of British descent from New Jersey, USA (Parry disease),3 and in a second family reported in Spain.4 More recently, a large American family with English ancestry (UCL563 in this study),5 another family from Alabama, USA (UCL562),6,7 and a third family from the Netherlands (N1)8 were presented. Common characteristics of affected individuals included generalized seizures, movement disorders, cognitive deterioration, and progressive dementia; the age of onset varied between 25 and 46 years.
In this work we describe a Czech family (P1) with autosomal-dominant ANCL in whom, by using a combination of linkage mapping, gene-expression analysis, and exome sequencing, we identified a unique heterozygous mutation in DNAJC5 encoding cysteine-string protein alpha (CSPα [MIM 611203]; information on CSPα is accessible in the National Center for Biotechnology Information [NCBI] Gene Entrez database under GeneID 54968). The same or a second heterozygous DNAJC5 mutation was found in four additional unrelated ANCL families that, together with altered palmitoylation-dependent sorting of mutant proteins in a cellular model and a reduced amount of CSPα in neuronal cells of affected individuals, confirmed the causality of CSPα mutations in autosomal-dominant ANCL.
Material and Methods
Subjects
The Czech family (P1) was ascertained at the Institute of Inherited Metabolic Disorders in Prague. Some families were described earlier—an American family from USA with English ancestry UCL563,5 a family from Alabama, USA (UCL562),6,7 and one from the Netherlands (N1)8. Previously unpublished data from families from the USA, France, the Netherlands, Belgium, Poland, Austria, Italy, and Germany were collected under the auspices of the Rare NCL Gene Consortium by Sara Mole. Enzyme assay or analysis of known genes in which mutations lead to NCL had excluded these mutations as the cause in some but not all subjects. Diagnosis of ANCL disease is very challenging, partly because of its rarity but also because for some cases it can only be verified by finding the characteristic pathology in the brain, and not all affected individuals undergo this procedure. The cases included here were diagnosed by clinicians in different countries over two decades. Because full documentation was not always accessible, some medical histories could not be reviewed. However, we chose to test as many likely cases as possible and to fully report negative findings. Investigations were approved by participating centers' institutional review boards and were conducted according to the Declaration of Helsinki principles. Written, informed consent was obtained from all subjects.
Genotyping and Linkage Analysis
Genomic DNA was isolated by standard technology. We genotyped DNA samples by using Affymetrix GeneChip Mapping 10K 2.0 arrays (Affymetrix, Santa Clara, CA) according to the manufacturer's protocol at the microarray core facility of the Institute of Molecular Genetics in Prague. We extracted raw feature intensities from the Affymetrix GeneChip Scanner 3000 7G images by using the GeneChip operating Software (GCOS) 1.4 and generated individual SNP calls by using Affymetrix Genotyping Analysis Software (GTYPE) 4.1.
We carried out multipoint parametric linkage analysis along with a determination of the most likely haplotypes by using affected-only analysis under the assumption of an autosomal-dominant mode of inheritance with a 0.99 constant, age-independent penetrance, 0.01 phenocopy rate, and 0.001 frequency of disease allele; the analysis was performed with version 1.1.2 of Merlin software.9 The results were visualized in the version 1.032 of the HaploPainter software10 and in version 2.9.2 of R-project statistical software.
Gene-Expression Analysis
We isolated leucocytes from freshly drawn blood by using a standard erythrocyte lysis protocol and isolated total RNA from freshly isolated cells by using TRIZOL solution (Invitrogen, Carlsbad, CA). RNA concentration was determined spectrophotometrically at A260 nm by NanoDrop (NanoDrop Technologies), and quality was checked on an Agilent 2100 Bioanalyser (Agilent Technologies). Aliquots of isolated RNA were stored at –80°C until analysis. Expression analysis was performed on the Illumina HumanRef-8_V2 BeadChip at the microarray core facility of the Institute of Molecular Genetics in Prague. Hybridized slides were scanned on an Illumina BeadArray Reader, and bead level data were summarized by Illumina BeadStudio Software v3. Bead summary data were imported into R-project statistical software v.2.9.2 and normalized with the quantile method in the Lumi package. Differential gene-expression analysis was performed with the Limma package and the lmFit function. A multiple testing correction was performed with the Benjamini and Hochberg method. Database for Annotation, Visualization and Integrated Discovery version 6.7 (DAVID) was used for functional annotation. Details on the experiment and raw expression data are available at the Gene Expression Omnibus (GEO) repository under accession GSE30369.
Copy-Number Analysis
DNA samples from seven individuals of family P1 (II.2, IV.1, IV.2, IV.3, IV.4, IV.7, and IV.8) were genotyped with Affymetrix GeneChip Mapping 6.0 array (Affymetrix, Santa Clara, CA) at the microarray core facility of the Institute of Molecular Genetics in Prague according to the manufacturer's protocol. Raw feature intensities were extracted from the Affymetrix GeneChip Scanner 3000 7G images with the GeneChip Control Console Software 2.01. We generated individual SNP calls by using Affymetrix Genotyping Console Software 3.02. Copy-number changes were identified in Affymetrix Genotyping Console Software (GTC version 3.02). We used data from both SNP and copy-number probes to identify copy-number aberrations relative to a built-in reference. Only regions larger than 10 Kb and containing at least five probes were reported.
Exome Sequencing
We performed DNA enrichment by using 3 μg of DNA from individual IV.7 and the SureSelect All Exome kit (Agilent, Santa Clara, USA) according to the manufacturer's protocol. DNA sequencing was performed on the captured DNA library with one-quarter of a SOLiD 4 slide (Applied Biosystems, Carlsbad, USA) at CeGaT (Tubingen, Germany). We aligned reads in color space to the reference genome (hg19) by using NovoalignCS version 1.01 (Novocraft, Malaysia) with the default parameters. Sequence variants in the analyzed sample were identified with the SAMtools package (version 0.1.8).11 The high-confidence variants list (SNP quality > 100 and indel quality > 50) was annotated with the SeattleSeq Annotation server (hg19). Sequence variants that were not annotated in the dbSNP or 1000 Genomes databases were prioritized for further analysis.
DNA Sequencing and Mutation Analysis
All exons and corresponding exon-intron boundaries of DNAJC5 (NM_025219.2), encoding CSPα, were amplified by PCR from genomic DNA of the probands and sequenced with version 3.1 Dye Terminator cycle sequencing kit (Applied Biosystems, Foster City, CA) with electrophoresis on an ABI 3500XL Avant Genetic Analyzer (Applied Biosystems). Data were analyzed with Sequencing Analysis software. Segregation of the candidate mutations was assessed by PCR and direct sequencing of the corresponding genomic DNA fragments. Primer sequences are available in Table S1, available online.
Homozygosity-Haplotype Analysis
DNAJC5 genomic fragments containing multiple SNPs with high-heterozygosity values were amplified by PCR from genomic DNA of probands and sequenced as described above. Genotypes for individual SNPs were obtained, and homozygous haplotypes were defined as described recently.12 We compared the resulting homozygous haplotypes across individuals to determine whether the chromosomal segments around the same identified mutations could be identical by descent.
Bioinformatic Analysis of the Cysteine-String Domain
Hydrophobicity of the wild-type and mutant cysteine-string domains were analyzed with a Kyte-Doolittle algorithm available at Expasy server. Potential effects of detected mutations on CSPα palmitoylation were assessed with the prediction program CSS-Palm 2.0. Obtained hydrophobicity values and palmitoylation score values were exported for each of the sequences and plotted with an Excel function. We assessed possible impacts of the p.Leu115Arg substitution on the structure and function of CSPα by using SIFT and PolyPhen-2 servers.
CSPα-Expression Vectors
DNAJC5/CSPα cDNA were amplified by RT-PCR from a control and an affected individuals' leucocytes with primers incorporating a BspEI site at the 5′ end of PCR products. Resulting PCR products were first cloned into pCR4 TOPO vector (Invitrogen) and, after sequencing verification, these were further subcloned in frame into a pEGFP-C1 vector with BspEI and ApaI restriction sites. The initiating methionine codon was removed from DNAJC5/CSPα in all enhanced green fluorescent protein (EGFP)-CSPα constructs.
Transient Expression of EGFP-CSPα
pEGFP-CSPα constructs were transfected into CAD-2A2D5 (CAD5) cells derived from Cath.a-differentiated (CAD) cells (provided by Sukhvir Mahal, The Scripps Research Institute, Jupiter, FL, USA). One day before transfection, 8 × 104 cells/cm2 were seeded with OptiMEM medium (OptiMEM; Invitrogen) containing 9% BGS (HyClone, Logan, UT), 90 units penicillin/ml, and 90 g of streptomycin/ml. Cells were transfected by either 0.8 μg or 4.5 μg of plasmid constructs with Lipofectamine 2000 (Invitrogen) in serum and antibiotics free OptiMEM medium according to the manufacturer's protocol. Transfection experiments were performed in more than five replicates.
Immunofluorescence Analysis
Cells were fixed 24 hr after transfection with 4% paraformaldehyde, permeabilized in 0.1% TRITON, washed, blocked with 5% bovine serum albumin (BSA), and incubated for 1 hr at 37°C with anti-protein disulfide isomerase (PDI) mouse monoclonal IgG1 (Stressgen, San Diego, CA) for endoplasmic reticulum (ER) localization, anti-GS28 mouse IgG1 (Stressgen, San Diego, CA) for Golgi localization, and anti-GFP rabbit polyclonal IgG (Abcam) for EGFP-CSPα detection. For fluorescence detection, corresponding species-specific secondary antibodies Alexa Fluor 488 and Alexa Fluor 555 (Molecular Probes, Invitrogen, Paisley, UK) were used. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Prepared slides were mounted in fluorescence mounting medium Immu-Mount (Shandon Lipshaw, Pittsburgh, PA) and analyzed by confocal microscopy.
Image Acquisition and Analysis
XYZ images sampled according to Nyquist criterion were acquired with a TE2000E C1si laser scanning confocal microscope, a Nikon PlanApo objective (40×, N.A.1.30), 488 nm and 543 nm laser lines, and 515 ± 15 nm and 590 ± 15 nm band-pass filters. Images were deconvolved with the classic maximum likelihood restoration algorithm in Huygens Professional software (SVI, Hilversum, The Netherlands).13 Colocalization maps employing single pixel overlap coefficient values ranging from 0–114 were created with Huygens Professional software. The resulting overlap coefficient values are presented as pseudocolor (the scale is shown in the corresponding lookup tables).
Immunoblot Analysis
Transfected CAD5 Cells
Cells were harvested in PBS; centrifuged at 500 g for 7 min; and resuspended in 10 mM Tris, 10 mM KCl, 2 mM EDTA, 4% glycerol, 1 mM DTT, and Complete Protease Inhibitor Cocktail (Roche); homogenized by sonication followed by centrifugation at 20,000 g for 15 min at 4°C; and assessed for protein content in the supernatant with the Bradford assay.
Brain Homogenates
Frozen autopsy materials were homogenized under liquid nitrogen; dissolved in 10 mM Tris, 10 mM KCl, 2 mM EDTA, 4% glycerol, 1 mM DTT, and Complete Protease Inhibitor Cocktail (Roche); centrifuged at 20,000 g for 15 min at 4°C; and assessed for protein content in the supernatant with the Bradford assay. Homogenate aliquots corresponding to 30 μg of total protein in brain homogenates or 20 μg of total protein in CAD5 cells were resolved on 12% SDS-PAGE under nonreducing or reducing conditions and transferred to the polyvinylidene fluoride (PVDF) membrane. Membranes were blocked by 5% BSA and 0.05% Tween 20 in PBS. CSPα or CSPα-EGFP protein was visualized by incubation with rabbit CSP antibody (Stressgen) at 1: 500 in 5% BSA and 0.05% Tween 20 in PBS for 90 min or rabbit GFP antibody (Abcam) at 1:5000 in 5% BSA and 0.05% Tween 20 in PBS for 90 min, followed by incubation with goat anti-rabbit HRP (Pierce) at 1:10000 in 0.05% Tween 20 in PBS for 60 min and detection by SuperSignal West Femto Maximum Sensitivity Substrate (Pierce). For depalmitoylation studies, samples were depalmitoylated prior to SDS-PAGE by treatment with neutral 1 M hydroxylamine or 1 M Tris as a control for 20 hr at room temperature.
Immunohistochemical and Histochemical Studies
Formaldehyde-fixed brain samples were analyzed. Immunodetection of CSPα on paraffin sections was performed with rabbit CSP antibody (Stressgen; diluted 1:750 in 5% BSA) in PBS. Synaptic regions were detected with monoclonal mouse IgG1 synaptobrevin antibody (Sigma, Saint Louis, USA; diluted 1:8000 in 5% BSA) in PBS, which was applied after heat-induced epitope retrieval at pH 6.0. Detection of the bound primary antibody was achieved with Dako EnVision + TM Peroxidase Rabbit kit (Dako, Glostrup, Denmark) with 3,3′-diaminobenzidine as substrate. The specificity of the antigen detection was always ascertained by omitting of the primary antibody-binding step.
Stored ceroid material was best detected because of its prominent autofluorescence via filter block with an excitation wavelength of 400–440 nm (fluorescence microscope Nikon E800, filter block BV-2A).
Results
Clinical Observations and Biochemical Findings
The diagnosis of ANCL in family P1 (Figure 1A) was based on clinical presentation and examination of proband III.6, who presented at age 30 with myoclonic epilepsy, generalized tonic-clonic seizures, and progressive cognitive deterioration with depression; these symptoms were followed by progressive motor neurological symptoms leading to death at age 37 years. There was normal activity of palmitoyl-protein-thioesterase 1 (PPT1) in leucocytes. Neuropathological examination of postmortem brain tissue showed characteristic neurolysosomal storage of autofluorescent material with ultrastructural appearance corresponding to granular osmiophilic deposits (GRODs) (Figures 1B and 1C). A skin biopsy was free of lysosomal storage at the ultrastructural level. An affected status in other family members was assigned if a very similar clinical course starting with myoclonic and/or generalized tonic-clonic seizures followed after 1–2 years by progressive cognitive deterioration and depressive symptomatology. All affected individuals showed generalized epileptic discharges in electroencephalograms and manifested brainstem and central pyramidal neurological symptomatology in the later period of disease. Other ANCL families analyzed in this study are described in Table 1. Previously unpublished families and cases with mutation in CSPα are described in more detail below.
Figure 1.

Pedigree and Neuropathology Findings in Family P1
(A) Pedigree of the Czech family. Black symbols denote affected individuals; open symbols denote unaffected individuals. Age of onset is shown above current age or age of death (indicated by †).
(B) Epifluorescence. Hippocampal pyramidal neurons with prominent lysosomal storage of autofluorescent material representing the general neurolysosomal storage pattern in the brain cortex. The autofluorescence was demonstrated with the filter block with an excitation wavelength of 400–440 nm.
(C) Electron micrograph. GROD-type ultrastructure of the storage lysosomes.
Table 1.
ANCL Families Analyzed in This Study
| Family No. | Mutation in CSPα | Country | Diagnosis | References |
|---|---|---|---|---|
| P1 | p.Leu116del | Czech Republic | ANCL, autosomal dominant | |
| N1 | p.Leu115Arg | The Netherlands | ANCL, autosomal dominant | 8,31,32 |
| UCL563 | p.Leu115Arg | USA | ANCL, autosomal dominant | 5 |
| UCL328 | p.Leu115Arg | USA, French-Canadian | Kufs | |
| UCL519 | p.Leu116del | USA | Kufs, autosomal dominant | |
| UCL417 | – | France | Kufs, autosomal dominant | |
| UCL562 | – | USA | Kufs, autosomal dominant | 6,7 |
| UCL572 | – | USA/Italy | Kufs, autosomal dominant? | |
| UCL327 | – | USA | Kufs, with ALS in extended family | |
| UCL385 | – | Belgium | Kufs Type A or atypical juvenile NCL, autosomal recessive | |
| UCL403 | – | France | Kufs Type B, autosomal recessive | |
| UCL450 | – | Poland | variant juvenile or ANCL, autosomal recessive (heterozygous change in CLCN6 already known) | 33 |
| UCL472 | – | Germany | variant juvenile or ANCL | 34 |
| UCL482 | – | The Netherlands | ANCL | |
| UCL508 | – | USA | Kufs | |
| UCL520 | – | USA | Kufs | |
| UCL522 | – | USA | Kufs | |
| UCL545 | – | Netherlands | Kufs | |
| UCL568 | – | Austria | Kufs | |
| UCL571 | – | Netherlands | Kufs |
Diagnosis is provided as reported by referring clinician. In all cases there was no visual failure, and no distinction was made according the mode of inheritance, if apparent.
The proband of family UCL328 was a male of European descent and in good health until his first generalized tonic-clonic seizure at age 34. This was followed by evidence of progressive confusion and dementia as well as more frequent, medically refractory generalized seizures. Long-term electroencephalography showed generalized periodic epileptiform discharges superimposed on a background of diffuse low-amplitude, high-frequency activity consistent with a dementing process. A brain MRI at age 38 showed prominence of cortical sulci and cerebellar folds and mild enlargement of the lateral ventricles consistent with diffuse cerebral and cerebellar atrophy. Concurrent neuropsychiatric testing showed a verbal IQ of 77, a performance IQ of 71, and a full-scale IQ of 73. Regression of gross and fine motor skills began at age 40, and there was ensuing evidence of ataxia and myoclonus. By age 45, the proband was wheelchair-bound and required nursing-home care. Visual function was normal. A frontal lobe brain biopsy revealed numerous neurons containing homogeneous eosinophilic material with a golden-brown hue. The pigmented material stained intensely by the periodic acid-Schiff reaction and was found to be autofluorescent. Ultrastructural examination showed multiple neurons distended by granular osmiophilic deposits. There was no family history of seizures, early-onset dementia, or other neurologic abnormality.
The proband of family UCL519 is one of at least five similarly affected individuals over three generations with apparent autosomal-dominant inheritance. He showed obsessive behavior starting in his mid-20s, and the first seizure occurred when he was in his early 30s. His speech regressed, his short-term memory became impaired, and he had difficulty in walking without an aid. No further details are available.
Identification of CSPα Mutation in Family P1 by a Combination of Linkage Analysis, Copy-Number Analysis, Gene-Expression Analysis, and Exome Sequencing
To map the disease locus, we used Affymetrix GeneChip Mapping 10K v2.0 arrays, genotyped all available and informative family members, and performed linkage analysis. We identified five candidate regions with positive LOD scores on chromosomes 1, 4, 15, 20, and 22 (Figure 2A). In parallel, we used Affymetrix GeneChip Mapping 6.0 array, genotyped seven individuals, and assessed copy-number changes; we found no indication for a potentially disease-causing deletion or duplication.
Figure 2.
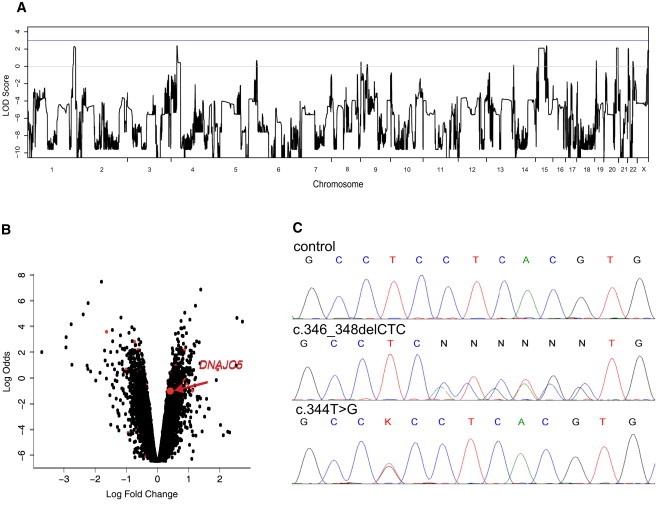
Identification of DNAJC5 Mutations
(A) A whole-genome parametric linkage analysis showing candidate regions reaching the theoretical maximum LOD scores of 2.1 attainable in this family on chromosomes 1 (1: 233,697,529–249,250,621), 4 (4: 23,561,661–28,920,119), 15 (15: 39,049,915–61,382,423; 65,139,935–67,296,086; 71,515,415–78,819,152), 20 (20:53,448,624–63,025,520), and 22 (22: 1–21,982,248). All coordinates refer to hg19.
(B) Gene-expression changes in leucocytes from four affected individuals compared to those of four controls. The logarithm of the probability that the gene is differentially expressed (log odds) is plotted as a function of the logarithm of the gene-expression fold change (log fold change) between the patient and control samples. Differentially expressed genes located in the candidate regions are shown as red dots, and DNAJC5 is specifically indicated. The list of differentially expressed genes located within the linked regions is, together with log fold changes and corresponding t test values, p-values and adjusted p-values, provided in Supplemental Data.
(C) Chromatograms of DNAJC5 genomic DNA sequences showing identified heterozygous mutations. (Upper panel) Sequence of an unaffected individual, (middle panel) sequence showing heterozygous mutation c.346_348delCTC in the proband from family P1, and (lower panel) sequence showing heterozygous mutation c.344T>G in the proband from family N1.
To identify a mutation that affected the amount of transcript, we compared gene-expression profiles in leucocytes isolated from four affected individuals to those from four age-matched controls by using Illumina HumanRef-8v2 Expression BeadChips. This analysis identified a set of 2131 differentially expressed genes, of which 65 were localized within candidate regions identified by linkage analysis (Figure 2B and Table S2). At the same time, we analyzed gene-expression changes by using gene-enrichment analysis and found that the identified profiles indicated significant dysregulation of spliceosome, upregulation of many components of respiratory chain complexes, altered expression of genes active in pathways involved in neurodegenerative diseases, and accelerated proteolysis (Table 2 and Figures S1–S7).
Table 2.
Functional Annotation of Gene-Expression Changes and KEGG Pathways Defined by Gene-Enrichment Analysis
| Term | Count | % | p Value | Population Hits | Population Total | Fold Enrichment | FDR |
|---|---|---|---|---|---|---|---|
| hsa03040: spliceosome | 41 | 2.48 | 2.3 ×−10 | 126 | 5085 | 2.92 | 2.9 ×−7 |
| hsa05016: Huntington disease | 46 | 2.78 | 7.7 ×−8 | 180 | 5085 | 2.29 | 9.5 ×−5 |
| hsa05010: Alzheimer disease | 43 | 2.60 | 8.5 ×−8 | 163 | 5085 | 2.37 | 1.1 ×−4 |
| hsa05012: Parkinson disease | 35 | 2.11 | 7.0 ×−7 | 128 | 5085 | 2.45 | 8.7 ×−4 |
| hsa00190: oxidative phosphorylation | 35 | 2.11 | 1.0 ×−6 | 130 | 5085 | 2.41 | 1.3 ×−3 |
| hsa00520: amino sugar and nucleotide sugar metabolism | 13 | 0.79 | 2.4 ×−3 | 44 | 5085 | 2.65 | 2.9 ×0 |
| hsa03050: proteasome | 12 | 0.73 | 1.2 ×−2 | 47 | 5085 | 2.29 | 1.4 ×1 |
| hsa04120: ubiquitin mediated proteolysis | 25 | 1.51 | 1.5 ×−2 | 137 | 5085 | 1.64 | 1.7 ×1 |
| hsa04662: B cell receptor signaling pathway | 16 | 0.97 | 1.7 ×−2 | 75 | 5085 | 1.91 | 1.9 ×1 |
| hsa04621: NOD-like receptor signaling pathway | 14 | 0.85 | 1.7 ×−2 | 62 | 5085 | 2.03 | 1.9 ×1 |
| hsa00052: galactose metabolism | 8 | 0.48 | 2.0 ×−2 | 26 | 5085 | 2.76 | 2.2 ×1 |
| hsa03010: ribosome | 17 | 1.03 | 2.9 ×−2 | 87 | 5085 | 1.75 | 3.0 ×1 |
To directly identify possible disease-causing mutation(s) among the candidate genes defined by this combination of linkage analysis and gene-expression profiling, we performed exome sequencing in individual IV.7. From the sequencing run we obtained 94.7 M sequencing reads, of which we were able to map 50.2 M on the human genome reference sequence. After removing PCR generated duplicate reads (23.6 M), we obtained 26.6 M unique reads, of which 19.5 M (73.3%) mapped on a targeted exome sequence and were 92% covered at least once. When the sequence of the proband was compared to the reference sequence, 22,617 single nucleotide variants (SNP quality > 100) and 2604 indels (indel quality > 50) were revealed in the proband, of which 957 (617 SNPs and 340 indels) were novel (e.g., were not present in the dbSNP and 1000 Genomes databases).
We intersected the results of exome sequencing with the mapping information and the gene-expression changes, and this analysis illuminated a single gene, DNAJC5, encoding the protein CSPα, located in the candidate region on chromosomal region 20q13.33, (DNAJC5 hg19 coordinates chr20:62526518–62565394) and showing a significant increase in transcript levels in affected individuals' leucocytes (Figure 2B), and had a unique heterozygous mutation c.346_348delCTC (p.Leu116del) compatible with autosomal-dominant inheritance of the disease (Table 3).
Table 3.
Exome Sequencing and a List of High-Confidence Novel Coding Variants Revealed by Exome Sequencing
| Chromosome | Position | Reference Base | Sample Alleles | Function Genome Variation Server | Amino Acids | Protein Position | Gene List |
|---|---|---|---|---|---|---|---|
| Single nucleotide variants | |||||||
| 1 | 235,715,488 | C | C/T | missense | ARG.GLN | 50/76 | GNG4 |
| 1 | 236,987,512 | C | C/T | synonymous | none | 286/1266 | MTR |
| 1 | 247,835,885 | G | C/G | synonymous | none | 153/308 | OR13G1 |
| 15 | 43,552,700 | G | G/T | missense | HIS.ASN | 30/721 | TGM5 |
| 15 | 43,900,153 | C | C/T | synonymous | none | 1234/1776 | STRC |
| 15 | 45,028,847 | G | G/T | utr-5 | none | NA | TRIM69 |
| 15 | 59,500,166 | A | A/G | missense | ILE.VAL | 343/382 | MYO1E |
| 15 | 65,555,518 | A | A/G | synonymous | none | 220/324 | PARP16 |
| 15 | 66,857,721 | C | C/T | utr-5 | none | NA | LCTL |
| 15 | 75,116,809 | G | A/G | missense | VAL.MET | 481/527 | LMAN1L |
| 20 | 60,884,827 | G | A/G | synonymous | none | 3631/3696 | LAMA5 |
| 22 | 20,097,643 | C | C/T | utr-3 | none | NA | DGCR8 |
| 22 | 21,138,487 | C | C/T | synonymous | none | 373/500 | SERPIND1 |
| Indels | |||||||
| 4 | 25,678,161 | TGC | −TGC | coding | none | NA | SLC34A2 |
| 20 | 62,562,227 | CTC | −CTC | coding | none | NA | DNAJC5 |
All coordinates refer to hg19. SNP qualilty > 100 and indel quality > 50. Only Variants located within the linkage candidate regions and not present in dbSNP or 1000 Genomes databases are shown.
CSPα Mutations Segregate with ANCL in Additional Families
Through sequence analysis of DNAJC5 genomic DNA, we found consistent segregation of the c.346_348delCTC mutation with the ANCL phenotype within the Czech family P1 (Figure 2C). Moreover, among 20 additional ANCL families and/or simplex cases tested (Table 1), we identified the same mutation in a previously unreported American family, UCL519, and a second heterozygous mutation (c.344T>G [p.Leu115Arg]) (Figure 1D) segregating with the phenotype in the Dutch family N18 and the American family UCL5635 and present in a previously unreported simplex case UCL328. Mutations were found in all 14 affected individuals (five Czech, six Dutch, and one in each of the other pedigrees) across these five families and were absent in all seven unaffected siblings (two Czech, six Dutch, and one from American family UCL563) from whom DNA was available for testing. In addition to this, the identified mutations were absent in 200 control samples of European descent and were not present in the dbSNP or 1000 Genomes databases.
Haplotypes segregating with ANCL phenotype in Czech family P1 and Dutch family N1 were obtained from genotypes generated with Affymetrix GeneChip Mapping 10K v2.0 arrays and are shown in Figures S8 and S9. For simplex cases, phased haplotypes could not be obtained. To reveal whether probands carrying the same mutation might be distantly related and share a mutation-carrying chromosomal segment from a common ancestor, we examined homozygosity haplotypes across the DNAJC5 genomic region (Table S3). The c.346_348delCTC (p.Leu116del) mutations in families P1 and UCL519 are present on two distinct haplotypes, indicating that these families are probably not related and that the mutations appeared independently. The mutations c.344T>G (p.Leu115Arg) are also present on two distinct haplotypes, one in UCL328 and one shared by family N1 and UCL563. This mutation therefore probably also appeared independently in two different lineages, but it is possible that families N1 and UCL563 are identical by descent.
Identified Mutations Affect Palmitoylation-Dependent Sorting and the Amount of CSPα in Neuronal Cells
Both identified mutations affect conserved dileucine residues located in the cysteine-string domain implicated in palmitoylation and membrane trafficking of CSPα16. Using in silico analysis, we found that p.Leu115Arg is predicted to decrease the hydrophobicity of the cysteine-string domain that is needed for initial binding of CSPα to the endoplasmic reticulum (ER) (Figure 3A), whereas p.Leu116del probably affects the efficiency of palmitoylation of adjacent cysteine residues (Figure 3B). SIFT analysis (score = 0.00) predicted that the p.Leu115Arg mutation affects protein function, and analysis with Polyphen (overall score = 0.782; sensitivity = 0.85; and specificity = 0.93) predicted that it is possibly damaging. No such predictions can be obtained for the identified deletion p.Leu116del.
Figure 3.
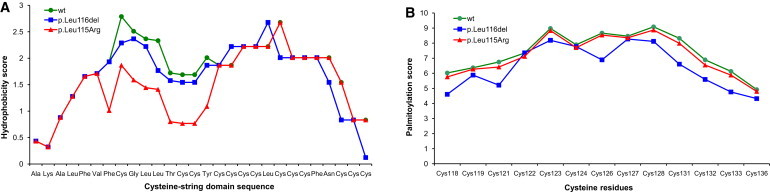
In Silico Analysis of Properties of the Cysteine-String Domain
(A) p.Leu115Arg mutation decreases the hydrophobicity of this domain, which is needed for initial binding to the ER.
(B) The p.Leu116del mutation decreases the palmitoylation score, that is, the confidence that cysteine residues adjacent to Leu116 might be efficiently palmitoylated.
To study an effect of the identified mutations, we transiently expressed wild-type EGFP-tagged CSPα or mutant protein containing either p.Leu115Arg or p.Leu116del in CAD5 neuronal cells. Using immunofluorescence analysis, we found wild-type EGFP-CSPα predominantly at the plasma membrane, whereas both mutated proteins showed diffuse intracellular staining and abnormal colocalization with markers for the ER and Golgi apparatus (Figure 4A). In addition, using immunoblot analysis of transfected cell lysates, we found that both mutated proteins were less efficiently palmitoylated than the wild-type protein (Figure 4B).
Figure 4.
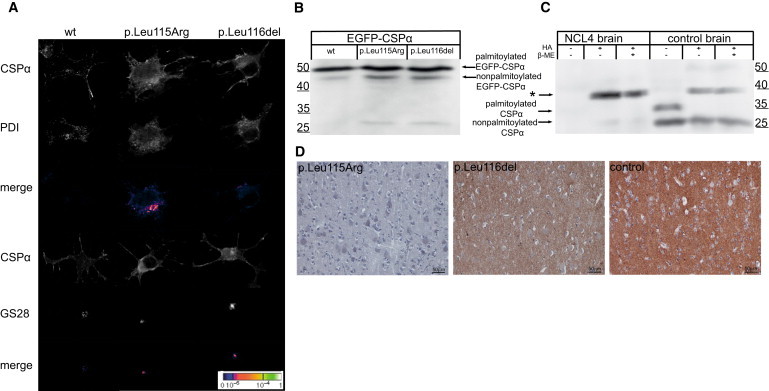
Characterization of Mutated CSPα
(A) Immunofluorescence analysis of transiently expressed EGFP-CSPα proteins in CAD5 cells showing prominent membrane localization of wild-type CSPα compared to the diffuse cytoplasmic staining and marked colocalization of mutated CSPα with endoplasmic reticulum represented by PDI and Golgi apparatus represented by Golgi-SNARE of 28 kDa (GS28).
(B) Immunoblot analysis of transiently expressed EGFP-CSPα proteins showing higher levels of nonpalmitoylated protein precursors for mutant proteins compared the wild-type (wt) protein.
(C) Immunoblot analysis of brain homogenates showing no soluble CSPα and the marked presence of CSPα-containing beta-mercaptoethanol (β-ME)-resistant aggregate (indicated by the asterisk) released upon hydroxylamine (HA) treatment in the affected individual (NCL4) compared to the brain homogenates of the control.
(D) Immunohistochemistry analysis of CSPα in gray matter of the cerebral cortex showing, at a low field, a significant decrease of CSPα in affected individuals compared to the strong CSPα staining in the age-matched control.
To correlate these observed effects with in vivo, we analyzed post-mortem brain specimens by immunoblotting. Although both palmitoylated and nonpalmitoylated CSPα were present in control brain lysates, we could not detect any CSPα in brain lysates from a Dutch case (family N1) with the p.Leu115Arg mutation. However, after chemical depalmitoylation, we detected a chemiluminescence signal, probably corresponding to an otherwise insoluble CSPα-containing aggregate, which appeared much stronger in brain lysate from the case, than in the control (Figure 4C). Using immunohistochemical staining of CSPα in paraffin-embedded brain sections, we consistently found an absence of CSPα staining in synaptic regions in both the cerebral and the cerebellar cortex of individuals with the p.Leu115Arg mutation and significantly reduced CSPα staining in the cerebral cortex of individuals with the p.Leu116del mutation when we compared these individuals to age-matched controls (Figures 4D and 5).
Figure 5.
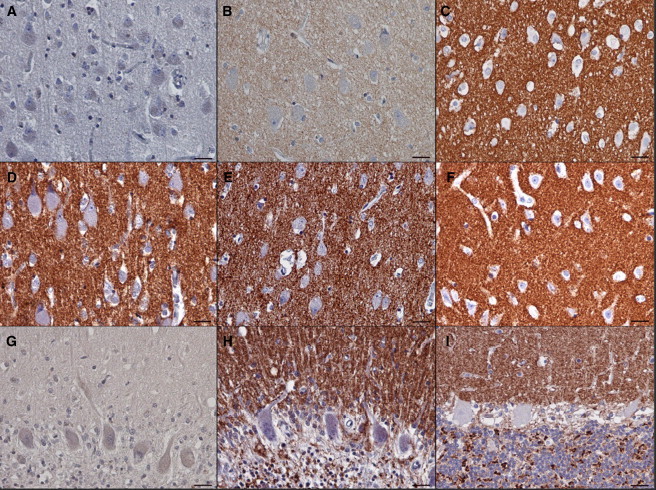
Brain Immunohistochemistry
(A–C) Detail of the CSPα staining in neuropil in the cerebral cortex that is absent in the individual with mutation p.Leu115Arg (A), decreased in the individual with mutation p.Leu116del (B), and strong in the age-matched control (C). Note the prominent neuronal storage, shown by large cell bodies, in both affected individuals.
(D–F) Staining pattern of the synaptic marker synaptobrevin in neuropil in the same regions in the individual with mutation p.Leu115Arg (D), in the individual with mutation p.Leu116del (E), and in the age-matched control (F).
(G and H) Cerebellar cortex of the case with p.Leu115Arg mutation. Similar to that in the cerebral cortex, CSPα staining is absent (G). This contrasts with a strong signal for synaptobrevin in the corresponding area in all three cerebellar cortical layers that are preserved adjacent to areas undergoing neurodegeneration (H).
(I) Strong CSPα staining in a control cerebellum. Note that the CSPα signal in the control (I) as well as the synaptobrevin signal in the individual with mutation p.Leu115Arg (H) are confined to the well defined synaptic regions (i.e., to the dendrites in the molecular layer, to the surface of the Purkinje cells, and to the synaptic glomeruli in the granular cell layer). The scale bars represent 25 μm.
Discussion
We carried out linkage mapping, gene-expression analysis, exome sequencing, and candidate-gene sequencing in affected individuals from 20 families and/or simplex cases of European descent suffering from autosomal-dominant adult-onset neuronal ceroid lipofuscinosis previously referred to as Parry disease. Using this approach, we identified in five of these families two recurrent mutations, c.346_348delCTC (p.Leu116del) and c.344T>G (p.Leu115Arg), in DNAJC5 encoding cysteine-string protein alpha (CSPα). To prove their causality, we performed haplotype analysis, which revealed that the mutations had to appear independently in at least four lineages, and by using targeted genotyping of seven unaffected siblings and 200 control individuals as well as searching the 1000 Genome and dbSNP databases, we found that the mutations are exclusively present in 14 affected individuals.
CSPα is a highly conserved protein with no amino acid sequence variant found in humans so far. The identified mutations affect evolutionary conserved dileucine residues located in the cysteine-string domain that is implicated in palmitoylation and membrane targeting of CSPα.15–17 Functional studies in transfected cell lines proved that these mutations affect palmitoylation and intracellular location of CSPα and thus decrease the level of the CSPα protein in the brain of affected individuals.
The molecular mechanisms underlying the dominant negative effect of the identified mutations on CSPα amounts in neuronal cells are not clear. It is known that CSPα forms detergent-resistant dimers18 and that the presence of these dimers correlates with an inhibition of synapse formation and synaptic transmission.19 Immunoblot analysis of brain lysate from affected individuals showed CSPα to be exclusively present in such an aggregate form. It is probable that the presence of mutant protein catalyzes accelerated aggregation and that the resulting aggregates will be composed equally of both mutant and wild-type proteins, and this will result in CSPα depletion. Another explanation of the dominant negative effect—nicely compatible with the observed lysosomal storage—would be a gradual accumulation of nondegradable CSPα aggregates in the lysosomal system. We followed this lead experimentally; however, we failed to identify CSPα in storage lysosomes by using immunohistochemistry analysis of fixed brain samples as well as in storage granules isolated from affected individuals' brains by using immunoblot analysis (data not shown).
CSPα associates with 70 kDa heat-shock cognate protein (Hsc70) and small glutamine-rich tetratricopeptide repeat domain protein (SGT) and forms an enzymatically active chaperone complex that is tethered to synaptic vesicles and ensures, in cooperation with other chaperones such as 40 kDa heat-shock protein (Hsp40),20 90 kDa heat-shock protein (Hsp90),21 Hsc70 interacting protein (HIP)22 and Hsp70 organizing protein (HOP),22 correct conformation of many proteins essential for the functionality of synapses. It was shown that CSPα deletion causes progressive neurodegeneration and reduced life span in Drosophila melanogaster23 and knockout mice.24,25 Depletion of CSPα interferes with SNARE complex formation and has a profound effect on presynaptic vesicle release and synaptic function.19,24,26–29 Thus, these CSPα mutations might lead to presynaptic dysfunction, explaining some of the neurological symptoms observed in affected individuals. In parallel, dysfunction of the CSPα/Hsc70/SGT chaperone complex might affect the folding quality of many client proteins and make them vulnerable to aggregation and degradation.30 This could, in the long term, lead to lysosomal accumulation of misfolded and proteolysis-resistant proteins in the form of characteristic ceroid deposits in neurons.
Our finding of neurodegenerative disease caused by mutations in DNAJC5 thus confirms a neuroprotective role for CSPα in humans and advocates detailed investigation of CSPα in the NCLs and other neurodegenerative diseases presenting with neuronal protein aggregation. It is interesting that there is no visual failure in the cases reported here, in contrast to the rapid loss of vision in mice completely lacking CSPα function.25
In this study we were able to explain ∼25% of ANCL cases tested, though not all were known to be autosomal-dominant and some could have been misdiagnosed. Those families that do not carry mutations in DNAJC5 or other known NCL genes provide a resource for identification of further genes whose disruption causes late-onset NCL.
In conclusion, we believe that our work represents an important step in the genetic dissection of a genetically heterogeneous group of ANCLs. From a clinical perspective, and in the absence of specific biochemical markers, our finding, together with the recent identification of CLN6 mutations in adult-onset recessive Kufs type A disease,2 provide essential information allowing efficient DNA-based testing in families as well as simplex cases with ANCL presentation.
Acknowledgments
This work was supported by the Grant Agency of Charles University of Prague (project 299911), the Ministry of Education of the Czech Republic (projects 1M6837805002 and MSM0021620806), Belgian Science Policy Office Interuniversity Attraction Poles program P6/43, Flemish Government Methusalem Excellence grant, Research Foundation Flanders (J.v.d.Z, postdoctoral fellowship), and the Batten Disease Support and Research Association. We thank clinical colleagues and families who contributed samples used in this study, especially John Morris and Joanne Porter (UCL563), David Sleat and the late Krystyna Wisniewski (UCL519), and Aristotle Siakotos (UCL328).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://www.1000genomes.org/
CSS-Palm 2.0, http://csspalm.biocuckoo.org/online.php
DAVID, Database for Annotation, Visualization and Integrated Discovery version 6.7, http://david.abcc.ncifcrf.gov/
ExPASy, http://expasy.org
Gene Expression Omnibus, http://www.ncbi.nlm.nih.gov/geo/
GeneReviews, Mole, S.E., and Williams, R.E. (2010). Neuronal Ceroid-Lipofuscinoses, www.ncbi.nlm.nih.gov/books/NBK1428
Online Mendelian Inheritance in man (OMIM), http://www.omim.org
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
R-project for Statistical Computing, http://www.r-project.org
SIFT BLink, http://sift.jcvi.org/www/SIFT_BLink_submit.html
Accession Numbers
Gene-expression data are available at the Gene Expression Omnibus (GEO) repository under accession GSE30369.
References
- 1.Mole S.E., Williams R.E., Goebel H.H. Oxford University Press; Oxford: 2011. The Neuronal Ceroid Lipofuscinoses (Batten Disease) [Google Scholar]
- 2.Arsov T., Smith K.R., Damiano J., Franceschetti S., Canafoglia L., Bromhead C.J., Andermann E., Vears D.F., Cossette P., Rajagopalan S. Kufs disease, the major adult form of neuronal ceroid lipofuscinosis, caused by mutations in CLN6. Am. J. Hum. Genet. 2011;88:566–573. doi: 10.1016/j.ajhg.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boehme D.H., Cottrell J.C., Leonberg S.C., Zeman W. A dominant form of neuronal ceroid-lipofuscinosis. Brain. 1971;94:745–760. doi: 10.1093/brain/94.4.745. [DOI] [PubMed] [Google Scholar]
- 4.Ferrer I., Arbizu T., Peña J., Serra J.P. A golgi and ultrastructural study of a dominant form of Kufs' disease. J. Neurol. 1980;222:183–190. doi: 10.1007/BF00313117. [DOI] [PubMed] [Google Scholar]
- 5.Josephson S.A., Schmidt R.E., Millsap P., McManus D.Q., Morris J.C. Autosomal dominant Kufs' disease: A cause of early onset dementia. J. Neurol. Sci. 2001;188:51–60. doi: 10.1016/s0022-510x(01)00546-9. [DOI] [PubMed] [Google Scholar]
- 6.Burneo J.G., Arnold T., Palmer C.A., Kuzniecky R.I., Oh S.J., Faught E. Adult-onset neuronal ceroid lipofuscinosis (Kufs disease) with autosomal dominant inheritance in Alabama. Epilepsia. 2003;44:841–846. doi: 10.1046/j.1528-1157.2003.39802.x. [DOI] [PubMed] [Google Scholar]
- 7.Sims K.B., Cole A.J., Sherman J.C., Caruso P.A., Snuderl M. Case records of the Massachusetts General Hospital. Case 8-2011. A 32-year-old woman with seizures and cognitive decline. N. Engl. J. Med. 2011;364:1062–1074. doi: 10.1056/NEJMcpc1013927. [DOI] [PubMed] [Google Scholar]
- 8.Nijssen P.C., Brusse E., Leyten A.C., Martin J.J., Teepen J.L., Roos R.A. Autosomal dominant adult neuronal ceroid lipofuscinosis: Parkinsonism due to both striatal and nigral dysfunction. Mov. Disord. 2002;17:482–487. doi: 10.1002/mds.10104. [DOI] [PubMed] [Google Scholar]
- 9.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 10.Thiele H., Nürnberg P. HaploPainter: A tool for drawing pedigrees with complex haplotypes. Bioinformatics. 2005;21:1730–1732. doi: 10.1093/bioinformatics/bth488. [DOI] [PubMed] [Google Scholar]
- 11.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang H., Orr A., Guernsey D.L., Robitaille J., Asselin G., Samuels M.E., Dubé M.P. Application of homozygosity haplotype analysis to genetic mapping with high-density SNP genotype data. PLoS ONE. 2009;4:e5280. doi: 10.1371/journal.pone.0005280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Landmann L. Deconvolution improves colocalization analysis of multiple fluorochromes in 3D confocal data sets more than filtering techniques. J. Microsc. 2002;208:134–147. doi: 10.1046/j.1365-2818.2002.01068.x. [DOI] [PubMed] [Google Scholar]
- 14.Manders E.M.M., Verbeek F.J., Aten J.A. Measurement of Colocalization of Objects in Dual-Color Confocal Images. Journal of Microscopy. 1993;169:375–382. doi: 10.1111/j.1365-2818.1993.tb03313.x. [DOI] [PubMed] [Google Scholar]
- 15.Greaves J., Salaun C., Fukata Y., Fukata M., Chamberlain L.H. Palmitoylation and membrane interactions of the neuroprotective chaperone cysteine-string protein. J. Biol. Chem. 2008;283:25014–25026. doi: 10.1074/jbc.M802140200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greaves J., Chamberlain L.H. Dual role of the cysteine-string domain in membrane binding and palmitoylation-dependent sorting of the molecular chaperone cysteine-string protein. Mol. Biol. Cell. 2006;17:4748–4759. doi: 10.1091/mbc.E06-03-0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chamberlain L.H., Burgoyne R.D. The cysteine-string domain of the secretory vesicle cysteine-string protein is required for membrane targeting. Biochem. J. 1998;335:205–209. doi: 10.1042/bj3350205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Swayne L.A., Blattler C., Kay J.G., Braun J.E. Oligomerization characteristics of cysteine string protein. Biochem. Biophys. Res. Commun. 2003;300:921–926. doi: 10.1016/s0006-291x(02)02964-9. [DOI] [PubMed] [Google Scholar]
- 19.Xu F., Proft J., Gibbs S., Winkfein B., Johnson J.N., Syed N., Braun J.E. Quercetin targets cysteine string protein (CSPalpha) and impairs synaptic transmission. PLoS ONE. 2010;5:e11045. doi: 10.1371/journal.pone.0011045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gibbs S.J., Barren B., Beck K.E., Proft J., Zhao X., Noskova T., Braun A.P., Artemyev N.O., Braun J.E. Hsp40 couples with the CSPalpha chaperone complex upon induction of the heat shock response. PLoS ONE. 2009;4:e4595. doi: 10.1371/journal.pone.0004595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakisaka T., Meerlo T., Matteson J., Plutner H., Balch W.E. Rab-alphaGDI activity is regulated by a Hsp90 chaperone complex. EMBO J. 2002;21:6125–6135. doi: 10.1093/emboj/cdf603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosales-Hernandez A., Beck K.E., Zhao X., Braun A.P., Braun J.E. RDJ2 (DNAJA2) chaperones neural G protein signaling pathways. Cell Stress Chaperones. 2009;14:71–82. doi: 10.1007/s12192-008-0056-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zinsmaier K.E., Eberle K.K., Buchner E., Walter N., Benzer S. Paralysis and early death in cysteine string protein mutants of Drosophila. Science. 1994;263:977–980. doi: 10.1126/science.8310297. [DOI] [PubMed] [Google Scholar]
- 24.Fernández-Chacón R., Wölfel M., Nishimune H., Tabares L., Schmitz F., Castellano-Muñoz M., Rosenmund C., Montesinos M.L., Sanes J.R., Schneggenburger R., Südhof T.C. The synaptic vesicle protein CSP alpha prevents presynaptic degeneration. Neuron. 2004;42:237–251. doi: 10.1016/s0896-6273(04)00190-4. [DOI] [PubMed] [Google Scholar]
- 25.Schmitz F., Tabares L., Khimich D., Strenzke N., de la Villa-Polo P., Castellano-Muñoz M., Bulankina A., Moser T., Fernández-Chacón R., Südhof T.C. CSPalpha-deficiency causes massive and rapid photoreceptor degeneration. Proc. Natl. Acad. Sci. USA. 2006;103:2926–2931. doi: 10.1073/pnas.0510060103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burgoyne R.D., Morgan A. Chaperoning the SNAREs: A role in preventing neurodegeneration? Nat. Cell Biol. 2011;13:8–9. doi: 10.1038/ncb0111-8. [DOI] [PubMed] [Google Scholar]
- 27.García-Junco-Clemente P., Cantero G., Gómez-Sánchez L., Linares-Clemente P., Martínez-López J.A., Luján R., Fernández-Chacón R. Cysteine string protein-alpha prevents activity-dependent degeneration in GABAergic synapses. J. Neurosci. 2010;30:7377–7391. doi: 10.1523/JNEUROSCI.0924-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burré J., Sharma M., Tsetsenis T., Buchman V., Etherton M.R., Südhof T.C. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sharma M., Burré J., Südhof T.C. CSPα promotes SNARE-complex assembly by chaperoning SNAP-25 during synaptic activity. Nat. Cell Biol. 2011;13:30–39. doi: 10.1038/ncb2131. [DOI] [PubMed] [Google Scholar]
- 30.Johnson J.N., Ahrendt E., Braun J.E. CSPalpha: The neuroprotective J protein. Biochem. Cell Biol. 2010;88:157–165. doi: 10.1139/o09-124. [DOI] [PubMed] [Google Scholar]
- 31.Nijssen P.C., Brekelmans G.J., Roos R.A. Electroencephalography in autosomal dominant adult neuronal ceroid lipofuscinosis. Clin. Neurophysiol. 2009;120:1782–1786. doi: 10.1016/j.clinph.2009.07.042. [DOI] [PubMed] [Google Scholar]
- 32.Nijssen P.C., Ceuterick C., van Diggelen O.P., Elleder M., Martin J.J., Teepen J.L., Tyynelä J., Roos R.A. Autosomal dominant adult neuronal ceroid lipofuscinosis: A novel form of NCL with granular osmiophilic deposits without palmitoyl protein thioesterase 1 deficiency. Brain Pathol. 2003;13:574–581. doi: 10.1111/j.1750-3639.2003.tb00486.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Poët M., Kornak U., Schweizer M., Zdebik A.A., Scheel O., Hoelter S., Wurst W., Schmitt A., Fuhrmann J.C., Planells-Cases R. Lysosomal storage disease upon disruption of the neuronal chloride transport protein ClC-6. Proc. Natl. Acad. Sci. USA. 2006;103:13854–13859. doi: 10.1073/pnas.0606137103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reif A., Schneider M.F., Hoyer A., Schneider-Gold C., Fallgatter A.J., Roggendorf W., Pfuhlmann B. Neuroleptic malignant syndrome in Kufs' disease. J. Neurol. Neurosurg. Psychiatry. 2003;74:385–387. doi: 10.1136/jnnp.74.3.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.