

Abstract
Holoprosencephaly (HPE), a common human congenital anomaly defined by a failure to delineate the midline of the forebrain and/or midface, is associated with diminished Sonic hedgehog (SHH)-pathway activity in development of these structures. SHH signaling is regulated by a network of ligand-binding factors, including the primary receptor PTCH1 and the putative coreceptors, CDON (also called CDO), BOC, and GAS1. Although binding of SHH to these receptors promotes pathway activity, it is not known whether interactions between these receptors are important. We report here identification of missense CDON mutations in human HPE. These mutations diminish CDON's ability to support SHH-dependent gene expression in cell-based signaling assays. The mutations occur outside the SHH-binding domain of CDON, and the encoded variant CDON proteins do not display defects in binding to SHH. In contrast, wild-type CDON associates with PTCH1 and GAS1, but the variants do so inefficiently, in a manner that parallels their activity in cell-based assays. Our findings argue that CDON must associate with both ligand and other hedgehog-receptor components, particularly PTCH1, for signaling to occur and that disruption of the latter interactions is a mechanism of HPE.
Introduction
Holoprosencephaly (HPE [MIM 236100]) comprises a clinical spectrum of related malformations of the brain and face that reflect gradations of disrupted developmental midline patterning.1 HPE occurs with a remarkably high frequency, in at least one in every 250 conceptions.2 Pathogenic causes include environmental factors, such as teratogens, and genetic factors, including clinically distinct syndromes, micro- and macro-cytogenetic alterations, and single gene mutations in at least nine genes.3 Single gene mutations are always heterozygous and, where tested, result in loss of function.3 The genetic heterogeneity of the HPE spectrum of disorders is considerable, as is the variable expressivity of pathogenic mutations, even within the same family. To a large extent, the molecular basis of this variability is unknown. However, the best understood mechanisms for HPE-like malformations are defects in various components of the Sonic hedgehog (SHH [MIM 600725])-signaling pathway.3,4
The binding of SHH to its primary receptor PTCH1 (MIM 601309) activates a signal transduction cascade that culminates in expression of pathway target genes via the GLI family of transcription factors.5,6 Among the direct target genes are PTCH1 and GLI1 (MIM 165220) themselves.5,6 Additional membrane-associated SHH-binding proteins exist, some of which appear to serve as coreceptors with PTCH1. For example, the related IgSF proteins CDON (MIM 608707) and BOC (MIM 608708) and the GPI-linked factor GAS1 (MIM 139185) each interact directly with SHH7–12 and individually associate with PTCH1.13 Although binding of SHH to these receptors promotes pathway activity, it is not known whether interactions between the receptors are important.
Cdon−/− mice display HPE with strain-specific severity and have phenotypes ranging from severe to mild depending on the genetic background.14,15 Although Boc−/− mice do not have HPE and Gas1−/− mice have mild HPE, Cdon−/−;Boc−/− and Cdon−/−;Gas1−/− double mutants display more severe forms of HPE than any single mutant, suggesting that these factors may cooperate to promote SHH-mediated patterning of the rostroventral midine.16,17 Because the mouse-strain-dependent variability in penetrance and expressivity of HPE phenotypes in Cdon−/− mice resembles the variability seen in human HPE, CDON is a strong candidate to be an HPE-associated factor. We report here the identification of missense CDON mutations in human HPE. The mutations result in diminished activity in SHH-signaling assays, consistent with a significant role for this coreceptor in HPE pathogenesis. Furthermore, we find that CDON associates with PTCH1 and GAS1, but the HPE-associated variants do so inefficiently in a manner that parallels their activity in cell-based assays. These results argue that CDON must associate with other hedgehog-receptor components for signaling to occur and that disruption of such interactions is a mechanism of HPE.
Material and Methods
Human DNA Samples and SNP Detection
We studied 282 unrelated individuals diagnosed with HPE spectrum disorders and 96 commercially available anonymous normal controls. Genomic DNA was extracted from peripheral blood or transformed lymphoblast cell lines by standard methods. All individuals with HPE were recruited into a National Human Genome Research Institute institutional-review-board-approved research protocol in accordance with their ethical guidelines and supervision. All probands and available parents were studied for relevant coding-region alterations and their inheritance in the four genes most commonly screened in HPE (SHH, ZIC2, SIX3, and TGIF) by bidirectional Big Dye version 3.1 terminator cycle sequencing on an ABI 3100 instrument (Applied Biosystems, Foster City, CA) as described.18
Mutation detection for CDON was performed by PCR-based denaturing high performance liquid chromatograhy (dHPLC) analysis followed by direct sequencing. PCR amplification, dHPLC analysis employing WAVE and WAVEMAKER (Transgenomic, Omaha, NE), amplicon purification with the QIAGEN PCR purification kit (QIAGEN, Valencia, CA), and DNA sequencing with the Big Dye version 3.1 terminator cycle sequencing on an ABI 3100 instrument were performed according to the manufacturers' instructions, essentially as previously described.19 We then compared our research findings to those described in publicly available databases (dbSNP and 1000 Genomes) as summarized in Table S1, available online.
For this study we used the reference sequence NM_016952.4 comprising 19 coding exons of human CDON. PCR was performed with primer pairs and conditions listed in Table S2. In brief, we performed amplification of genomic DNA in 35 μl reaction volumes by using 60–100 ng of genomic DNA, 200 μM dNTP, 20 pmol of each primer, 1× PCR buffer (Invitrogen, Carlsbad, CA), 0.5× enhancer (Invitrogen), 1.5 mM MgSO4 (Invitrogen), and 2.5 U of AmpliTaq (Applied Biosystems). All reactions were performed in a PTC-225 thermocycler (MJ Research, Waltham, MA). PCR-cycling parameters were (1) 95°C for 4 min; (2) 50 cycles of 95°C for 30 s, an annealing temperature (Table S2) for 30 s, and 72°C for 1 min; and (3) a final step of 72°C for 7 min. After amplification, PCR products were denatured at 96°C for 1 min, followed by gradual reannealing to 65°C over a period of 30 min to form homo- and/or heteroduplexes. Products were automatically loaded on a DNA sep column and eluted according to the manufacturer's instructions at a flow rate of 1.5 ml/min with a mixture of buffer A (0.1 mM TEAA) and buffer B (0.1 mM TEAA and 25% acetonitrile); the amount of buffer B was increased 10% per minute for 2.5 min. Samples were detected by an ultraviolet C system. We determined the oven temperature(s) for optimal heteroduplex separation under partial DNA denaturation for each amplified fragment by using the WAVEMaker software (version 4.1) and adjusted empirically. Because the dHPLC approach is based on the differential retention of homo- and heteroduplex DNA fragments by ion-pair chromatography under conditions of partial heat denaturation, optimal discrimination of double-stranded combinations depends on the temperature at which partial denaturation of heteroduplexes occur. Most amplicons were composed of different melting domains; thus more than one elution temperature was needed to detect the variants. The data profiles of all samples were analyzed by visual inspection, and those with clear or suggestive variant peaks were sequenced with the same primers as used for PCR. The dHPLC screening method detects at least 96% of the sequence variation of a test amplicon20 but is not as specific as Sanger sequencing for allele discrimination.
Cell-Culture and Expression Vectors
C2C12, 293T, Cos7, RD, and 10T1/2 cells were cultured as previously described.21,22 Mouse embryo fibroblast lines derived from Cdon−/− mice were derived and cultured with standard protocols.23 For SHH-signaling assays, cells were reverse transfected with plasmids diluted in Opti-MEM with Lipofectamine 2000 (Invitrogen) in individual wells of 6-well plates. Cells were added to 80%–90% confluence; transfection efficiency was 80%–90%. Plasmids were either previously published expression vectors encoding rat CDON,24 human BOC,25 human PTCH1,26 and mouse Cdon siRNA15 or a mouse GAS1 expression vector; the mouse Gas1 cDNA was cloned by RT-PCR. Vectors encoding rat CDON variants were constructed by PCR mutagenesis27 with pBabePuro-rCDON as a template and were verified by sequencing (primers used are described in Table S4). Vectors encoding CDON-Fc and CDON proteins with specific ectodomain deletions were described previously.25,28
Immunoblot Analysis and Immunoprecipitation
Immunoblot analyses were carried out as previously described.29 Briefly, cells were lysed in lysis buffer (10 mM Tris-HCl [pH 7.2], 150 mM NaCl, 1% Triton X-100, and 1 mM EDTA) containing Complete Protease Inhibitor Cocktail (Roche Diagnostics), followed by SDS-PAGE. Primary antibodies used in this study were anti-CDON, anti-BOC, anti-GAS1 (R&D Systems), anti-β-tubulin (Zymed), anti-flag (Sigma Aldrich), anti-human Fc region of IgG (Jackson Immunoresearch), and anti-PTCH1 (Santa Cruz). Coimmunoprecipitations were performed as described previously.29 Quantification of coimmunoprecipitations was performed with Image-Gauge software (Fuji Film, Tokyo, Japan). Results are expressed as mean ± standard deviation (SD) from at least three independent experiments and analyzed by Student's t test using SPSS (version 12.0; SPSS, Chicago, IL, USA).
RNA Extraction and Quantitative RT-PCR
For analysis of Ptch1 and Gli1 mRNA levels after SHH treatment, 10T1/2 cells or Cdon−/− mouse embryonic fibroblasts (MEFs) transfected with the indicated expression vectors were grown in Dulbecco modified Eagle medium (DMEM) containing 10% fetal bovine serum. Twenty-four hours after transfection, near confluent cultures were changed to DMEM plus 2% horse serum and treated with recombinant SHH (R&D Systems) (400 ng/ml) for 24 hr. Total RNA was extracted using easy-Blue reagent (iNtRON Biotechnology, Kyunggi-do, South Korea). The cDNAs were reverse-transcribed from one μg of total RNA with oligo-dT primer and SuperScript II reverse transcriptase (Invitrogen). PCR reactions were performed with 5% of the reverse transcription (RT) reaction, 250 nM of each primer, and SYBR Premix Ex Taq polymerase (Takara). PCR reactions were performed on an ABI PRISM 7000 Sequence Detection System (Applied Biosystems). Expression levels of Gapdh were used to normalize the expression levels of each sample. Primer sequences used were as described previously.15
Determination of CDON Protein Half-Lives
We transfected Cdon−/− MEFs with expression vectors encoding wild-type (WT) CDON and CDON variants by using Lipofectamine 2000. Forty-eight hours after transfection, cells were treated with 10 μM cycloheximide and harvested at various time points thereafter. Cell lysates were analyzed by immunoblotting for CDON, and, as a loading control, β-actin. CDON levels were quantified by densitometry with ImageJ software, normalized to actin levels, and compared to untreated samples. Half-lives reported are averages of three experiments. Data were analyzed by Student's t test.
SHH-N::AP Cell-Surface-Binding Assays
Experiments were performed as previously described.11 In brief, Cos7 cells were transfected with expression vectors for WT CDON or CDON variants with Lipofectamine 2000. Forty-eight hours after transfection, cells were washed with PBS and incubated with increasing concentrations of ShhN::AP for 90 min at room temperature. Cells were washed extensively with PBS, fixed with PFA, and bound AP activity was measured with AP yellow liquid substrate (Sigma). Saturation binding curves and Scatchard analyses were performed with GraphPad Prism software. Experiments were performed at least three times and Kd values were determined by fitting each data set to the one-site-specific binding model by nonlinear regression.
Results
We analyzed 282 unrelated individuals with HPE spectrum disorders and 96 anonymous controls for sequence variations of CDON. A total of 44 distinct sequence variations were identified within the amplicons tested (Tables S1 and S2), of which 24 were not previously described; one variant was also seen in a cohort with a different anomaly.30 We chose to focus on the rare variants that were not detected in public databases or in our anonymous control cohort. Six heterozygous missense mutants found only in HPE cases were chosen for further study: (1) c.2051C>G (p.Thr684Ser), (2) c.2065C>G (p.Pro689Ala), (3) c.2071G>A (p.Val691Met), (4) c.2339T>A (p.Val780Glu), (5) c.2368A>G (p.Thr790Ala), and (6) c.2818A>C (p.Ser940Arg) (see the clinical summary in Table 1 and Figure 1A). Although information on parental genotypes was limited, at least one mutation was a de novo occurrence in the affected individual (p.Thr790Ala; Table 1).
Table 1.
Clinical and Molecular Findings of Individuals with CDON Mutations
| Patient Number | CDON Variation | Amino Acid | Inheritance | Clinical Findings |
|---|---|---|---|---|
| 5410 | c.2051C>G | p.Thr684Ser | maternal; both parents tested | aborted fetus, referred for HPE findings. |
| 7190 | c.2065C>G | p.Pro689Ala | unknown; parents unavailable | agenesis of the corpus callosum, hypotelorism, growth hormone deficiency, global developmental delay; dark, thick eyebrows with synophrys. |
| 5308 | c.2071G>A | p.Val691Met | maternal by clinical history; parents not available | suspected autosomal dominant HPE, microcephaly, developmental delay; half-sib died at two months of semi-lobar HPE and biliary atresia. |
| 6864 | c.2339T>A | p.Val780Glu | unknown; mother negative, father unavailable | HPE-like, midline cyst of falx cerebri. |
| 5288 | c.2368A>G | p.Thr790Ala | de novo; both parents tested | agenesis of the corpus callosum, alobar HPE, hypotelorism, mild proptosis, median cleft lip/palate, absent columella, cryptorchidism. at autopsy: incomplete separation of the frontal lobes, absent pituitary, adrenal atrophy, absent corpus callosum, optic tracts with single cerebral artery. hepatic cholestasis and polysplenia noted. |
| 7321 | c.2818A>C | p.Ser940Arg | unknown; mother negative, father unavailable | alobar HPE findings. |
Figure 1.
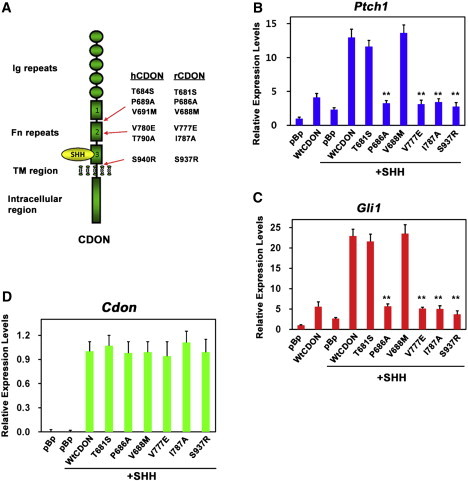
CDON HPE Variants Do Not Support SHH-Dependent Gene Expression
(A) Schematic diagram of CDON and the location of HPE-associated variants. The missense CDON variants from human HPE cases (hCDON) and the corresponding residues engineered into a rat Cdon (rCDON) cDNA are shown at the right. The p.Thr684Ser and p.Thr681Ser (T684S and T681S), p.Pro689Ala and p.Pro686Ala (P689A and P686A), and p.Val691Met and p.Val688Met (V691M and V688M) variants are in the linker region between Fn repeats 1 and 2. The p.Val780Glu and p.Val777Glu (V780E and V777E) and the p.Thr790Ala and p.Ile787Ala (T790A and I787A) variants are in Fn repeat 2. The p.Ser940Arg and p.Ser937Arg (S940R and S937R) variants are in the linker region between Fn repeat 3 and the transmembrane (TM) region. SHH binds to Fn repeat 3.
(B and C) qRT-PCR analysis of Ptch1 (B) and Gli1 (C) expression in Cdon−/− MEFs transfected with 2 μg of the indicated CDON vectors plus or minus treatment with SHH. Expression was normalized to Gapdh.
(D) qRT-PCR analysis of expression of CDON variants in Cdon−/− MEFs from same lysates analyzed in (B and C).
Error bars represent the means of triplicate determinations ±SD. ∗∗p < 0.01 as compared to wild-type CDON.
To assess the functional consequences of the six HPE-associated CDON mutations, we engineered equivalent mutations into a rat Cdon cDNA and performed cell-based assays (the corresponding human and rat mutations and their positions within CDON are shown in Figure 1A). Expression of WT CDON in Cdon−/− MEFs conferred SHH-dependent induction of the endogenous Ptch1 and Gli1 (Figures 1A and 1C). Expression of the p.Pro686Ala, p.Val777Glu, p.Ile787Ala, and p.Ser937Arg variants failed to support induction of Ptch1 and Gli1 in this system, revealing them to be defective in ligand-initiated pathway activity; in contrast, the p.Thr681Ser and p.Val688Met variants were as active as WT CDON in this assay (Figures 1B–1D). Similar results were obtained with 10T1/2 cells, except that the p.Ile787Ala substitution was only partially defective (Figure S1). p.Thr681Ser may correspond to a rare human SNP;30 p.Val688Met may also be a rare, benign variant, or it may affect CDON function in a way not revealed by this assay. The ability of the defective variants to promote SHH signaling was dose dependent, because transfection of 2.5 times the amount of plasmid used in Figure 1B–1D and Figure S1 revealed that when expressed at this level, the p.Val777Glu, p.Ile787Ala, and p.Ser937Arg substitutions possessed 35%–75% the activity of WT CDON in Cdon−/− MEFs; p.Pro686Ala was still largely inactive even at the higher dose (Figure 2). Again, similar results were seen with 10T1/2 cells (Figure S2). Therefore, these mutations appear to be loss-of-function but not null. We also analyzed this cohort for changes in the four genes most commonly studied in HPE18 (SHH, SIX3 [MIM 603714], ZIC2 [MIM 603073],and TGIF [MIM 602630]), and of those individuals with CDON mutations, only one had a mutation in these genes (Table S3); our study subject with the p.Val691Met variation (p.Val688Met in the rat construct that behaved normally in functional assays) had an alanine tract expansion in ZIC2 that is most likely a pathological alteration.31,32 Therefore, the four individuals with loss-of-function CDON mutations did not harbor additional mutations in the genes most often associated with HPE.
Figure 2.
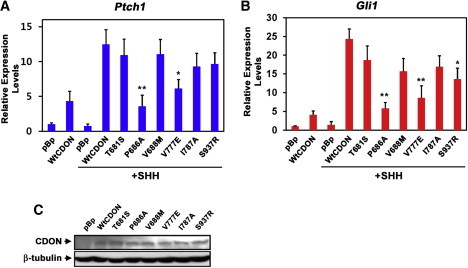
Overexpression Reveals Partial Loss of Function in Some CDON Variants
(A and B) qRT-PCR analysis of Ptch1 (A) and Gli1 (B) expression in Cdon−/− MEFs transfected with 5 μg of the indicated CDON vectors plus or minus treatment with SHH. Expression was normalized to Gapdh. Error bars represent the means of triplicate determinations ±SD. ∗∗p < 0.01, ∗p < 0.05 as compared to WT CDON.
(C) Immunoblot analysis of CDON expression in 10T1/2 cells under the conditions used in (A and B).
CDON variants are designated as in Figure 1.
To begin to address the molecular properties of the HPE-associated variant CDON proteins, their half-lives and ability to bind SHH were quantified. The half-lives of WT CDON and the p.Thr681Ser, p.Pro686Ala, p.Val688Met, and p.Ile787Ala variants were all ∼2 hr, whereas the half-lives of p.Val777Glu and p.Ser937Arg were ∼34% shorter than that (Table 2). Furthermore, when the CDON variants were immunoprecipitated under conditions of limited proteolysis, p.Val777Glu and p.Ser937Arg revealed distinct products not seen with WT CDON or the variants with a normal half-life (Figure S3). These results suggest that p.Val777Glu and p.Ser937Arg have an altered conformation that may partially destabilize them and contribute to their loss of function.
Table 2.
Properties of HPE-Associated CDON Variants
| CDON Variant | Half-Life (hr) |
Dissociation Constant (Kd) for ShhN::AP (nM) |
|---|---|---|
| WT CDON | 2.03 ± 0.23 | 5.52 ± 1.13 |
| p.Thr681Ser | 2.22 ± 0.21 | 3.12 ± 1.16 |
| p.Pro686Ala | 2.22 ± 0.51 | 3.14 ± 1.18 |
| p.Val688Met | 2.89 ± 0.65 | 2.03 ± 0.19 |
| p.Val777Glu | 1.36 ± 0.21a | 9.26 ± 3.16 |
| p.Ile787Ala | 2.23 ± 0.47 | 1.54 ± 0.63 |
| p.Ser937Arg | 1.34 ± 0.21a | 4.05 ± 1.71 |
p < 0.05.
To determine the ability of CDON HPE variants to bind SHH ligand, a soluble SHH-N::AP fusion protein (consisting of the N-terminal portion of SHH with alkaline phosphatase fused to its carboxy terminus) was used in saturation binding experiments with cells that expressed WT or variant forms of CDON. Dissociation constants (Kd) for binding of SHH-N::AP to WT CDON and all variants were in the low nanomolar range (Table 2 and Figure S4), similar to the findings in a previous report.11 Moreover, the variation in Kd that existed among the CDON variants did not correlate with their abilities to promote SHH-initiated induction of Ptch1 or Gli1 in Cdon−/− MEFs or 10T1/2 cells as shown in Figure 2 and Figure S2 (R = 0.17–0.24; Figure S5). This is consistent with the fact that none of the mutations occurred in Fn(3), the SHH-binding repeat of CDON11,12 (Figure 1A), and indicates that the variants' defective ability to support SHH-dependent gene expression is unlikely to be due to diminished ability to associate with ligand.
Because the CDON HPE variants were not defective in binding to SHH, we assessed their ability to bind other components of the signal reception machinery, including PTCH1, BOC, GAS1, and WT CDON itself, in coimmunoprecipitation experiments. CDON and PTCH1 coimmunoprecipitated both endogenously and in transfectants (Figure 3A and Figure S6). Three of the four variants defective in promoting SHH signaling (p.Pro686Ala, p.Val777Glu, and p.Ser937Arg) displayed significantly diminished ability to coprecipitate with PTCH1 (Figures 3A and 3B). These experiments require a level of transfected vector similar to that used in the Cdon−/−-MEF-based and 10T1/2-cell-based functional assays shown in Figure 2 and Figure S2, where partial loss of function of the HPE-associated variants was observed. In contrast to the lack of correlation with SHH binding, the diminished activities of the variants in these assays correlated very well with their reduced level of binding to PTCH1 (R = 0.88–0.99, p = 0.008–< 0.001; Figure S5). CDON has also been shown to interact with itself and with BOC,25,33 but the CDON HPE variants did not display any defect in binding to WT CDON (Figure S7) and only p.Pro686Ala showed decreased ability to associate with BOC in coimmunoprecipitations (Figures 3C and 3D).
Figure 3.
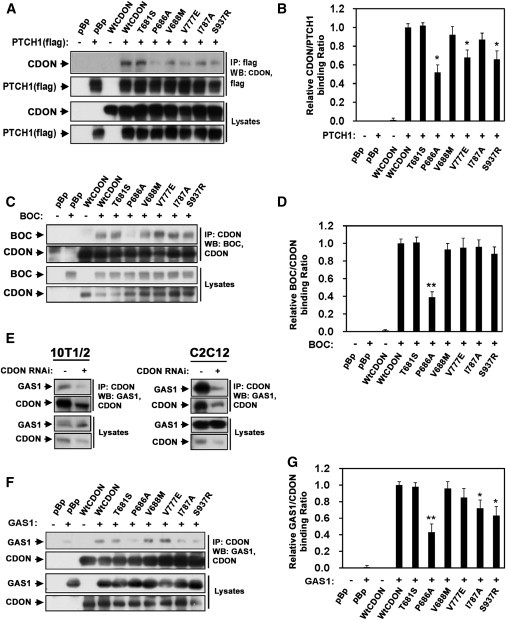
CDON Variants Display Diminished Interaction with Other SHH-Binding Proteins
(A) Cos7 cells were transfected with an expression vector encoding flag-tagged PTCH1 (+) or a control vector (−) and expression vectors encoding CDON, the indicated CDON variants, or a control vector (pBp) as indicated. Lysates were immunoprecipitated with antibodies to flag epitope and immunoblotted with antibodies to flag or CDON. Lysates were also blotted as a control.
(B) Results from three independent experiments were quantified for the amount of CDON that immunoprecipitated with PTCH1.
(C) 293T cells were transfected with an expression vector encoding BOC (+) or a control vector (−) and expression vectors encoding CDON, the indicated CDON variants, or a control vector (pBp) as indicated. Lysates were immunoprecipitated with antibodies to the CDON intracellular region and immunoblotted with antibodies to CDON or BOC. Lysates were also blotted as a control.
(D) Results from three independent experiments were quantified for the amount of BOC that immunoprecipitated with CDON.
(E) 10T1/2 or C2C12 cell lysates were immunoprecipitated with antibodies to CDON and immunoblotted with antibodies to CDON and GAS1. Lysates were immunoblotted as a control.
(F) 293T cells were transfected with an expression vector encoding GAS1 (+) or a control vector (−) and expression vectors encoding CDON, the indicated CDON variants, or a control vector (pBp) as indicated. Lysates were immunoprecipitated with antibodies to the CDON intracellular region and immunoblotted with antibodies to CDON or GAS1. Lysates were also blotted as a control.
(G) Results from three independent experiments were quantified for the amount of GAS1 that immunoprecipitated with CDON.
CDON variants are designated as in Figure 1. Error bars represent the means of triplicate determinations ±SD. ∗∗p < 0.01, ∗p < 0.05 as compared to wild-type CDON.
CDON has not previously been shown to associate with GAS1. Endogenous GAS1 coimmunoprecipitated with endogenous CDON and the presence of GAS1 in the precipitate was strongly diminished by depletion of CDON by RNAi, indicating the specificity of the interaction (Figure 3E and Figure S8). GAS1 also coimmunoprecipitated with BOC (Figure S8). The amount of GAS1 that coprecipitated with CDON was not significantly altered by pretreatment of the cells with SHH-N (Figure S8). Coimmunoprecipitation was also observed when GAS1 was ectopically coexpressed with the CDON or BOC ectodomains in 293T cells (Figure S8). The ability of HPE-associated CDON variants to interact with GAS1 was therefore assessed in cotransfectants. Three of the four variants defective in promoting SHH signaling (p.Pro686Ala, p.Ile787Ala, and p.Ser937Arg) displayed significantly diminished ability to coprecipitate GAS1 (Figures 3F and 3G), and the reduction in GAS1 binding correlated with their reduction in activity in the Cdon−/−-MEF-based and 10T1/2-cell-based assays (R = 0.73–0.90, p = 0.06–0.005; Figure S5), though not as strongly as PTCH1 binding did.
If the diminished binding to GAS1 (and to BOC for p.Pro686Ala) was critical to the CDON variants' loss of function, these receptors might be predicted to synergize to promote SHH ligand-dependent pathway activity. However, coexpression of combinations of any two or all three coreceptors in Cdon−/− MEFs or 10T1/2 cells revealed largely additive effects (Figure 4 and Figure S9). Furthermore, expression of CDON HPE variants did not inhibit the activity of GAS1 plus BOC to stimulate SHH-dependent induction of Gli1 or Ptch1 in 10T1/2 cells or in Cdon−/− MEFs, suggesting that these variants do not possess a dominant inhibitory activity toward WT (endogenous) CDON or the other coreceptors (Figure 4 and Figure S9).
Figure 4.
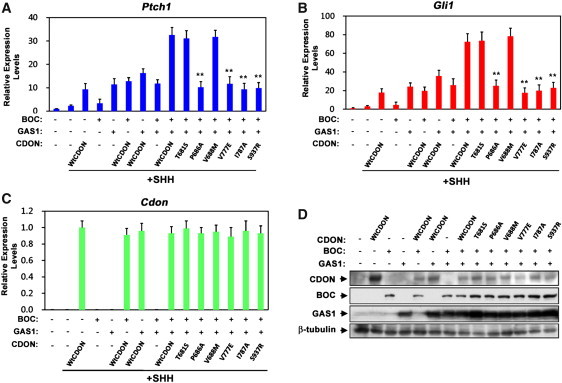
CDON, BOC, and GAS1 Have Roughly Additive Activity in Promoting SHH-Dependent Gene Expression
(A and B) qRT-PCR analysis of Ptch1 and Gli1 expression in Cdon−/− MEFs transfected with 2 μg of the indicated CDON, BOC, and GAS1 expression vectors plus or minus treatment with SHH. Expression was normalized to Gapdh.
(C) qRT-PCR analysis of expression of CDON variants in Cdon−/− MEFs from same RNA samples analyzed in (A and B).
(D) Immunoblot analysis of CDON, BOC, and GAS1 expression in Cdon−/− MEFs transfected with 5 μg of CDON, BOC, and GAS1 expression vectors.
CDON variants are designated as in Figure 1. Error bars represent the means of triplicate determinations ±SD. ∗∗p < 0.01 as compared to wild-type CDON.
CDON binds to PTCH1 via both the Fn(1) and Fn(2) repeats in its ectodomain.13 So that we could determine the specific CDON ectodomain repeat(s) involved in binding to BOC, GAS1, and CDON itself, cells were cotransfected to express a series of secreted CDON extracellular region-Fc fusion proteins and either BOC, GAS1, or WT CDON. The CDON-Fc proteins were pulled down and immunoblotted for Fc and either BOC, GAS1, or CDON. In these experiments, we found that Fn(2) bound to GAS1; Fn(2) and, to a lesser extent, Fn(3) bound to BOC; and full-length CDON associated efficiently with a CDON-Fc construct containing all three Fn repeats but more weakly with the individual Fn(2) and Fn(3) repeat constructs (Figures 5A–5D). Fn(3), which is necessary and sufficient for binding SHH, is required for CDON activity.11,12 We investigated the requirements of Fn(1) and Fn(2), involved in CDON binding to PTCH1 and GAS1, through use of CDON deletion mutants that lacked either or both domains. Removal of Fn(1) reduced CDON's ability to promote SHH-dependent induction of Gli1 and Ptch1 in Cdon−/− MEFs by less than 50%, whereas loss of Fn(2) reduced activity by ∼70% and removal of both Fn(1) and Fn(2) reduced activity by ∼85% (Figures 5E–5G). These results indicate that the Fn repeats that interact with PTCH1 and other SHH receptors, particularly Fn(2), are important for CDON function.
Figure 5.
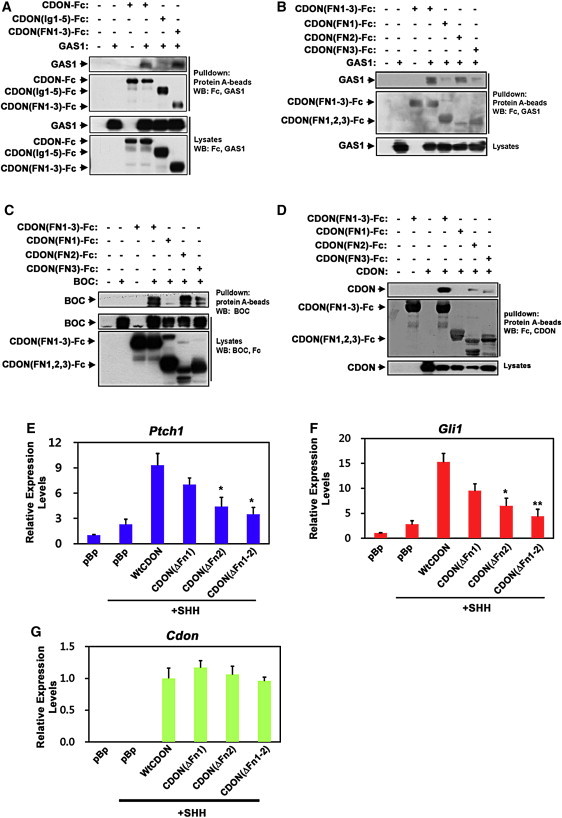
CDON Fn Repeats Bind GAS1, BOC, and CDON Itself
(A) Cos7 cells were transfected with plasmids encoding CDON-Fc fusion proteins containing the entire ectodomain (CDON-Fc), the 5 Ig repeats (CDONIg1-5)-Fc) or the 3 Fn repeats (CDON(Fn1-3)-Fc) and GAS1 as indicated; lysates were pulled down with protein A-agarose and immunoblotted with antibodies to Fc or GAS1.
(B) As in (A), except CDON-Fc proteins contained the indicated CDON Fn repeats.
(C) 293T cells were transfected with plasmids encoding CDON-Fc fusion proteins containing the indicated CDON Fn repeats and BOC as indicated, lysates were pulled down with protein A-agarose and immunoblotted with antibodies to Fc or BOC.
(D) 293T cells were transfected with plasmids encoding CDON-Fc fusion proteins containing the indicated CDON Fn repeats and full-length CDON as indicated; lysates were pulled down with protein A-agarose and immunoblotted with antibodies to Fc or CDON.
(E and F) qRT-PCR analysis of Ptch1 (E) and Gli1 (F) expression in Cdo−/− MEFs transfected with 2 μg of vectors encoding WT CDON or CDON deletion variants lacking the indicated Fn domains plus or minus treatment with SHH. Expression was normalized to Gapdh.
(G) qRT-PCR analysis of expression of CDON and CDON deletion mutants in the same lysates analyzed in (E and F).
Error bars represent the means of triplicate determinations ±SD. ∗∗p < 0.01, ∗p < 0.05 as compared to wild-type CDON.
Discussion
We report here the identification of CDON mutations in individuals with HPE. The HPE spectrum of defects is characterized by a very broad range of phenotypic severity. Studies with Cdon mutant mice recapitulate these characteristics because these animals display HPE with strain-dependent severity.14,15 The CDON mutations identified are typical of those generally associated with HPE in that they are heterozygous and result in a loss of ability to promote SHH signaling. Additionally, at least one such mutation was a de novo occurrence in the affected individual (Table 1). Taken together, these results argue persuasively that CDON mutations are important contributing factors to HPE.
Defects in the SHH pathway are the most common known cause of HPE, and our analysis of HPE-associated CDON mutations provides insight into mechanisms of hedgehog signaling. The mutations reported here did not occur in Fn(3), the CDON ectodomain repeat that binds directly to SHH, and they did not result in obvious defects in ability to confer cell-surface binding of a recombinant form of SHH. In contrast, we find that these CDON variants associated inefficiently with two other proteins that bind SHH to the cell-surface, the primary receptor PTCH1 and the coreceptor GAS1, in a manner that paralleled their diminished activity in SHH-signaling assays. This was specific as the variants associated normally with two different SHH coreceptors, BOC and WT CDON itself (the one exception was a decrease in the ability of the p.Pro686Ala substitution to associate with BOC). These results indicate that interaction between proteins that bind SHH to the cell-surface is an important aspect of SHH signal reception.
Studies with mice carrying mutations in Cdon, Boc, and Gas1, singly or in combination, lead to the conclusion that: (1) these factors have both specific and overlapping functions; (2) no single factor is essential for hedgehog-pathway activity; and (3) they are collectively required for hedgehog-pathway function in that combined loss of all three factors results in a nearly complete loss of pathway activity in the early embryo.16,17,34 Furthermore, a SHH point mutant that binds PTCH1 but not CDON, BOC, or GAS1 is unable to activate hedgehog-pathway signaling.13 These studies are consistent with a model whereby SHH must engage PTCH1 and at least one of these coreceptors to activate signaling. However, whether the coreceptors need to interact with PTCH1 and/or each other has not been addressed. The selectively reduced ability of the HPE-associated CDON variants to interact with PTCH1 and GAS1 argues that interactions between SHH receptors are important for signaling. Taken together, the results are consistent with the notion that CDON (and presumably BOC and GAS1) must associate with both ligand and other hedgehog-receptor components, particularly PTCH1, for signaling to occur. The fact that mice lacking any two of CDON, BOC, or GAS1 still display a significant, though very obviously diminished, level of SHH-dependent embryonic patterning argues that these factors share at least some redundant or compensatory activity.16,17,34 Our finding that these three factors function largely additively to promote SHH signaling in transfectants is consistent with this conclusion. The diminished binding to PTCH1 displayed by the HPE-associated CDON variants is therefore likely to be a more important functional defect than diminished binding to GAS1 in their loss of activity, though the latter may also contribute. In summary, it is concluded that mutation-induced disruption of interactions between components of the hedgehog-reception machinery is a mechanism of HPE.
Acknowledgments
We are grateful to M. Lu for help with data analysis, B. Allen and F. Charron for helpful discussions, R. Toftgard and A. McMahon for reagents, and P. Wassarman and B. Gelb for critical reading of the manuscript. This work was supported by a grant from the March of Dimes (R.S.K.); by grants from the Korea Healthcare Technology R and D Project, Ministry for Health, Welfare and Family Affairs, Republic of Korea (A090176); and the National Research Foundation of Korea (NRF) funded by the Korea government (MEST; 2011-0017315) (J.-S.K.); and by the Division of Intramural Research, National Human Genome Research Institute, National Institutes of Health, Department of Health and Human Services (M.M.).
Contributor Information
Jong-Sun Kang, Email: kangj01@skku.edu.
Maximilian Muenke, Email: mamuenke@mail.nih.gov.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://www.1000genomes.org/
BLAST, NCBI, http://blast.ncbi.nlm.nih.gov/Blast.cgi
COBALT, NCBI, http://www.ncbi.nlm.nih.gov/tools/cobalt/
dbSNP, NCBI, http://www.ncbi.nlm.nih.gov/projects/SNP/
Mutalyzer 2.0 β-8, http://www.mutalyzer.nl/2.0/
Nomenclature for the description of sequence variants (Human Genome Variation Society [HGVS]), http://www.hgvs.org/mutnomen/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Reference Sequence Annotation, NCBI, http://www.ncbi.nlm.nih.gov/RefSeq/
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Muenke M., Beachy P.A. Holoprosencephaly. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., editors. The Metabolic & Molecular Bases of Inherited Disease. McGraw-Hill; New York: 2001. pp. 6203–6230. [Google Scholar]
- 2.Yamada S., Uwabe C., Fujii S., Shiota K. Phenotypic variability in human embryonic holoprosencephaly in the Kyoto Collection. Birth Defects Res. A Clin. Mol. Teratol. 2004;70:495–508. doi: 10.1002/bdra.20048. [DOI] [PubMed] [Google Scholar]
- 3.Roessler E., Muenke M. The molecular genetics of holoprosencephaly. Am. J. Med. Genet. C. Semin. Med. Genet. 2010;154C:52–61. doi: 10.1002/ajmg.c.30236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schachter K.A., Krauss R.S. Murine models of holoprosencephaly. Curr. Top. Dev. Biol. 2008;84:139–170. doi: 10.1016/S0070-2153(08)00603-0. [DOI] [PubMed] [Google Scholar]
- 5.Jiang J., Hui C.C. Hedgehog signaling in development and cancer. Dev. Cell. 2008;15:801–812. doi: 10.1016/j.devcel.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Varjosalo M., Taipale J. Hedgehog: Functions and mechanisms. Genes Dev. 2008;22:2454–2472. doi: 10.1101/gad.1693608. [DOI] [PubMed] [Google Scholar]
- 7.Beachy P.A., Hymowitz S.G., Lazarus R.A., Leahy D.J., Siebold C. Interactions between Hedgehog proteins and their binding partners come into view. Genes Dev. 2010;24:2001–2012. doi: 10.1101/gad.1951710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martinelli D.C., Fan C.M. Gas1 extends the range of Hedgehog action by facilitating its signaling. Genes Dev. 2007;21:1231–1243. doi: 10.1101/gad.1546307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McLellan J.S., Zheng X., Hauk G., Ghirlando R., Beachy P.A., Leahy D.J. The mode of Hedgehog binding to Ihog homologues is not conserved across different phyla. Nature. 2008;455:979–983. doi: 10.1038/nature07358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Okada A., Charron F., Morin S., Shin D.S., Wong K., Fabre P.J., Tessier-Lavigne M., McConnell S.K. Boc is a receptor for sonic hedgehog in the guidance of commissural axons. Nature. 2006;444:369–373. doi: 10.1038/nature05246. [DOI] [PubMed] [Google Scholar]
- 11.Tenzen T., Allen B.L., Cole F., Kang J.-S., Krauss R.S., McMahon A.P. The cell surface membrane proteins Cdo and Boc are components and targets of the Hedgehog signaling pathway and feedback network in mice. Dev. Cell. 2006;10:647–656. doi: 10.1016/j.devcel.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 12.Yao S., Lum L., Beachy P. The ihog cell-surface proteins bind Hedgehog and mediate pathway activation. Cell. 2006;125:343–357. doi: 10.1016/j.cell.2006.02.040. [DOI] [PubMed] [Google Scholar]
- 13.Izzi L., Lévesque M., Morin S., Laniel D., Wilkes B.C., Mille F., Krauss R.S., McMahon A.P., Allen B.L., Charron F. Boc and gas1 each form distinct shh receptor complexes with ptch1 and are required for shh-mediated cell proliferation. Dev. Cell. 2011;20:788–801. doi: 10.1016/j.devcel.2011.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cole F., Krauss R.S. Microform holoprosencephaly in mice that lack the Ig superfamily member Cdon. Curr. Biol. 2003;13:411–415. doi: 10.1016/s0960-9822(03)00088-5. [DOI] [PubMed] [Google Scholar]
- 15.Zhang W., Kang J.-S., Cole F., Yi M.J., Krauss R.S. Cdo functions at multiple points in the Sonic Hedgehog pathway, and Cdo-deficient mice accurately model human holoprosencephaly. Dev. Cell. 2006;10:657–665. doi: 10.1016/j.devcel.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 16.Allen B.L., Tenzen T., McMahon A.P. The Hedgehog-binding proteins Gas1 and Cdo cooperate to positively regulate Shh signaling during mouse development. Genes Dev. 2007;21:1244–1257. doi: 10.1101/gad.1543607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang W., Hong M., Bae G.-U., Kang J.-S., Krauss R.S. Boc modifies the holoprosencephaly spectrum of Cdo mutant mice. Dis. Model Mech. 2011;4:368–380. doi: 10.1242/dmm.005744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pineda-Alvarez D.E., Dubourg C., David V., Roessler E., Muenke M. Current recommendations for the molecular evaluation of newly diagnosed holoprosencephaly patients. Am. J. Med. Genet. C. Semin. Med. Genet. 2010;154C:93–101. doi: 10.1002/ajmg.c.30253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schimmenti L.A., de la Cruz J., Lewis R.A., Karkera J.D., Manligas G.S., Roessler E., Muenke M. Novel mutation in sonic hedgehog in non-syndromic colobomatous microphthalmia. Am. J. Med. Genet. A. 2003;116A:215–221. doi: 10.1002/ajmg.a.10884. [DOI] [PubMed] [Google Scholar]
- 20.Xiao W., Oefner P.J. Denaturing high-performance liquid chromatography: A review. Hum. Mutat. 2001;17:439–474. doi: 10.1002/humu.1130. [DOI] [PubMed] [Google Scholar]
- 21.Cole F., Zhang W., Geyra A., Kang J.-S., Krauss R.S. Positive regulation of myogenic bHLH factors and skeletal muscle development by the cell surface receptor CDO. Dev. Cell. 2004;7:843–854. doi: 10.1016/j.devcel.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 22.Takaesu G., Kang J.S., Bae G.U., Yi M.J., Lee C.M., Reddy E.P., Krauss R.S. Activation of p38α/β MAPK in myogenesis via binding of the scaffold protein JLP to the cell surface protein Cdo. J. Cell Biol. 2006;175:383–388. doi: 10.1083/jcb.200608031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun H., Taneja R. Analysis of transformation and tumorigenicity using mouse embryonic fibroblast cells. Methods Mol. Biol. 2007;383:303–310. doi: 10.1007/978-1-59745-335-6_19. [DOI] [PubMed] [Google Scholar]
- 24.Kang J.-S., Gao M., Feinleib J.L., Cotter P.D., Guadagno S.N., Krauss R.S. CDO: An oncogene-, serum-, and anchorage-regulated member of the Ig/fibronectin type III repeat family. J. Cell Biol. 1997;138:203–213. doi: 10.1083/jcb.138.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kang J.-S., Mulieri P.J., Hu Y., Taliana L., Krauss R.S. BOC, an Ig superfamily member, associates with CDO to positively regulate myogenic differentiation. EMBO J. 2002;21:114–124. doi: 10.1093/emboj/21.1.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kogerman P., Krause D., Rahnama F., Kogerman L., Undén A.B., Zaphiropoulos P.G., Toftgård R. Alternative first exons of PTCH1 are differentially regulated in vivo and may confer different functions to the PTCH1 protein. Oncogene. 2002;21:6007–6016. doi: 10.1038/sj.onc.1205865. [DOI] [PubMed] [Google Scholar]
- 27.Makarova O., Kamberov E., Margolis B. Generation of deletion and point mutations with one primer in a single cloning step. Biotechniques. 2000;29:970–972. doi: 10.2144/00295bm08. [DOI] [PubMed] [Google Scholar]
- 28.Kang J.-S., Yi M.-J., Zhang W., Feinleib J.L., Cole F., Krauss R.S. Netrins and neogenin promote myotube formation. J. Cell Biol. 2004;167:493–504. doi: 10.1083/jcb.200405039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bae G.U., Kim B.G., Lee H.J., Oh J.E., Lee S.J., Zhang W., Krauss R.S., Kang J.S. Cdo binds Abl to promote p38α/β MAPK activity and myogenic differentiation. Mol. Cell. Biol. 2009;29:4130–4143. doi: 10.1128/MCB.00199-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jehee F.S., Alonso L.G., Cavalcanti D.P., Kim C.H., Wall S.A., Mulliken J.B., Sun M., Jabs E.W., Boyadjiev S.A., Wilkie A.O., Passos-Bueno M.R. Mutational screening of FGFR1, CER1, and CDON in a large cohort of trigonocephalic patients. Cleft Palate Craniofac. J. 2006;43:148–151. doi: 10.1597/04-206.1. [DOI] [PubMed] [Google Scholar]
- 31.Brown L.Y., Odent S., David V., Blayau M., Dubourg C., Apacik C., Delgado M.A., Hall B.D., Reynolds J.F., Sommer A. Holoprosencephaly due to mutations in ZIC2: Alanine tract expansion mutations may be caused by parental somatic recombination. Hum. Mol. Genet. 2001;10:791–796. doi: 10.1093/hmg/10.8.791. [DOI] [PubMed] [Google Scholar]
- 32.Brown L., Paraso M., Arkell R., Brown S. In vitro analysis of partial loss-of-function ZIC2 mutations in holoprosencephaly: Alanine tract expansion modulates DNA binding and transactivation. Hum. Mol. Genet. 2005;14:411–420. doi: 10.1093/hmg/ddi037. [DOI] [PubMed] [Google Scholar]
- 33.Kang J.-S., Feinleib J.L., Knox S., Ketteringham M.A., Krauss R.S. Promyogenic members of the Ig and cadherin families associate to positively regulate differentiation. Proc. Natl. Acad. Sci. USA. 2003;100:3989–3994. doi: 10.1073/pnas.0736565100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Allen B.L., Song J.Y., Izzi L., Althaus I.W., Kang J.-S., Charron F., Krauss R.S., McMahon A.P. Overlapping roles and collective requirement for the co-receptors Gas1, Cdo and Boc in Shh pathway function. Dev. Cell. 2011;20:775–787. doi: 10.1016/j.devcel.2011.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.