

Abstract
Human sexual determination is initiated by a cascade of genes that lead to the development of the fetal gonad. Whereas development of the female external genitalia does not require fetal ovarian hormones, male genital development requires the action of testicular testosterone and its more potent derivative dihydrotestosterone (DHT). The “classic” biosynthetic pathway from cholesterol to testosterone in the testis and the subsequent conversion of testosterone to DHT in genital skin is well established. Recently, an alternative pathway leading to DHT has been described in marsupials, but its potential importance to human development is unclear. AKR1C2 is an enzyme that participates in the alternative but not the classic pathway. Using a candidate gene approach, we identified AKR1C2 mutations with sex-limited recessive inheritance in four 46,XY individuals with disordered sexual development (DSD). Analysis of the inheritance of microsatellite markers excluded other candidate loci. Affected individuals had moderate to severe undervirilization at birth; when recreated by site-directed mutagenesis and expressed in bacteria, the mutant AKR1C2 had diminished but not absent catalytic activities. The 46,XY DSD individuals also carry a mutation causing aberrant splicing in AKR1C4, which encodes an enzyme with similar activity. This suggests a mode of inheritance where the severity of the developmental defect depends on the number of mutations in the two genes. An unrelated 46,XY DSD patient carried AKR1C2 mutations on both alleles, confirming the essential role of AKR1C2 and corroborating the hypothesis that both the classic and alternative pathways of testicular androgen biosynthesis are needed for normal human male sexual differentiation.
Introduction
Male sexual determination is initiated by Y-chromosomal SRY, which activates a cascade of genes that lead the embryonic gonad to develop into a testis. Fetal testicular Sertoli cells then produce Mullerian inhibitory substance, which is responsible for the involution of the Mullerian ducts. These ducts otherwise develop into the uterus, fallopian tubes, and cervix. Fetal testicular Leydig cells produce testosterone from cholesterol by the sequential actions of enzymes encoded by CYP11A1 (MIM 118485), CYP17A1 (MIM 609300), HSD3B2 (MIM 201810), and HSD17B3 (MIM 605573) (Figure 1).1 Subsequent differentiation of male external genitalia also requires the action of dihydrotestosterone (DHT), produced from testicular testosterone by the action of 5α-reductase, type 2 (encoded by SRD5A2 [MIM 607306]) in genital skin.2 Genetic males with disorders in the enzymes in this classic pathway of androgen biosynthesis have disordered sexual development with incompletely developed (“ambiguous”) external genitalia, but some hormonal disorders of male development remain unexplained, and this fact suggests that not all relevant factors have been identified.3 Fetal male marsupials produce DHT by an alternative biosynthetic pathway without the intermediacy of testosterone (Figure 1)4–6. Two disorders of human steroidogenesis suggest that this alternative pathway might be involved in human sexual development. First, fetuses with severe 21-hydroxylase deficiency (CAH1 [MIM 201910]) produce amounts of 17-hydroxyprogesterone (17OHP) that are typically elevated 100-fold,7 but 17OHP is not readily converted to androstenedione and testosterone by the classic pathway,8 yet affected females become severely virilized in utero, and this suggests conversion of 17OHP to dihydrotestosterone via the alternative pathway.9 Second, patients with mutations in POR encoding P450 oxidoreductase (encoded by POR [MIM 124015, 613571, and 201750]), which donates electrons to steroid 21-hydroxylase (CYP21A2), 17α-hydroxylase/17,20 lyase (P450c17, CYP17A1), and aromatase (CYP19A1 [MIM 107910]), typically have ambiguous genitalia10,11 and excrete urinary steroid metabolites that suggest involvement of the alternate pathway.12–16 We explored the potential role of the alternative pathway in human male development in five patients from two families with genital ambiguity and disordered androgen biosynthesis after mutations in the classic pathway of steroid biosynthesis had been excluded.
Figure 1.
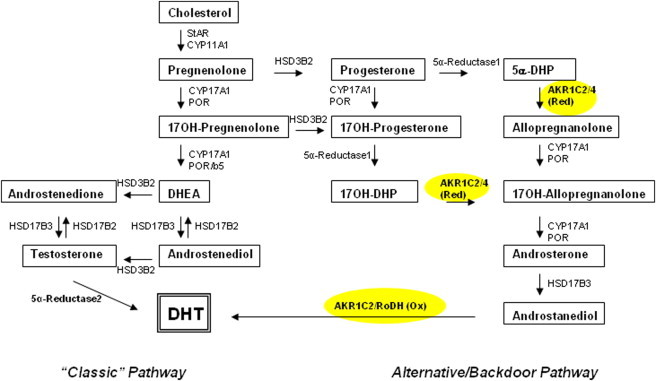
Synthesis of Dihydrotestosterone via the Classic and Alternative (Backdoor) Pathways
The classic pathway of steroidogenesis leading to dihydrotestosterone is shown on the left, and the alternative pathway is shown on the right. The factors in the classic pathway are CYP11A1 (cholesterol side-chain cleavage enzyme, P450scc), StAR (steroidogenic acute regulatory protein), CYP17A1 (17α-hydroxylase/17,20-lyase, P450c17), HSD3B2 (3β-hydroxysteroid dehydrogenase, type 2), HSD17B3 (17β-HSD3 [17β-hydroxysteroid dehydrogenase, type 3] and 5α-reductase, type 2 [5a-reductase 2, encoded by SRD5A2]). The alternative pathway is characterized by the presence of additional enzymes: 5α-reductase, type 1 (5a-reductase 1, encoded by SRD5A1), AKR1C2 3 (3α-reductase, type 3) and possibly AKR1C4 (3α-reductase, type 1) and RoDH (3-hydroxyepimerase, encoded by HSD17B6). Most steroids are identified by their trivial names; 17-hydroxy-dihydroprogesterone (17OH-DHP) is 5α-pregnane-17α-ol-3,20-dione; 17-hydroxy-allopregnanolone (17OH-allo) is 5α-pregnan-3α,17α-diol-20-one; 5α-dihydroprogesterone (5α-DHP) is 5α-pregnane-3,20-dione, and allopregnanolone is 3α-hydroxy-dihydroprogesterone (3α-OH-DHP) or 5α-pregnane-3α-ol-20-one.
Subjects and Methods
Clinical Material
Family 1
A Swiss family that included three persons with ambiguous genitalia was first described in 1972 as having “17,20-desmolase deficiency”17. Two 46,XY cousins (Figure 2A, individuals III.3 and III.4) had undervirilized external genitalia and cryptorchidism. Analyses of urinary steroids at that time showed increased excretion of pregnenetriol (a urinary metabolite of 17-hydroxypregnenolone) and pregnanetriolone (a metabolite of 21-deoxycortisol), which was also hyperresponsive to stimulation with either chorionic gonadotropin or corticotropin. Excretion of pregnanetriol (a metabolite of 17-hydroxyprogesterone) was modestly elevated, hyperresponsive to corticotrophin, and unresponsive to chorionic gonadotropin. Normal total excretion of etiocholanolone, androsterone, and other steroids requiring 5α-reduction (e.g., 5α-tetrahydrocortisol), and their ratios excluded 5α-reductase deficiency. Metabolites of cortisol and cortisone were normal, but urinary dehydroepiandrosterone was unmeasureable after stimulation with chorionic gonadotropin or corticotropin. A maternal aunt (II.5) had tall stature, slightly virilized genitalia, and primary amenorrhea. Her karyotype was 46,XY; a laparatomy revealed no uterus, and gonadectomy revealed normal testicular tissue.
Figure 2.
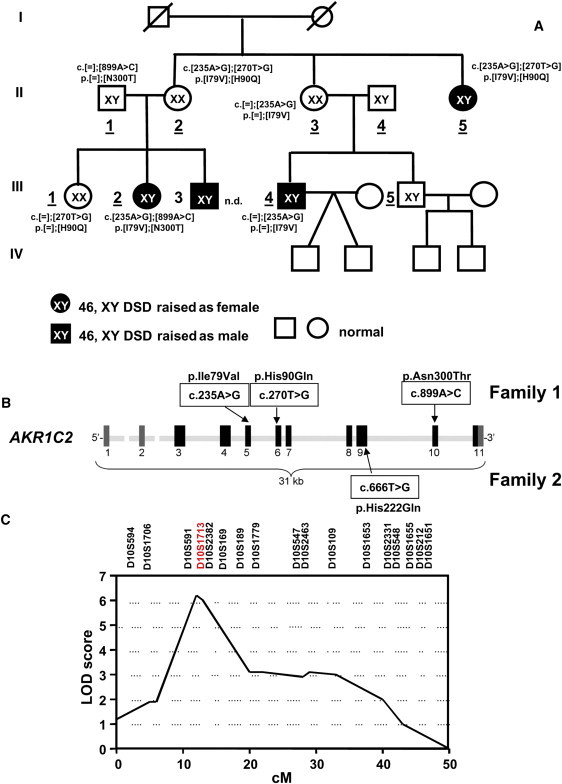
Identification of AKR1C2 Mutations in Families 1 and 2
(A) The pedigree of the index family (family 1). The AKR1C2 genotype and the amino acid exchange are given adjacent to the symbols. Individuals II.4 and III.5 had a normal genotype, which is not indicated. Family generations are indicated in Roman numerals, and relevant individuals within each generation are numbered. Individuals whose numbers are underlined were analyzed. Individual III.3 refused genetic testing (n.d. is an abbreviation for not done). The sexes indicated (a square for a male and a circle for a female) correspond to the sex of rearing. The filled symbols represent individuals with a disorder of sexual development. The proband is individual III.2.
(B) Diagram of AKR1C2 on chromosomal region 10p14-p15 encoding 3α-HSD3. Coding exons 3–11 are shown as black boxes, noncoding exons 1 and 2 are shown in gray. The locations of the four identified missense mutations are shown.
(C) High-resolution multipoint linkage analysis by GENEHUNTER (the model is based on autosomal-recessive transmission). For markers D10S1713 and D10S2382, the maximum LOD scores were Zmax = 6.3 and 6.2, respectively.
Individual III.1 was SRY-negative and had normal female external genitalia and a sonographically detectable uterus. Individual III.2 had normal female external genitalia but had steroidal responses to chorionic gonadotropin and corticotropin that were similar to individuals III.3 and III.4 suggesting she had the same defect in steroidogenesis. Her karyotype was 46,XY, and she had no uterus. She was gonadectomized before puberty and histology showed normal (not dysgenetic) testicular tissue. No family members had signs or steroidal findings of adrenal insufficiency. Individuals II.5 and III.2 underwent gonadectomy in the 1970s and declined further investigations of adrenal steroidogenesis.
Family 2
This Swiss patient was diagnosed with 46,XY disordered sexual development (DSD) during surgery for bilateral inguinal hernias at the age of 7 weeks. Normal looking testes were found intraoperatively in a completely feminized subject without evidence of Müllerian structures. Chromosome analysis revealed a 46,XY karyotype. There was no evidence of additional 46,XY DSD cases in the family. Laparascopic gonadectomy was carried out at 2 years of age. No further hormonal assessment was performed in this patient. The phenotype of 46,XY patients of both families is summarized in Table S1, available online.
Mutation Analysis
After obtaining informed consent from the studied subjects, genomic DNA was extracted from peripheral blood leukocytes (PBL) (QIAGEN DNA blood and cell culture kit; QIAGEN GmbH, Hilden, Germany). The genes for steroidogenic factor 1 (NR4A1, NM_004959.4 [MIM 184757]), androgen receptor (AR, NM_000044.2, NM_00101165.1 [MIM 313700]), 17α-hydroxylase/17,20 lyase (CYP17A1, NM_000102.3), P450 oxidoreductase (POR,NM_000941.2), and 3-hydroxyepimerase (RoDH/HSD17B6, NM_003725.2 [MIM 606623]) were sequenced in all patients.9 The genes AKR1C1–4 and AKR1CL1 are clustered on chromosome 1018 (NCBI entry NC_000010.10) and localize as follows: AKR1C1, 5005454-5020158 (MIM 600449); AKR1C2, 5042103-5046053 (MIM 600450); AKR1C3, 5136568-5149878 (MIM 603966); AKR1CL1, 5227144-5236924 ; AKR1C4, 5238798-5260912 (MIM 600451). The genes were amplified and sequenced with specific primers designed according to these entries. Three different human AKR1C2 mRNA transcripts are listed in the NCBI database, but two of them (variant 1 NM_001354.4 and variant 2 NM_205845.1) differ only in their 5′ untranslated regions and encode the same protein (NP_001345.1, UniProt P52895). The primer sequences are reported in Tables S2 and S3. The third splicing variant (NM_001135241.1) encodes the shorter protein (isoform 2, NP_001128713.1, UniProt B4DKR9) with a different C terminus; it has minimal 3α-hydroxysteroid dehydrogenase (3α-HSD) activity and was not considered. The exon positions and intron-exon boundaries of the AKR1C genes were checked by BLAT analysis. For AKR1C2, DNA from 200 unrelated, healthy individuals (180 white, ten Asians; eight blacks; two Hispanics) and the 1000 Genome database were used as a control for polymorphisms. AKR1CL1 (NR_027916.1) produces a noncoding RNA and was not considered for this study. Total RNA was extracted from PBL of all family members (RNAeasy, QIAGEN GmbH, Hilden, Germany) and used to perform end-point reverse transcriptase PCR (RT-PCR) of AKR1C1–4 full-length cDNAs. RNAs from 50 unrelated individuals were used as control.
For the patient in family 2, an additional long-range PCR method was developed for the coding exons, with an AKR1C1 (NM_001353.5,) or an AKR1C2-specific forward primer (Table S2) and the Expand 20kbPlus PCR System (Roche Applied Sciences). Given the identity shared between the two genes in their 3′ regions, the reverse primer is not specific. The two genes differ in the number of EcoRI sites so that the EcoRI digestion of the PCR product of AKR1C1 (3 sites) results in four main bands of 6.7, 5.5, 4.0, and 2.7 Kb, whereas digestion of AKR1C2 (4 sites) results in five main bands of 6.0, 4.0, 3.1, 2.72, and 1.8 Kb (Figure 3).
Figure 3.
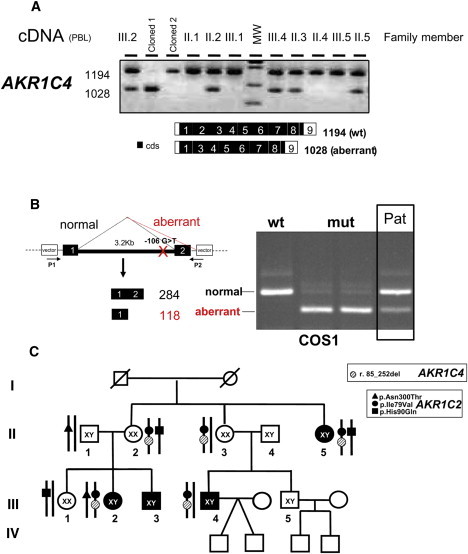
Analysis of AKR1C4 Transcripts
(A) The end-point reverse-transcriptase PCR analysis of AKR1C4 transcript from PBL of all available family members. In the affected members of the family, the normally spliced AKR1C4 band (1194 bp) is present together with a shorter band (1028 bp) that was shown by sequencing to lack all of exon 2 (r.[ = ];[r.85_252del]).
(B) An exon-trapping assay, demonstrating that the mutation found in IVS1 (c.85-106G>T) reproduced in COS1 cells, is responsible for the aberrant splicing. The following abbreviations are used: wt: cloned WT; mut: cloned mutant in duplicate; Pat: patient: RT-PCR generated cDNAs from PBL of Individual III.4. Black boxes represent the AKR1C4 exons; the white boxes are vector sequences.
(C) Summary of the genetic findings in the index family. Given the simultaneous presence of the p.Ile79Val mutation in AKR1C2 and the AKR1C4 splice mutation in all the cases, these two mutations are expected to segregate together.
Splicing-Reporter Analysis
DNA fragments encompassing AKR1C4 (NM_001818.2 [MIM 600451]) exons 1 and 2 were amplified from patients' and control DNA and cloned into pCR3.1 (Invitrogen). Reporter constructs were transfected into HeLA cells with the Fugene transfection reagent. Forty-eight hours after transfection, cells were harvested, and total RNA was extracted with an RNAeasy kit (QIAGEN). The cDNAs were amplified with vector-specific primers.19
Microsatellite Analysis
Microsatellite analysis for members of family 1 was performed with the ABI Prism Linkage mapping set v 2.5 (811 markers, Applied Biosystems) and a custom-made set of 17 microsatellite markers for chromosome 10. Haplotypes were reconstructed with the GENEHUNTER package (Version 1.2). The disease gene frequency in the general population was set at 0.0001.20 Two-point and multipoint linkage analyses were performed by GENEHUNTER with the affected-only allele-sharing method. SLink simulations were done by the FASTLINK package21.
Computational Modeling and Docking Studies
We studied the effect of AKR1C2 mutations by structural analysis to understand the consequences of specific amino acid replacements on steroidogenic activities. A model of AKR1C2 (NP_001345.1) in a complex with 3α-androstanediol (3αDiol) was built with YASARA22–24 and WHAT IF25. First, a 3D model of 3αDiol was built with ChemDraw and Chem3D and optimized by energy minimization and molecular-dynamics (MD) simulation in YASARA with the AMBER03 force field.26 The optimized 3αDiol was docked into the AKR1C2 structure with the program Patchdock.27 We refined the docked structures by performing MD simulations with YASARA dynamics.23,24 We ran MD simulations with AMBER03 force field at 298 K and 0.9% NaCl in the simulation cell for 500 ps to refine the docked structure. The positions of amino acids changed by mutations were evaluated for possible contact with steroids or cofactors as well as structural instabilities. Effects of mutations on binding of steroid substrates to AKR1C2 were also evaluated by in silico analysis. To identify the potential routes for binding of the steroid substrate, nicotinamide adenine dinucleotide phosphate (NADP+) and the acetate or citrate ions that sit between steroid and nicotinamide adenine dinucleotide phosphate (NADPH) and facilitate the electron transfer, we probed the AKR1C2 structure without the bound steroids with the program Caver, which identifies tunnels in the protein structures by connecting internal cavities with the solvent.28 The shape of tunnels in AKR1C2 was approximated as a pipeline originating from the acetate ion that was used as the starting point for calculations. Distinct routes for binding of steroid substrate and NADPH and a potential route for solvent access for acetate or citrate ions was identified. Tunnels were depicted with a surface representation of the empty space and the models were edited with the program Pymoland and depicted as ray-traced images with POVRAY.
A 3D crystal structure of AKR1C4 (NP_001809.2) in complex with NADPH is available (Protein Data Bank [PDB] number 2FVL) from the structural genomics consortium but has not been described in a publication. We used the crystal structure of AKR1C4 to make a structural model of alternatively spliced variant of AKR1C4 lacking exon 2. We built the model with YASARA by using the structural alignment with full-length AKR1C4 and independently modeling the N-terminal helix and joining it with the rest of the structure. We modeled one loop (lndhFMPvlgfg) separately by scanning the database of known loop structures. The model was refined by MD simulation with the AMBER03 force field as previously described.22–24,29
Expression and Purification of Wild-Type and Mutant AKR1C2
The bacterial expression vector pGEX-AKR1C2 containing wild-type (WT) human AKR1C2 cDNA with an N-terminal glutathione-S-transferase tag (GST) was a generous gift of Rock Breton, Oncology and Molecular Endocrinology Research Center, Laval University Medical Center, Quebec, Canada.30 Both the N and C termini of AKR1C2 are important for steroid and cofactor binding; therefore, the 972 bp AKR1C2 cDNA encoding the full-length protein (NP_001345.1, 323 amino acids) was used in our studies. Vectors for expression of the mutants were generated by PCR-based site-directed mutagenesis with specific primers; all mutations were confirmed by direct sequencing. Proteins were expressed in E. coli BL21(DE3)pLysS and then purified by glutathione affinity chromatography as described.30 As a negative control, GST was expressed from the empty pGEX vector and processed similarly. The purity of the prepared proteins was assessed by SDS-polyacrylamide gel electrophoresis; Coomassie blue staining showed single bands, and the purified proteins were quantitated colorimetrically (Bio-Rad protein assay, Bio-Rad Laboratories GmbH, Munich, Germany).
In Vivo Activity of WT and Aberrantly Spliced AKR1C4 in Cell Culture
WT or mutant AKR1C4 cDNA with an N-terminal Myc tag were inserted into a pCMV6 vector and constructs were transfected into ∼80% confluent COS1 cells (ATCC CRL-1650) with TransFast transfection reagent (Promega; 9 μl Transfast/μg DNA), and an empty vector was used as the control. Transfection efficiency was measured by transfecting with a vector containing pSV-β-Galactosidase. Forty-eight hours after transfection, two 15 cm plates were pooled for microsomal preparation as previously described.31 Total protein content of the microsomal preparations was measured with UniCel DxC600 (Beckman Coulter). Twelve micrograms of microsomal protein (WT, Mut, and an empty vector) were incubated with 0.0, 0.1, 1, 10, and 100 μM 5α-dihydroprogesterone (5α-DHP; Steraloids). In a second series of experiments, 12 μg microsomal protein was incubated with 100 μM DHP in the presence of 1, 10, and 100 μM and 1 mM NADPH or nicotinamide adenine dinucleotide (NAD) for 45 min as described.31 Subsequently, steroids were extracted with 0.8 ml heptane and ethyl acetate (1:1, vol/vol), concentrated by evaporation with N2 at 64°C, and assayed by gas chromatography followed by mass spectrometry (GC/MS). The specific ion monitored for 3αDiol (IS1) was 331.3; for stigmasterol (IS2), 394.3; for cholesterol butyrate (IS3), 368.3; for 5α-DHP, 686; and for allopregnanolone, 474. Immunoblot analysis was performed with standard procedures and an antibody against the Myc epitope (9E10, dilution 1:75).
Activity Assays of Human Recombinant WT and Mutant AKR1C2
The catalytic activities of WT AKR1C2 and the three missense mutations found in family 1 were assayed with two different substrates. Oxidation of 3αDiol was assessed by incubating 10 μg recombinant AKR1C2 with 0.3–1000 μM [3H]3αDiol (80,000cpm/reaction) and 1 mM NAD+ in 50 mM KiP buffer (K2HPO4/KH2PO4, pH7.4) in a final volume of 200 μl for 120 min at 37°C. Reduction of 5α-DHP was assessed with 3 μg recombinant 3α-HSD3, 0.1–100 μM [carbon 4-14C]5α-DHP (15,000cpm/reaction), and 1 μM NADPH in the KiP buffer for 10–30 min at 37°C. Radiolabeled [3H]3αDiol was purchased from PerkinElmer AG (Schwerzenbach, Switzerland). Radiolabeled [carbon 4-14C]5α-DHP was a kind gift of Prof. Synthia H. Mellon (University of California San Francisco, San Francisco, CA). The reactions were stopped and the steroids were extracted and separated by thin-layer chromatography as described8 and quantified by PhosphorImaging (FLA-7000, Fujifilm AG, Dielsdorf, Switzerland). The Michaelis constant (Km) and maximal velocity (Vmax) were calculated, and Lineweaver-Burk plots were drawn computationally with Prism (GraphPad Software, San Diego, CA, USA).
WT AKR1C2 and the c.666T>G (p.His222Gln) AKR1C2 mutant found in family 2 were constructed in pCMV6 and transfected in COS1 cells as described above for AKR1C4. Enzymatic activity in microsomes was assessed with 0.0. 0.1, 1, 10, and 100 μM 3αDiol (Steraloids). For the GC/MS measurements, ion monitoring was 131.3 for 3αDiol and 347 for DHT. In this case, 3αDiol was not used as internal standard.
Fetal and Adult Expression of AKR1C1–4, AKR1CL1, SRD5A1/2, and RoDH
Human fetal testes and adrenal cDNA were purchased from Biochain (Amsbio, Lugano Switzerland, C1244260-10 and C1244004, respectively). Normal adult testes and adrenals were kindly provided by Prof. George Thalmann, University of Bern. cDNAs from these tissues were prepared by total RNA extraction by the Trizol method (Invitrogen, Basel, Switzerland) and reverse transcription with the Improm RNA Transcriptase kit (Promega, Madison, WI, USA) according to the manufacturer's protocols. Qualitative and quantitative real-time PCR experiments were then performed on these cDNAs with primers specifically recognizing the cDNAs of AKR1C1–4 and AKR1CL1 (AKR1C1, NM_001353.5; AKR1C2, NM_001354.4; AKR1C3, NM_003739.4; AKR1C4, NM_001818.3; AKR1CL1 alias AKR1E2, NR_027916), SRD5A1-2 (SRD5A1, NM_001047.2 [MIM 184753]; SRD5A2, NM_00348.3 [MIM 607306]) and RoDH (HSD17B6, NM_003725.2). The identities of the end-point reverse transcriptase-PCR products were confirmed by sequencing. Quantitative real-time PCR was performed with the same primers and quantified as described.32 In brief, real-time PCR, performed with an ABI 7000 Sequence Detection System (Applera Europe) and PCR products were quantified fluorometrically with the SYBR Green Core Reagent kit. The reference mRNA cyclophillin was used for normalizing the data (the primers are listed in Table S4). The relative expression was calculated with the 2−ΔΔCt method. Statistical analysis was performed with a two-tailed t test (confidence interval 95%) with GraphPad Prism software.
Results
Clinical Genetics in Family 1
In 1972 a Swiss family with a 17,20-desmolase deficiency17, now termed 17,20 lyase deficiency”(MIM 202110), was described. Two 46,XY cousins (Figure 2, III.3, III.4) had cryptorchidism and undervirilized external genitalia at birth and hormonal evidence of defective androgen synthesis. The maternal aunt (II.5) had 46,XY sex reversal and similar steroid hormone abnormalities. We sought to determine the genetic defect in these individuals; the family agreed to genetic testing but refused all further hormonal testing, hence the only hormonal data available are those from 1972. Steroid 17α-hydroxylase and 17,20 lyase activities are catalyzed by P450c17 and encoded by CYP17A1;33–35 the 17,20 lyase activity of P450c17 requires electron donation from P450 oxidoreductase and the allosteric assistance of cytochrome b5.8 The hormonal findings of isolated 17,20-lyase deficiency might result from specific mutations in CYP17A136 or POR37 and by absence of cytochrome b5.38 Sequencing of CYP17A1, POR, NR5A1 (steroidogenic factor-1), CYB5 (cytochrome b5), and NR3C4 (an AR) revealed no mutations in these subjects, and the available hormonal data suggested no defects in 17β-hydroxysteroid dehydrogenase or 5α-reductase activities. Therefore, we considered whether mutations in genes for factors specific to the alternative pathway for production of DHT might be involved. This pathway entails both oxidative and reductive 3α-HSD reactions, which are not components of the classic pathway (Figure 1). Available evidence indicates that the reductive reaction is catalyzed by AKR1C2,39,40 although the involvement of other members of the AKR1C family is also possible. The enzyme catalyzing the oxidative reaction is less clear, although 3-hydroxyepimerase (HSD17B6, also known as RoDH) has oxidative 3α-HSD activities and converts androstanediol to DHT in the prostate;41 another candidate might be AKR1C4.42,43
Sequencing of the exons of the 31 kb AKR1C2 gene identified missense mutations in all affected individuals (Figures 2A and 2B). Individual II.5 was a compound heterozygote for the AKR1C2 missense mutations c.235A>G (p.Ile79Val) and c.270T>G (p.His90Gln), and individual III.2 was compound heterozygous for c.235A>G and c.899A>C (p.Asn300Thr); individual III.3 declined genetic testing (Figure 2A). All affected individuals had a 46,XY karyotype; 46,XX individual II.2 was a compound heterozygote for AKR1C2 mutations c.235A>G and c.270T>G (p.His90Gln), as was affected individual II.5, yet she was phenotypically normal and bore three children. Thus, in this family, the consequences of AKR1C2 mutations were a sex-limited autosomal-recessive trait. These genetics are consistent with androgens playing no essential role in female sexual development. AKR1C2 heterozygosity in 46,XY individual II.1 was associated with normal genital development and normal reproductive capacity, and heterozygous 46,XX individual II.3 was phenotypically normal and fertile. The external genitalia in 46,XY individuals II.5 and III.2 were severely undervirilized, and these individuals had been assigned a female sex at birth. By contrast, heterozygous individual III.4 was reported to be moderately undervirilized (Prader 3 genitalia with undescended testes and a microphallus) and to be hormonally similar to individual III.3,17 who refused genetic testing. To address the possibility that these missense mutations were polymorphisms, we sequenced AKR1C2 in 200 unrelated, healthy individuals (180 whites, ten Asians, eight blacks, and two Hispanics) and found no mutations in these 400 alleles. None of the mutations is present in the 1000 Genomes database (location browsed was chromosome 10: 5042103-5046053).
To confirm that the mutations in AKR1C2 are responsible for the phenotype in family 1, we scanned the whole genomes of nine individuals in generations II and III with 811 microsatellite markers (Figure S1). Multipoint affected-only allele-sharing methods suggested potential linkage to chromosomes 10 and 22 with LOD scores higher than 2.2 (genome-wide suggestive linkage LOD 2.2). Therefore, we re-examined these chromosomes with custom-made primers to probe microsatellites on 10p and 22q at higher resolution. On chromosomal region 22q12, D22S1167 and D22S1144 gave LOD scores of 3.2; D22S1167 had no cross references, whereas D22S1144 is linked to an insulinoma tumor suppressor gene (ITS [MIM 606960]), a sensorineural hearing loss gene (DFNB4 [MIM 600791]), and a gene for familial partial epilepsy syndrome (FPEVF [MIM 604364]). None of these genes appears to be involved with steroidogenesis. On chromosomal region 10p15, D10S1713 and D10S2382 gave scores of 6.3 and 6.2, respectively. These two markers span a region of about 0.5 Mb containing AKR1C1–4. No other signals were detected; in particular, signals were not found at the chromosomal loci encoding microsomal cytochrome b5 (CYB5A [MIM 613218]) (18q23), mitochondrial b5 (CYB5B [MIM 611964]) (16q22.1), 5α-reductase type 1 (SRD5A1; 5p15), and RoDH (HSD17B6) (12q13.3) (Figure S1), confirming our identification of a disordered AKR1C enzyme in this family (Figure 2C and Figure S1). Because other members of the AKR1C family might also catalyze 3α-HSD reactions and the microsatellite analysis could not distinguish these closely linked genes, we also sequenced AKR1C1, AKR1C3, AKR1C4, and AKR1CL1 from individual III.2 but found no mutations in the coding regions.
Splicing Analysis
Because the exonic sequencing could not exclude splicing mutations, we studied the splicing pattern of these five AKR1Cs by analyzing cDNA amplified from reverse transcribed mRNAs extracted from PBL of all the family members. AKR1C1, AKR1C2, AKR1C3, and AKR1CL1 were normally spliced, whereas AKR1C4 showed an aberrantly spliced fragment in individuals II.2, II.3, II.5, III.2, and III.4, in addition to normally spliced mRNAs (cDNA) (r.[ = ];[85_252del], Figure 3A). Direct sequencing showed that exon 2 was completely absent from the aberrantly spliced AKR1C4. Sequencing of the flanking introns (IVS) identified a heterozygote G to T substitution 106 bp upstream of exon 2 (c.[ = ]; [85-106G>T]). An exon trapping assay demonstrated that this IVS mutation resulted in loss of exon 2 (c.85_252del) (Figure 3B). The splicing mutation is in frame and is expected to delete AKR1C4 residues 29–84 (p.Val29_Lys84del). No aberrant splicing was found in 50 normal controls. Given the simultaneous presence of the p.Ile79Val mutation in AKR1C2 and the AKR1C4 splice mutation in all the cases, these two mutations are expected to segregate together (Figure 3C).
Clinical Genetics in Family 2
Family 2 was studied after the AKR1C mutations had been identified in family 1 so that investigation quickly focused on the AKR1C locus in family 2. This patient carried a complex rearrangement that was apparently due to an unequal crossover between AKR1C2 and the neighboring AKR1C1, which share about 90% sequence identity. Long-range PCR of AKR1C1 and AKR1C2 followed by EcoRI digestion of the 18 kb products distinguished these two genes (Figure 4A). The normal array of EcoRI products is seen in the two parents, but the patient has bands typical of AKR1C1 and AKR1C2 (Figure 4B), suggesting a recombination between the two genes in which a hybrid AKR1C1/AKR1C2 containing the first three coding exons of AKR1C1 was fused to the 3′-terminal exons of AKR1C2. This was confirmed by reamplification and sequencing of the AKR1C2-specific 18 kb band with AKR1C1-specific internal reverse primers (Figure 4C). Thus the patient has a single AKR1C1/AKR1C2 hybrid on allele 1 and paternal AKR1C1, AKR1C1/AKR1C2, and maternal AKR1C2 genes on allele 2; this cluster spans >85 kb (Figure 4D). The intact maternal AKR1C2 on allele 2 carries a missense mutation in exon 9 (the sixth coding exon) (c.666T>G), and the paternal copy of AKR1C1 carries no mutation (Figures 4B and 4D). On allele 1, no mutation was found in the exons of either the AKR1C1 or AKR1C2. AKR1C4 carried no mutations in this patient (not shown). A diagram of the putative unequal crossover leading to the rearrangement found in the patient is shown in Figure 4E.
Figure 4.
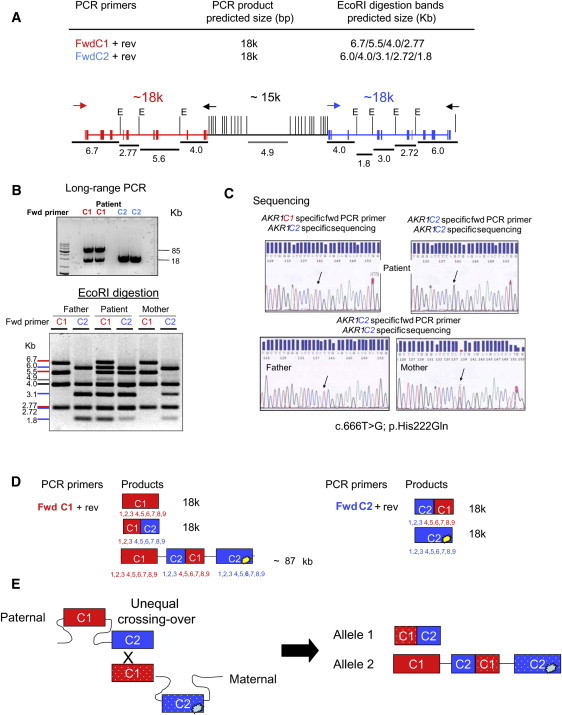
Identification of AKR1C2 Mutations in Family 2
(A) A schematic representation of the genomic organization of AKR1C1 (red), the intervening sequence (black), and AKR1C2 (blue) on chromosome 10. The vertical lines represent EcoRI (E) recognition sites. The expected EcoRI fragments and their relative locations are represented by black lines. Primers are indicated by arrows: red for AKR1C1-specific, blue for AKR1C2-specific, and black for nonspecific.
(B) Upper Panel: long-range PCR. The PCR products using the AKR1C1 primers yield an unexpected band of 85 kb in addition to the expected band of 18 kb, as also seen with the AKR1C2 primers. The 85 kb product suggests a complex recombination between AKR1C1 and AKR1C2 in the patient. Lower Panel: EcoRI digestion of the long-range PCR. The colored markers to the left indicate the bands derived from AKR1C1 (red), AKR1C2 (blue), or both (black). The 4.9 bp band indicated by the gray line likely derives from the AKR1C1/AKR1C2 junction. Because the PCR used an end-point amplification method, the intensity of the bands does not allow any quantitative (copy-number) conclusion.
(C) Sequencing demonstrating the presence of the missense mutation c.666T>G in the patient's AKR1C2 gene inherited from the mother.
(D) Putative scheme of long-range PCR and EcoRI digestion of PCR products from genomic DNA with either AKR1C1-specific (red) or AKR1C2-specific (blue) primers as derived by the results in (B).
(E) A diagram of the putative unequal crossover leading to the rearrangement found in the patient. The p.His222Gln mutation is indicated by the bright yellow lesion in the downstream portion of AKR1C2.
AKR1C2 Modeling Studies
The AKR family of enzymes have very similar amino acid sequences, and most catalytically important residues are conserved (Figure 5A and Figure S2). AKR1C2 acts on structurally diverse steroids, such as androgens and progestins, and can bind numerous nonsteroidal molecules through an induced-fit mechanism.30 To investigate its functional plasticity, we built a structural model of AKR1C2 based on the crystallographic structure of human AKR1C244–48 and docked 3αDiol into the structure (Figure 5B). The substrate-binding pocket of AKR1C2 consists mainly of loops and is therefore quite flexible, allowing the binding of different substrates. Two of the amino acids, Ile79 and Asn300, that were mutated in our patients are at the beginning of loops that make up the steroid-binding pocket (Figure 5B); amino acid changes at these locations might disturb the Lys84, Trp86, and Leu308 residues further down the loops that interact with the substrates at the catalytic site. The His90 residue that was found to be mutated is in close proximity (5.2Å) to His117 and in contact with Ile116 and Phe118, which are part of catalytic center. In our analysis, the His117-NADPH contact was broken in the His90Gln variant, potentially from the additional interactions of Gln90 with side chains of neighboring residues. His117 is essential for the activity in all AKR family members (Figure S2); it interacts with a central acidic ion to act as a bridge between steroid substrate and NADPH. A change in interaction between NADPH and His117 will likely affect cofactor binding and electron transfer.
Figure 5.
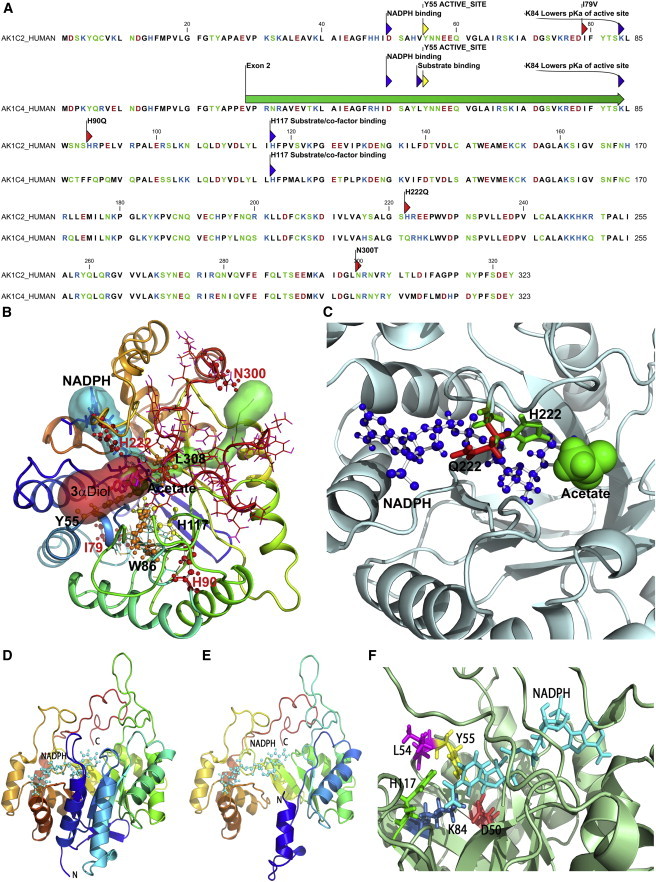
Structural Biology of AKR1C2 and AKR1C4
(A) Amino acid sequences of human AKR1C2 and AKR1C4. Mutations causing amino acid changes (p.Ile79Val, p.His90Gln, and p.Asn300Thr) in AKR1C2 are marked with red arrows and positions of active site residues are marked in gray (Asp50, Tyr55, Lys84, and His117). For AKR1C4, the major active site residues are marked and the position of amino acids encoded by exon 2 is shown with a green bar. Amino acids are colored based on charge; acidic residues (Asp, Glu) are in red, basic residues (His, Arg, Lys) are in blue and the rest of the polar amino acids (Asn, Cys, Ser, Thr, Tyr) are in green.
(B) A model of AKR1C2 with docking of 3αDiol. An acetate molecule between 3αDiol and NADPH facilitates electron transfer. Tunnels or binding pockets for NADPH (blue), 3αDiol (red) and acetate (green) are shown as transparent surface models. The amino acid mutations found in the patients are depicted as ball-and-stick models (red); the docked 3αDiol (magenta) and NADPH (blue) are shown as stick models and the acetate molecule is shown as a sphere model (green). The substrate-binding pocket of AKR1C consists of mainly flexible loops. Changing amino acids Ile79 and Asn300, which are at the beginning of the loops that comprise the steroid-binding pocket, might disturb the orientation of Trp86 and Leu308 (shown as orange ball-and-stick models) that interact with a steroid at the catalytic site. Residue His222 is involved in interaction with the steroid substrates, and the cofactors and its alteration to Gln222 leads to the side-chain of Gln222 blocking the NADPH-binding site. The catalytically important Tyr55 and His117 are shown as yellow ball-and-stick models. The AKR1C2 structure is shown as a ribbon model with rainbow colors starting from violet at the N terminus and ending with red at the C terminus.
(C) A detailed view of the side chains of His222 and Gln222. The His222Gln mutation in the AKR1C2 structure was produced in silico and the mutated structure was aligned with the WT structure. The His222Gln mutation causes flipping of the side chain in Gln222 that blocks the path of the NADPH-binding tunnel. The His222 (green) and Gln222 (red) residues are shown as stick models, whereas NADPH (blue) is shown as a ball-and-stick model and the central acetate ion found in the AKR1C2 structure is shown as a sphere model (green).
(D) The crystal structure of AKR1C4 (PDB number 2fvl chain c) shown as a ribbons model. The crystal structure is quite rigid and access to the substrate-binding site is blocked. However, most of the substrate and/or cofactor-binding pocket is made of flexible loops that showed variable positions during MD simulations.
(E) A model of the product of AKR1C4 lacking exon 2. The model was based on the crystal structure of AKR1C4 chain c. In the model lacking exon 2, most of the core structure is still intact, but the first two N-terminal helices and the loop joining the helices are missing and have been replaced by a single helix at the start of the N terminus.
(F) A close-up of the active site of AKR1C4. The catalytically important residues are shown as stick models. Three of the four residues of the catalytic tetrad (Asp50, Tyr55, and Leu84) plus the active site residue Leu54 that is involved in substrate orientation are missing in AKR1C4 lacking exon 2. Catalytic center components are shown as sticks with active site Tyr55 in yellow, Asp50 in red, Leu54 in magenta, Lys84 in blue, His117 in green, and NADPH is in cyan.
The p.His222Gln mutation elicited intriguing structural changes in the AKR1C2 structure. His222 is situated in the middle of a loop that appears to be involved in binding both the steroid substrate and the cofactors. Earlier analysis comparing the rat and human AKR1C2 structures has shown that change of His222 to Ser222 in the AKR1C2 causes structural changes by pushing the side-chain of neighboring Trp227, resulting in structural constraints on steroid binding at the catalytic center.47 We found that the substitution of His for Gln, as found in our patient, does not shift the side-chain of Trp227, so that the steroid-binding pocket remains unchanged. However, analysis with Caver program indicates that flipping of the side-chain of Gln222 pushed it into the NADPH-binding tunnel, potentially blocking NADPH entry (Figures 5B and 5C and Figure S2). In human AKR1C3 and AKR1C4, His222 is naturally replaced Gln222 (Figure S2), but unlike in AKR1C2, this change is accommodated in both of these enzymes by other amino acid replacements (as compared to AKR1C2) that could alter the cofactor-binding patterns (Figure S2). A major factor in AKR1C2 reactions is the release of cofactors after catalysis; minor changes in charge caused by p.Asn300Thr could influence how a particular steroid or cofactor could exit the catalytic site. An enzyme with slower release of product or cofactor will impair binding of the next molecule of substrate or the cofactors.
Effect of Alternate Splicing on Structure of AKR1C4
The amino acid sequence of AKR1C4 is very similar to AKR1C2, and most of the catalytically important residues (Asp50, Tyr55, Lys84, and His117) are found in similar positions in both enzymes (Figure 5A). Binding and release of both NADPH and steroid substrates require significant flexibility in the AKR1C4 structure (Figure 5D). Analysis of MD simulation revealed movements of short flexible fragments that comprise amino acids 9–14 and 50–59, and open and close access to the catalytic center. To study the effect of deleting exon 2, we made a structural model of AKR1C4 lacking the 56 amino acids (Val29-Lys84) encoded by exon 2. Deletion of exon 2 results in a stable structure that should bind NADPH (Figure 5E), but several catalytically essential residues are missing, which would render the truncated mutant inactive. In both AKR1C2 and AKR1C4 an active site tyrosine (Tyr55) acts as catalytic center that is missing in the alternatively spliced variant of AKR1C4 (Figure 5E). In addition, three other amino acids that are part of the catalytic center, Asp50, Leu54, and Lys84 are missing from the truncated AKR1C4 mutant. In AKR1C4, Leu54 is potentially involved in determining substrate specificity, and Asp50 and Lys84 together balance the pKa of the active site, which is necessary for transferring electrons from NADPH to the steroid substrate (Figure 5F). The missing residues in the AKR1C4 catalytic center would prohibit steroid substrates from binding in the correct orientation for the enzymatic reaction, rendering the exon 2 deficient variant of AKR1C4 catalytically inactive or grossly impaired compared to the WT enzyme.
Enzymology of the Identified AKR1C2 Mutants
WT and mutant AKR1C2 proteins were expressed in bacteria, purified to apparent homogeneity, and their activities were characterized in vitro with substrates that are part of the alternative pathway but not the classic pathway (Figure 6). AKR1C enzymes catalyze both oxidative and reductive reactions, depending on cofactor availability; oxidative reactions require NAD+, whereas reductive reactions require NADPH (Figure 6A).49 In the alternative pathway, AKR1C2 catalyzes the reduction of 5α-DHP (for systematic names, see the legend to Figure 1) to allopregnanolone (allo) and the reduction of 17-hydroxy-dihydroprogesterone (17OH-DHP) to 17OH-allo; AKR1C2 might also hypothetically catalyze the oxidation of 3α-androstanediol (3αDiol) to dihydrotestosterone (DHT). However, the AKR1C enzymes have very high affinity for NADP(H) (Kd = 100–200 nM) and low affinity for NAD(H) (Kd = 200 μM), making NADP(H) the preferred cofactor in vivo, so that AKR1C2 should function primarily as a reductase. When transfected into COS-1 cells and forced to use the prevailing concentration of the cofactor, AKR1C2 functions only as a 3-ketosteroidreductase,40 and when 1 mM NAD+ is supplied to recombinant AKR1C2 in vitro so that it permits oxidation of 3αDiol to DHT, the reaction can be blocked by only 10 μM NADPH.40,50 Thus AKR1C2 is expected to function almost exclusively as a reductive 3α-HSD. Consistent with this cofactor requirement, other members of the AKR1C family do not oxidize 3αDiol effectively,42 although in vitro AKR1C4 can oxidize 3αDiol to DHT.43,51 The AKR1C2 mutants Ile79Val, His90Gln and Asn300Thr had reduced activities in our functional assays. In the presence of 1 mM NAD+, the capacity (Vmax/Km) of AKR1C2 to oxidize 3αDiol to DHT was reduced to 28%–49% of WT control for the four mutants; in the presence of 1 μM NADPH, the capacity of AKR1C2 to reduce 5α-DHP to allo was reduced to 22%–82% of WT control (Figure 6 and Table 1).
Figure 6.
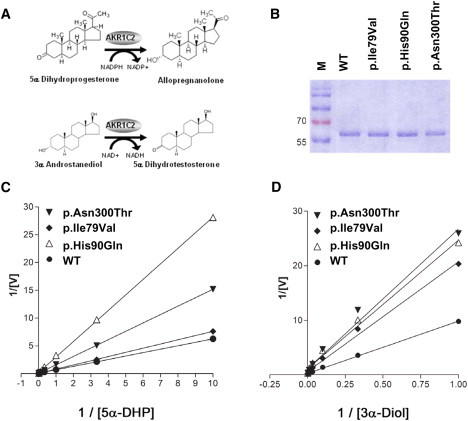
Assessment of AKR1C2 Activities
(A) Enzymatic reactions catalyzed by AKR1C2 that were assayed in vitro. AKR1C2 catalyzes the reduction of 5α-DHP (5α-pregnane-3,20-dione) to allopregnanolone (5α-pregnane-3α-ol-20-one) in the alternative pathway. AKR1C2 also supports the oxidation of 3αDiol to dihydrotestosterone in vitro.
(B) SDS-PAGE of the bacterially expressed, purified proteins used for enzymatic assays.
(C and D) Lineweaver-Burk plots of activities of WT and mutant AKR1C2 proteins. (C) Reduction of different concentrations of radiolabeled 5α-DHP to allopregnanolone in the presence of 1 μM NADPH. (D) Oxidation of androstanediol to DHT in the presence of 1 mM NAD+.
Table 1.
Activities of WT and Mutant AKR1C2
| Enzyme | Substrate | Km | Vmax | Intrinsic clearance Vmax/Km (% of WT) | Reaction Type |
|---|---|---|---|---|---|
| WT | 3αDiol | 38.17 ± 0.49 | 3.89 ± 0.8 | 0.104 (100) | oxidation |
| p.Ile79Val | 3αDiol | 32.52 ± 1.10 | 1.62 ± 0.71 | 0.050 (48) | oxidation |
| p.His90Gln | 3αDiol | 20.56 ± 0.83 | 0.87 ± 0.29 | 0.042 (41) | oxidation |
| p.Asn300Thr | 3αDiol | 22.23 ± 1.24 | 0.87 ± 0.43 | 0.039 (28) | oxidation |
| WT | 5α-DHP | 11.62 ± 0.06 | 18.7 ± 4.2 | 1.61 (100) | reduction |
| p.Ile79Val | 5α-DHP | 15.31 ± 0.08 | 20.3 ± 7.9 | 1.32 (82) | reduction |
| p.His90Gln | 5α-DHP | 15.17 ± 0.11 | 5.4 ± 2.5 | 0.36 (22) | reduction |
| p.Asn300Thr | 5α-DHP | 24.22 ± 0.09 | 16.0 ± 6.0 | 0.66 (41) | reduction |
Km is measured in μM, and Vmax is measured in nmol/min/mg protein. The following abbreviations are used: 3αDiol, androstanediol; 5α-DHP, 5α-pregnane-3,20-dione (5α-dihydroprogesterone).
GC/MS assays of steroids produced by cells expressing WT and the His222Gln mutant showed that WT readily converted 100 μM 3αDiol to DHT, but cells expressing the mutant His222Gln had dramatically reduced ability to produce DHT (Figure 7A). Immunoblotting suggested that there were no differences in the quantity or stability of the WT and truncated proteins (Figure 7B, inset). In microsomes from COS1 cells, the mutant had no detectable activity, whereas the WT had a Michaelis constant (Km) of 5.4 μM and a maximum velocity (Vmax) of 350 pg/min/mg protein (Figure 7C). Thus, the identified AKR1C2 mutations partially impair the 3α-HSD activity of AKR1C2 but not to the degree typically associated with recessive disorders of steroidogenesis.
Figure 7.
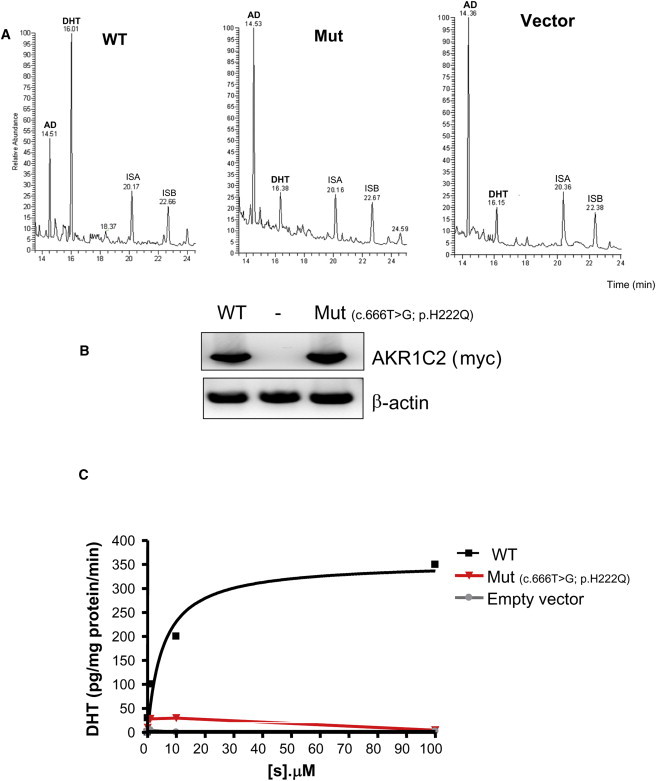
Assessment of AKR1C2 Mutant Enzymatic Activity in Family 2
(A) Steroid identification by gas chromatography. Extracts from COS1 cells transiently transfected with WT or mutant AKR1C2 cDNAs were challenged with 100 μM 3αDiol for 6 hr, and the resulting steroids in the culture medium were identified by mass spectrometry. The following abbreviations are used: AN, 3αDiol; DHT, dihydrotestosterone; internal standard A (ISA) = pregnanediol (PD); Internal standard B (ISB) = tetrahydro-11-deoxycortisol (THS).
(B) Immunoblotting of WT and p.His222Gln AKR1C2.
(C) Catalytic activity of the AKR1C2 p.His222Gln mutant. Quantification of DHT in microsomes from COS1 cells transiently transfected with WT or mutant AKR1C2 cDNAs and exposed to different concentrations of 3αDiol as in (A). The calculated Vmax and Km are depicted (mean ± standard deviation [SD] data from three independent experiments).
Enzyme Activity of the Identified Aberrantly Spliced AKR1C4 In Vivo
The activity of AKR1C4 to convert 5α-DHP to allo was determined in transiently transfected COS1 cells. As assayed by GC/MS, cells expressing WT AKR1C4 readily converted 100 μM 5α-DHP to allo (Figure 8A), but cells expressing the truncated mutant had only about 10% of WT activity (Figures 8B and 8C). Immunoblotting suggested that there were no differences in the quantity or stability of the WT and truncated proteins, although the mutant protein was shorter, as expected (Figure 8C, inset). Conversion of 5α-DHP to allo was also measured in microsomes from COS1 cells expressing the WT or mutant AKR1C4; this permitted calculation of the apparent Michaelis constant and maximum velocity and showed the mutant was essentially devoid of activity (Figure 8D). Addition of NADPH (but not NAD) increased DHP reduction by the WT AKR1C4 in a dose-dependent manner, but addition of NADPH or NAD had no effect on the truncated mutant (Figure 8E). We found no detectable oxidation of allopregnanolone to 5α-DHP by WT AKR1C4 (not shown).
Figure 8.
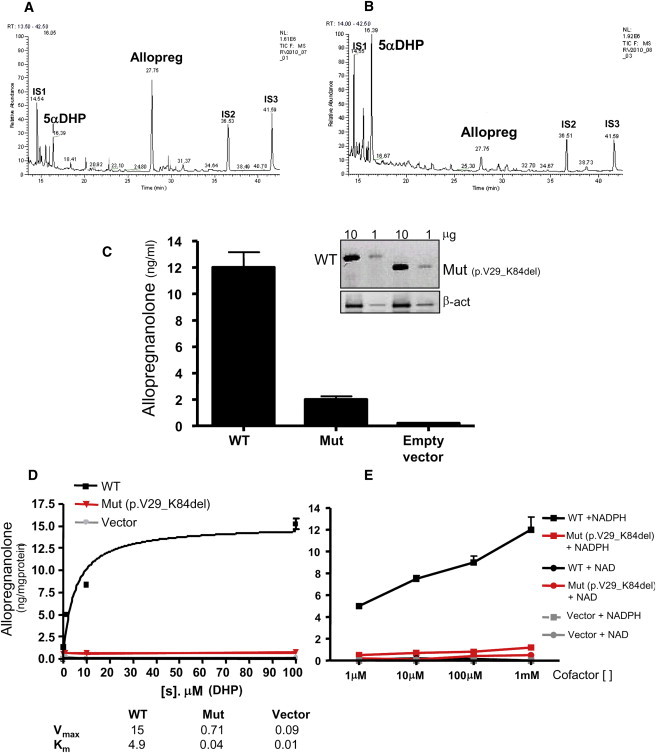
Assessment of AKR1C4 Reductive Activity
(A and B) Steroid identification by gas chromatography. Extracts from COS1 cells transiently transfected with WT (A) or mutant (B) AKR1C4 cDNAs were challenged with 100 μM 5α-DHP for 6 hr. Individual steroids were identified by mass spectrometry; internal standard (IS): 1 = androstanediol; 2 = stigmasterol; 3 = cholesterol butyrate. A representative experiment is shown.
(C) Quantification of the product allopregnanolone in intact COS1 cells transfected with normal (WT), mutant (aberrantly spliced, Mut), and empty vectors exposed to 100 μM 5α-DHP. Inset: Immunoblotting showing no differences in the amount or stability of WT and Mut AKR1C4 loaded in two different amounts (one representative experiment out of three is shown). The mutant protein, lacking 56 AA, is shorter than the WT. β-act is an abbreviation for β-actin. The data are from three independent experiments (mean ± SD).
(D) Quantification of allopregnanolone in microsomes of COS1 cells treated as in (C) and exposed to different concentrations of 5α-DHP. The calculated Vmax and Km are depicted (mean ± SD data from three independent experiments).
(E) Allopregnanolone formation by microsomes from COS1 cells transfected with vectors for either WT (black) or mutant (red) AKR1C4 or empty vector (gray). The microsomes were incubated with 100 μM 5α-DHP and exposed to increasing amounts of NADPH (reductive) or NAD (oxidative) cofactors. Data are mean ± SD of three independent experiments.
Tissue Distribution
To characterize the role of AKR1C2 in fetal DHT production, we performed quantitative RT-PCR expression profiling of AKR1C and SRD5A in normal fetal and adult testes and normal fetal and adult adrenal tissues (Figure 9). AKR1C2 cDNA could be readily amplified from fetal but not adult testes (Figure 9A), whereas SRD5A2 was expressed at lower levels in fetal than in adult testes (Figure 9B). AKR1C4 is also expressed in fetal (and adult) testes although at lower levels. RoDH was abundantly expressed in the control human liver HepG2 cells, and low but detectable amounts of RoDH were detected in the fetal testes (Figure 9C). Thus the role of RoDH as a potential oxidative enzyme converting 3α-HSD to DHT in the testicular alternative pathway to DHT is probable but not proven. Adult adrenals expressed markedly more AKR1C2 than fetal adrenals (Figure 9D), so that the alternative pathway to DHT production might not be a significant pathway in the fetal adrenal but might be important in the adult adrenal, especially in situations where 17OHP concentrations are high, such as in congenital adrenal hyperplasia from 21-hydroxylase deficiency. Taken together, these data support a role of AKR1C2 and AKR1C4 in DHT production in the fetal testis.
Figure 9.
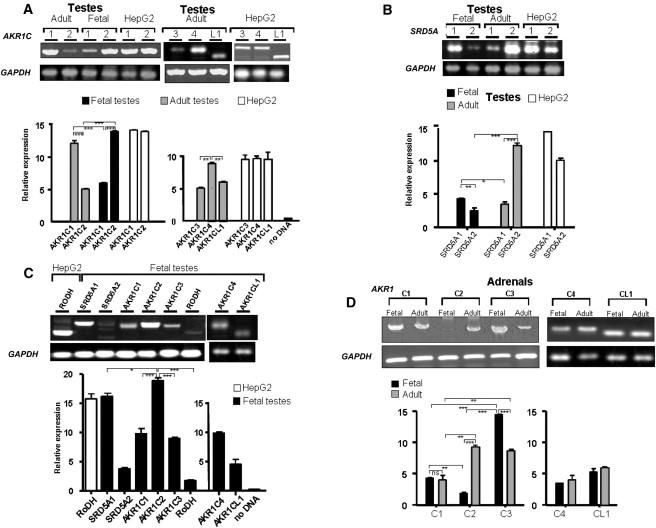
Quantitative RT-PCR Assessment of AKR1C1–4 and AKR1CL1, SRD5A1–2, and RoDH Expression in Normal Fetal and Adult Testes and Adrenals
(A) Expression of AKR1C1 and C2 in normal fetal versus adult testes and in human liver HepG2 cells (the positive control). AKR1C2 is expressed at high levels in fetal but very low levels in adult testes. In adult testes, AKR1C1 seems to be the predominant AKR1C, followed by AKR1C4, whereas C2, C3, and CL1 are expressed at similar lower levels.
(B) SRD5A2 is expressed at low levels in fetal testes but is highly expressed in adult testes. SRD5A1 expression is lower in adult than fetal testes.
(C) Compared to AKR1C2, RoDH is expressed at very low levels in the fetal testes, even lower than SRD5A2. The other four AKR1Cs (1, 3, and 4 and AKR1CL1) are expressed at similar levels but are significantly lower than AKR1C2.
(D) Normal fetal adrenals express very little AKR1C2; low levels of AKR1C1, AKR1C3, and AKR1CL1; and abundant AKR1C4. By contrast, the adult adrenals express all AKR1Cs; AKR1C1 at a lower level; and AKR1C2, AKR1C3, AKR1C4, and AKR1CL1 at similar levels. ∗∗∗p < 0.0005, ∗∗p < 0.005, ∗p < 0.05. ns is an abbreviation for not significant.
Discussion
AKR1C1–4 on chromosomal region 10p14-p15 encode four enzymes belonging to a family of 15 aldo-keto reductases. These four AKR1Cs share high sequence similarity and act as ketosteroid reductases and hydroxysteroid oxidases.18,52–54 Whereas all four AKR1Cs act as 3-, 17-, and 20-ketosteroid reductases, only AKR1C2 and AKR1C4 can oxidize 3αDiol to DHT in vitro,55 and AKR1C2 is more efficient.51 AKR1C2 uses NAD+ as its cofactor for oxidative reactions and NADPH for reductive reactions. AKR1C2 can bind steroids in multiple orientations, providing exceptional flexibility for multiple substrates and types of reactions catalyzed. The relative abundances of the cofactors available in different tissues thus governs the direction of AKR1C2 catalysis. In most cells, NAD and NADH are ∼10-fold more abundant than NADP and up to 1000-fold higher than NADPH,56,57 and this makes AKR1C2 primarily a reductive enzyme. Low levels of NADP and NADPH should favor oxidative reactions of AKR1C2 and conversion of 3αDiol to DHT. Of the AKR1C enzymes, AKR1C4 is catalytically the most efficient, is most abundant in the adult liver, and is thought to work in concert with the 5α/5β reductases to protect the liver from excess steroid hormones.51 AKR1C4 is also expressed in rat testis and adrenals.58
In a family having several members with 46,XY DSD, we found linkage to the region of chromosome 10 containing the five AKR1Cs and found mutations in the coding region of AKR1C2 and a splicing mutation in AKR1C4. Inheritance was sex-limited recessive, as 46,XY individuals had DSD but the genetically affected 46,XX individuals were phenotypically normal and fertile. These observations confirm that androgens, especially DHT, are not necessary for female sexual development; these findings are similar to those in women with SRD5A2 mutations. Two observations were puzzling. First, the AKR1C2 mutations caused a milder reduction in activity than that usually found in recessive disorders of steroidogenesis. Second, heterozygous 46,XY individuals had divergent phenotypes: individual II.1 was normal and fertile, whereas III.4 had DSD. This suggested the presence of a second hit. After excluding coding mutations in the other linked AKR1Cs on chromosome 10, we sought aberrant splicing, which could account for up to 50% of all functional mutations.59 AKR1C4 was aberrantly spliced in the affected individuals, leading to loss of key amino acids in the translated protein that reduced enzyme activity as assessed in expression studies. The presence of loss-of-function mutations in two closely linked genes indicates that mutations in both loci were needed to cause disease in family 1. Similar examples of such digenic inheritance are known, including holoprosencephaly (MIM 236100)60 and cortisone reductase deficiency (MIM 604931), a triallelic, digenic defect of steroidogenesis.61 Thus family 1 appears to exhibit synergistic heterozygosity62 in which mutations in more than one gene in a pathway give rise to a phenotype, whereas a defect in only one gene is insufficient to cause disease.60 Thus the effect of these mutations is cumulative, as the phenotype is more severe with an increasing number of mutations (see individual III.2 versus individual III.4).
Although it appears that a phenotype occurs in family 1 only if mutations in both AKR1C2 and AKR1C4 are present, all mutations identified in this family retained partial activity, so that the relative importance of AKR1C2 and AKR1C4 was uncertain. The presence of AKR1C2 mutations without AKR1C4 mutations in the patient with 46,XY DSD from family 2, and the absence of AKR1C4 mutations in 33 other patients with 46,XY DSD (M.M-B and A.B-L, unpublished data) indicates that AKR1C2 mutation is sufficient for disease manifestation and suggests that AKR1C2 could serve a more important role than AKR1C4 for this form of DSD.
Genetic 46,XY males lacking AR activity have female external genitalia, establishing the essential role of androgen action in male genital development.63 Two androgens are involved, testosterone and its more potent derivative, DHT, acting through a single AR.64 Testosterone is produced by fetal and adult testicular Leydig cells and is converted to DHT in target tissues, such as fetal genital skin, by SRD5A2.2,65 The complete absence of SRD5A2 activity does not yield the female phenotype seen when AR is absent but instead leads to incompletely developed male genitalia;66 this partial virilization has been attributed to the action of testicular testosterone because DHT synthesis was thought to be confined to extratesticular sites.67 Our finding that AKR1C2 and AKR1C4 are expressed in fetal testes but are barely detectable in adult testes is consistent with a crucial role for these AKR1C enzymes in fetal male development, despite the miniscule quantities of DHT found in adult spermatic vein blood.68 It appears that both AKR1C2 and AKR1C4 function as reductive 3α-HSD enzymes, and HSD17B6 functions as the oxidative enzyme; however, further work is needed to delineate the precise roles of these enzymes in the human fetus. Assuming that wallaby, mouse, and human pathways are comparable, 5α-DHP, which needs to be reduced to allopregnanolone for the synthesis of 3αDiol, is the key intermediate in the alternative/backdoor pathway. Thus, both AKR1C4 and AKR1C2 appear to be needed for synthesis of DHT in the backdoor pathway of the fetal testis. The roles of the relative abundances of oxidative NAD and reductive NADPH in the fetal testis remain unclear. The regulation of NAD kinase, which converts NAD to NADP,69 might control this ratio.
Thus fetal deficiencies of AKR1C2 and AKR1C4 cause a phenotype similar to that associated with deficiencies of SRD5A2 or P450c17 (17,20 lyase activity), indicating that the fetal testis secretes DHT synthesized via the alternative pathway without the intermediacy of testosterone. The presence of the alternative pathway in the marsupial,6,70 rodent,71 and human fetal testes indicates that the use of two pathways of androgen biosynthesis is a general mammalian feature. Because defects in both the classical and alternative pathways of DHT synthesis cause similar phenotypes, it now appears that both pathways are essential to normal human male genital development.
Acknowledgments
This work was supported by the grants from the Swiss National Science Foundation (31003A-130710 to C.E.F. and 320000-116636 to A.B.L.). We thank Rock Breton for providing the human WT AKR1C2 expression vector, Synthia H Mellon for the generous gift of radiochemicals and George Thalmann for providing normal testes and adrenal tissues. We thank Gaby Hofer for excellent technical assistance.
Contributor Information
Christa E. Flück, Email: christa.flueck@dkf.unibe.ch.
Anna Biason-Lauber, Email: anna.lauber@kispi.uzh.ch.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genome, http://www.1000genomes.org
Aldo-keto Reductase Superfamily Homepage, http://www.med.upenn.edu/akr
Human BLAT Search (BLAT), http://www.genome.ucsc.edu/cgi-bin/hgBlat
National Center for Biotechnology Information (NCBI), http://www.ncbi.nlm.nih.gov/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
POVRAY, http://www.povray.org
Protein Data Bank (PDB), http://www.rcsb.org/
Pymol, http://www.pymol.org
UCSC human genome browser, http://genome.cse.ucsc.edu/cgi-bin/hgGateway
Uniprot, http://www.uniprot.org
References
- 1.Miller W.L., Auchus R.J. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 2011;32:81–151. doi: 10.1210/er.2010-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wilson J.D., Griffin J.E., Russell D.W. Steroid 5 α-reductase 2 deficiency. Endocr. Rev. 1993;14:577–593. doi: 10.1210/edrv-14-5-577. [DOI] [PubMed] [Google Scholar]
- 3.Mendonca B.B., Domenice S., Arnhold I.J., Costa E.M. 46,XY disorders of sex development (DSD) Clin. Endocrinol. (Oxf.) 2009;70:173–187. doi: 10.1111/j.1365-2265.2008.03392.x. [DOI] [PubMed] [Google Scholar]
- 4.Auchus R.J. The backdoor pathway to dihydrotestosterone. Trends Endocrinol. Metab. 2004;15:432–438. doi: 10.1016/j.tem.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 5.Wilson J.D. The role of androgens in male gender role behavior. Endocr. Rev. 1999;20:726–737. doi: 10.1210/edrv.20.5.0377. [DOI] [PubMed] [Google Scholar]
- 6.Wilson J.D., Auchus R.J., Leihy M.W., Guryev O.L., Estabrook R.W., Osborn S.M., Shaw G., Renfree M.B. 5α-androstane-3α,17β-diol is formed in tammar wallaby pouch young testes by a pathway involving 5α-pregnane-3α,17α-diol-20-one as a key intermediate. Endocrinology. 2003;144:575–580. doi: 10.1210/en.2002-220721. [DOI] [PubMed] [Google Scholar]
- 7.Speiser P.W., Azziz R., Baskin L.S., Ghizzoni L., Hensle T.W., Merke D.P., Meyer-Bahlburg H.F., Miller W.L., Montori V.M., Oberfield S.E., Endocrine Society Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: An Endocrine Society clinical practice guideline. J. Clin. Endocrinol. Metab. 2010;95:4133–4160. doi: 10.1210/jc.2009-2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Auchus R.J., Lee T.C., Miller W.L. Cytochrome b5 augments the 17,20-lyase activity of human P450c17 without direct electron transfer. J. Biol. Chem. 1998;273:3158–3165. doi: 10.1074/jbc.273.6.3158. [DOI] [PubMed] [Google Scholar]
- 9.Ghayee H.K., Auchus R.J. Clinical implications of androgen synthesis via 5α-reduced precursors. Endocr. Dev. 2008;13:55–66. doi: 10.1159/000134780. [DOI] [PubMed] [Google Scholar]
- 10.Flück C.E., Tajima T., Pandey A.V., Arlt W., Okuhara K., Verge C.F., Jabs E.W., Mendonça B.B., Fujieda K., Miller W.L. Mutant P450 oxidoreductase causes disordered steroidogenesis with and without Antley-Bixler syndrome. Nat. Genet. 2004;36:228–230. doi: 10.1038/ng1300. [DOI] [PubMed] [Google Scholar]
- 11.Huang N., Agrawal V., Giacomini K.M., Miller W.L. Genetics of P450 oxidoreductase: Sequence variation in 842 individuals of four ethnicities and activities of 15 missense mutations. Proc. Natl. Acad. Sci. USA. 2008;105:1733–1738. doi: 10.1073/pnas.0711621105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arlt W., Walker E.A., Draper N., Ivison H.E., Ride J.P., Hammer F., Chalder S.M., Borucka-Mankiewicz M., Hauffa B.P., Malunowicz E.M. Congenital adrenal hyperplasia caused by mutant P450 oxidoreductase and human androgen synthesis: Analytical study. Lancet. 2004;363:2128–2135. doi: 10.1016/S0140-6736(04)16503-3. [DOI] [PubMed] [Google Scholar]
- 13.Fukami M., Nishimura G., Homma K., Nagai T., Hanaki K., Uematsu A., Ishii T., Numakura C., Sawada H., Nakacho M. Cytochrome P450 oxidoreductase deficiency: Identification and characterization of biallelic mutations and genotype-phenotype correlations in 35 Japanese patients. J. Clin. Endocrinol. Metab. 2009;94:1723–1731. doi: 10.1210/jc.2008-2816. [DOI] [PubMed] [Google Scholar]
- 14.Homma K., Hasegawa T., Nagai T., Adachi M., Horikawa R., Fujiwara I., Tajima T., Takeda R., Fukami M., Ogata T. Urine steroid hormone profile analysis in cytochrome P450 oxidoreductase deficiency: Implication for the backdoor pathway to dihydrotestosterone. J. Clin. Endocrinol. Metab. 2006;91:2643–2649. doi: 10.1210/jc.2005-2460. [DOI] [PubMed] [Google Scholar]
- 15.Shackleton C., Marcos J., Arlt W., Hauffa B.P. Prenatal diagnosis of P450 oxidoreductase deficiency (ORD): A disorder causing low pregnancy estriol, maternal and fetal virilization, and the Antley-Bixler syndrome phenotype. Am. J. Med. Genet. A. 2004;129A:105–112. doi: 10.1002/ajmg.a.30171. [DOI] [PubMed] [Google Scholar]
- 16.Fukami M., Horikawa R., Nagai T., Tanaka T., Naiki Y., Sato N., Okuyama T., Nakai H., Soneda S., Tachibana K. Cytochrome P450 oxidoreductase gene mutations and Antley-Bixler syndrome with abnormal genitalia and/or impaired steroidogenesis: Molecular and clinical studies in 10 patients. J. Clin. Endocrinol. Metab. 2005;90:414–426. doi: 10.1210/jc.2004-0810. [DOI] [PubMed] [Google Scholar]
- 17.Zachmann M., Völlmin J.A., Hamilton W., Prader A. Steroid 17,20-desmolase deficiency: A new cause of male pseudohermaphroditism. Clin. Endocrinol. (Oxf.) 1972;1:369–385. doi: 10.1111/j.1365-2265.1972.tb00407.x. [DOI] [PubMed] [Google Scholar]
- 18.Khanna M., Qin K.N., Klisak I., Belkin S., Sparkes R.S., Cheng K.C. Localization of multiple human dihydrodiol dehydrogenase (DDH1 and DDH2) and chlordecone reductase (CHDR) genes in chromosome 10 by the polymerase chain reaction and fluorescence in situ hybridization. Genomics. 1995;25:588–590. doi: 10.1016/0888-7543(95)80066-u. [DOI] [PubMed] [Google Scholar]
- 19.Královicová J., Vorechovsky I. Global control of aberrant splice-site activation by auxiliary splicing sequences: Evidence for a gradient in exon and intron definition. Nucleic Acids Res. 2007;35:6399–6413. doi: 10.1093/nar/gkm680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vollmer M., Jung M., Rüschendorf F., Ruf R., Wienker T., Reis A., Krapf R., Hildebrandt F. The gene for human fibronectin glomerulopathy maps to 1q32, in the region of the regulation of complement activation gene cluster. Am. J. Hum. Genet. 1998;63:1724–1731. doi: 10.1086/302162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kruglyak L., Daly M.J., Reeve-Daly M.P., Lander E.S. Parametric and nonparametric linkage analysis: A unified multipoint approach. Am. J. Hum. Genet. 1996;58:1347–1363. [PMC free article] [PubMed] [Google Scholar]
- 22.Krieger E., Darden T., Nabuurs S.B., Finkelstein A., Vriend G. Making optimal use of empirical energy functions: Force-field parameterization in crystal space. Proteins. 2004;57:678–683. doi: 10.1002/prot.20251. [DOI] [PubMed] [Google Scholar]
- 23.Krieger E., Joo K., Lee J., Lee J., Raman S., Thompson J., Tyka M., Baker D., Karplus K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins. 2009;77(Suppl 9):114–122. doi: 10.1002/prot.22570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krieger E., Nielsen J.E., Spronk C.A., Vriend G. Fast empirical pKa prediction by Ewald summation. J. Mol. Graph. Model. 2006;25:481–486. doi: 10.1016/j.jmgm.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 25.Vriend G. WHAT IF: A molecular modeling and drug design program. J. Mol. Graph. 1990;8:52–56, 29. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- 26.Case D.A., Cheatham T.E., 3rd, Darden T., Gohlke H., Luo R., Merz K.M., Jr., Onufriev A., Simmerling C., Wang B., Woods R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schneidman-Duhovny D., Inbar Y., Polak V., Shatsky M., Halperin I., Benyamini H., Barzilai A., Dror O., Haspel N., Nussinov R., Wolfson H.J. Taking geometry to its edge: Fast unbound rigid (and hinge-bent) docking. Proteins. 2003;52:107–112. doi: 10.1002/prot.10397. [DOI] [PubMed] [Google Scholar]
- 28.Damborský J., Petrek M., Banás P., Otyepka M. Identification of tunnels in proteins, nucleic acids, inorganic materials and molecular ensembles. Biotechnol. J. 2007;2:62–67. doi: 10.1002/biot.200600208. [DOI] [PubMed] [Google Scholar]
- 29.Pandey A.V., Kempná P., Hofer G., Mullis P.E., Flück C.E. Modulation of human CYP19A1 activity by mutant NADPH P450 oxidoreductase. Mol. Endocrinol. 2007;21:2579–2595. doi: 10.1210/me.2007-0245. [DOI] [PubMed] [Google Scholar]
- 30.Couture J.F., de Jésus-Tran K.P., Roy A.M., Cantin L., Côté P.L., Legrand P., Luu-The V., Labrie F., Breton R. Comparison of crystal structures of human type 3 3α-hydroxysteroid dehydrogenase reveals an “induced-fit” mechanism and a conserved basic motif involved in the binding of androgen. Protein Sci. 2005;14:1485–1497. doi: 10.1110/ps.051353205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang L.H., Rodriguez H., Ohno S., Miller W.L. Serine phosphorylation of human P450c17 increases 17,20-lyase activity: Implications for adrenarche and the polycystic ovary syndrome. Proc. Natl. Acad. Sci. USA. 1995;92:10619–10623. doi: 10.1073/pnas.92.23.10619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Biason-Lauber A., Konrad D., Meyer M., DeBeaufort C., Schoenle E.J. Ovaries and female phenotype in a girl with 46,XY karyotype and mutations in the CBX2 gene. Am. J. Hum. Genet. 2009;84:658–663. doi: 10.1016/j.ajhg.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakajin S., Hall P.F. Microsomal cytochrome P-450 from neonatal pig testis. Purification and properties of A C21 steroid side-chain cleavage system (17 α-hydroxylase-C17,20 lyase) J. Biol. Chem. 1981;256:3871–3876. [PubMed] [Google Scholar]
- 34.Chung B.C., Picado-Leonard J., Haniu M., Bienkowski M., Hall P.F., Shively J.E., Miller W.L. Cytochrome P450c17 (steroid 17 α-hydroxylase/17,20 lyase): Cloning of human adrenal and testis cDNAs indicates the same gene is expressed in both tissues. Proc. Natl. Acad. Sci. USA. 1987;84:407–411. doi: 10.1073/pnas.84.2.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Picado-Leonard J., Miller W.L. Cloning and sequence of the human gene for P450c17 (steroid 17 α-hydroxylase/17,20 lyase): Similarity with the gene for P450c21. DNA. 1987;6:439–448. doi: 10.1089/dna.1987.6.439. [DOI] [PubMed] [Google Scholar]
- 36.Geller D.H., Auchus R.J., Mendonça B.B., Miller W.L. The genetic and functional basis of isolated 17,20-lyase deficiency. Nat. Genet. 1997;17:201–205. doi: 10.1038/ng1097-201. [DOI] [PubMed] [Google Scholar]
- 37.Hershkovitz E., Parvari R., Wudy S.A., Hartmann M.F., Gomes L.G., Loewental N., Miller W.L. Homozygous mutation G539R in the gene for P450 oxidoreductase in a family previously diagnosed as having 17,20-lyase deficiency. J. Clin. Endocrinol. Metab. 2008;93:3584–3588. doi: 10.1210/jc.2008-0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kok R.C., Timmerman M.A., Wolffenbuttel K.P., Drop S.L., de Jong F.H. Isolated 17,20-lyase deficiency due to the cytochrome b5 mutation W27X. J. Clin. Endocrinol. Metab. 2010;95:994–999. doi: 10.1210/jc.2008-1745. [DOI] [PubMed] [Google Scholar]
- 39.Jin Y., Penning T.M. Multiple steps determine the overall rate of the reduction of 5alpha-dihydrotestosterone catalyzed by human type 3 3alpha-hydroxysteroid dehydrogenase: Implications for the elimination of androgens. Biochemistry. 2006;45:13054–13063. doi: 10.1021/bi060591r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rizner T.L., Lin H.K., Peehl D.M., Steckelbroeck S., Bauman D.R., Penning T.M. Human type 3 3α-hydroxysteroid dehydrogenase (aldo-keto reductase 1C2) and androgen metabolism in prostate cells. Endocrinology. 2003;144:2922–2932. doi: 10.1210/en.2002-0032. [DOI] [PubMed] [Google Scholar]
- 41.Biswas M.G., Russell D.W. Expression cloning and characterization of oxidative 17β- and 3α-hydroxysteroid dehydrogenases from rat and human prostate. J. Biol. Chem. 1997;272:15959–15966. doi: 10.1074/jbc.272.25.15959. [DOI] [PubMed] [Google Scholar]
- 42.Dufort I., Soucy P., Labrie F., Luu-The V. Molecular cloning of human type 3 3 α-hydroxysteroid dehydrogenase that differs from 20 α-hydroxysteroid dehydrogenase by seven amino acids. Biochem. Biophys. Res. Commun. 1996;228:474–479. doi: 10.1006/bbrc.1996.1684. [DOI] [PubMed] [Google Scholar]
- 43.Lin H.K., Jez J.M., Schlegel B.P., Peehl D.M., Pachter J.A., Penning T.M. Expression and characterization of recombinant type 2 3 α-hydroxysteroid dehydrogenase (HSD) from human prostate: Demonstration of bifunctional 3 α/17 β-HSD activity and cellular distribution. Mol. Endocrinol. 1997;11:1971–1984. doi: 10.1210/mend.11.13.0026. [DOI] [PubMed] [Google Scholar]
- 44.Hoog S.S., Pawlowski J.E., Alzari P.M., Penning T.M., Lewis M. Three-dimensional structure of rat liver 3 α-hydroxysteroid/dihydrodiol dehydrogenase: A member of the aldo-keto reductase superfamily. Proc. Natl. Acad. Sci. USA. 1994;91:2517–2521. doi: 10.1073/pnas.91.7.2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bennett M.J., Schlegel B.P., Jez J.M., Penning T.M., Lewis M. Structure of 3 α-hydroxysteroid/dihydrodiol dehydrogenase complexed with NADP+ Biochemistry. 1996;35:10702–10711. doi: 10.1021/bi9604688. [DOI] [PubMed] [Google Scholar]
- 46.Bennett M.J., Albert R.H., Jez J.M., Ma H., Penning T.M., Lewis M. Steroid recognition and regulation of hormone action: Crystal structure of testosterone and NADP+ bound to 3 α-hydroxysteroid/dihydrodiol dehydrogenase. Structure. 1997;5:799–812. doi: 10.1016/s0969-2126(97)00234-7. [DOI] [PubMed] [Google Scholar]
- 47.Nahoum V., Gangloff A., Legrand P., Zhu D.W., Cantin L., Zhorov B.S., Luu-The V., Labrie F., Breton R., Lin S.X. Structure of the human 3α-hydroxysteroid dehydrogenase type 3 in complex with testosterone and NADP at 1.25-A resolution. J. Biol. Chem. 2001;276:42091–42098. doi: 10.1074/jbc.M105610200. [DOI] [PubMed] [Google Scholar]
- 48.Jin Y., Stayrook S.E., Albert R.H., Palackal N.T., Penning T.M., Lewis M. Crystal structure of human type III 3α-hydroxysteroid dehydrogenase/bile acid binding protein complexed with NADP(+) and ursodeoxycholate. Biochemistry. 2001;40:10161–10168. doi: 10.1021/bi010919a. [DOI] [PubMed] [Google Scholar]
- 49.Penning T.M., Pawlowski J.E., Schlegel B.P., Jez J.M., Lin H.K., Hoog S.S., Bennett M.J., Lewis M. Mammalian 3 α-hydroxysteroid dehydrogenases. Steroids. 1996;61:508–523. doi: 10.1016/s0039-128x(96)00093-1. [DOI] [PubMed] [Google Scholar]
- 50.Steckelbroeck S., Jin Y., Gopishetty S., Oyesanmi B., Penning T.M. Human cytosolic 3α-hydroxysteroid dehydrogenases of the aldo-keto reductase superfamily display significant 3β-hydroxysteroid dehydrogenase activity: Implications for steroid hormone metabolism and action. J. Biol. Chem. 2004;279:10784–10795. doi: 10.1074/jbc.M313308200. [DOI] [PubMed] [Google Scholar]
- 51.Penning T.M., Burczynski M.E., Jez J.M., Hung C.F., Lin H.K., Ma H., Moore M., Palackal N., Ratnam K. Human 3α-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo-keto reductase superfamily: Functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochem. J. 2000;351:67–77. doi: 10.1042/0264-6021:3510067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Winters C.J., Molowa D.T., Guzelian P.S. Isolation and characterization of cloned cDNAs encoding human liver chlordecone reductase. Biochemistry. 1990;29:1080–1087. doi: 10.1021/bi00456a034. [DOI] [PubMed] [Google Scholar]
- 53.Qin K.N., New M.I., Cheng K.C. Molecular cloning of multiple cDNAs encoding human enzymes structurally related to 3 α-hydroxysteroid dehydrogenase. J. Steroid Biochem. Mol. Biol. 1993;46:673–679. doi: 10.1016/0960-0760(93)90308-j. [DOI] [PubMed] [Google Scholar]
- 54.Dufort I., Labrie F., Luu-The V. Human types 1 and 3 3 α-hydroxysteroid dehydrogenases: Differential lability and tissue distribution. J. Clin. Endocrinol. Metab. 2001;86:841–846. doi: 10.1210/jcem.86.2.7216. [DOI] [PubMed] [Google Scholar]
- 55.Penning T.M., Jin Y., Steckelbroeck S., Lanisnik Rizner T., Lewis M. Structure-function of human 3 α-hydroxysteroid dehydrogenases: Genes and proteins. Mol. Cell. Endocrinol. 2004;215:63–72. doi: 10.1016/j.mce.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 56.Glock G.E., McLean P. Levels of oxidized and reduced diphosphopyridine nucleotide and triphosphopyridine nucleotide in animal tissues. Biochem. J. 1955;61:388–390. doi: 10.1042/bj0610388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Friedrich W. Walter de Gruyter; New York: 1988. Niacin, nicotinic acid, nicotinamide, NAD(P) [Google Scholar]
- 58.Barbaccia M.L., Roscetti G., Trabucchi M., Purdy R.H., Mostallino M.C., Concas A., Biggio G. The effects of inhibitors of GABAergic transmission and stress on brain and plasma allopregnanolone concentrations. Br. J. Pharmacol. 1997;120:1582–1588. doi: 10.1038/sj.bjp.0701046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Desmet F.O., Hamroun D., Lalande M., Collod-Béroud G., Claustres M., Béroud C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:e67. doi: 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ming J.E., Muenke M. Multiple hits during early embryonic development: Digenic diseases and holoprosencephaly. Am. J. Hum. Genet. 2002;71:1017–1032. doi: 10.1086/344412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Draper N., Walker E.A., Bujalska I.J., Tomlinson J.W., Chalder S.M., Arlt W., Lavery G.G., Bedendo O., Ray D.W., Laing I. Mutations in the genes encoding 11beta-hydroxysteroid dehydrogenase type 1 and hexose-6-phosphate dehydrogenase interact to cause cortisone reductase deficiency. Nat. Genet. 2003;34:434–439. doi: 10.1038/ng1214. [DOI] [PubMed] [Google Scholar]
- 62.Vockley J., Rinaldo P., Bennett M.J., Matern D., Vladutiu G.D. Synergistic heterozygosity: Disease resulting from multiple partial defects in one or more metabolic pathways. Mol. Genet. Metab. 2000;71:10–18. doi: 10.1006/mgme.2000.3066. [DOI] [PubMed] [Google Scholar]
- 63.Quigley C.A., Friedman K.J., Johnson A., Lafreniere R.G., Silverman L.M., Lubahn D.B., Brown T.R., Wilson E.M., Willard H.F., French F.S. Complete deletion of the androgen receptor gene: Definition of the null phenotype of the androgen insensitivity syndrome and determination of carrier status. J. Clin. Endocrinol. Metab. 1992;74:927–933. doi: 10.1210/jcem.74.4.1347772. [DOI] [PubMed] [Google Scholar]
- 64.Grino P.B., Griffin J.E., Wilson J.D. Testosterone at high concentrations interacts with the human androgen receptor similarly to dihydrotestosterone. Endocrinology. 1990;126:1165–1172. doi: 10.1210/endo-126-2-1165. [DOI] [PubMed] [Google Scholar]
- 65.Russell D.W., Wilson J.D. Steroid 5 α-reductase: Two genes/two enzymes. Annu. Rev. Biochem. 1994;63:25–61. doi: 10.1146/annurev.bi.63.070194.000325. [DOI] [PubMed] [Google Scholar]
- 66.Imperato-McGinley J., Miller M., Wilson J.D., Peterson R.E., Shackleton C., Gajdusek D.C. A cluster of male pseudohermaphrodites with 5 α-reductase deficiency in Papua New Guinea. Clin. Endocrinol. (Oxf.) 1991;34:293–298. doi: 10.1111/j.1365-2265.1991.tb03769.x. [DOI] [PubMed] [Google Scholar]
- 67.Ito T., Horton R. The source of plasma dihydrotestosterone in man. J. Clin. Invest. 1971;50:1621–1627. doi: 10.1172/JCI106650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hammond G.L., Ruokonen A., Kontturi M., Koskela E., Vihko R. The simultaneous radioimmunoassay of seven steroids in human spermatic and peripheral venous blood. J. Clin. Endocrinol. Metab. 1977;45:16–24. doi: 10.1210/jcem-45-1-16. [DOI] [PubMed] [Google Scholar]
- 69.Pollak N., Niere M., Ziegler M. NAD kinase levels control the NADPH concentration in human cells. J. Biol. Chem. 2007;282:33562–33571. doi: 10.1074/jbc.M704442200. [DOI] [PubMed] [Google Scholar]
- 70.Wilson J.D., George F.W., Shaw G., Renfree M.B. Virilization of the male pouch young of the tammar wallaby does not appear to be mediated by plasma testosterone or dihydrotestosterone. Biol. Reprod. 1999;61:471–475. doi: 10.1095/biolreprod61.2.471. [DOI] [PubMed] [Google Scholar]
- 71.Mahendroo M., Wilson J.D., Richardson J.A., Auchus R.J. Steroid 5α-reductase 1 promotes 5α-androstane-3α,17β-diol synthesis in immature mouse testes by two pathways. Mol. Cell. Endocrinol. 2004;222:113–120. doi: 10.1016/j.mce.2004.04.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.