

Abstract
We report eight unrelated individuals with intellectual disability and overlapping submicroscopic deletions of 8q21.11 (0.66–13.55 Mb in size). The deletion was familial in one and simplex in seven individuals. The phenotype was remarkably similar and consisted of a round face with full cheeks, a high forehead, ptosis, cornea opacities, an underdeveloped alae, a short philtrum, a cupid's bow of the upper lip, down-turned corners of the mouth, micrognathia, low-set and prominent ears, and mild finger and toe anomalies (camptodactyly, syndactyly, and broadening of the first rays). Intellectual disability, hypotonia, decreased balance, sensorineural hearing loss, and unusual behavior were frequently observed. A high-resolution oligonucleotide array showed different proximal and distal breakpoints in all of the individuals. Sequencing studies in three of the individuals revealed that proximal and distal breakpoints were located in unique sequences with no apparent homology. The smallest region of overlap was a 539.7 kb interval encompassing three genes: a Zinc Finger Homeobox 4 (ZFHX4), one microRNA of unknown function, and one nonfunctional pseudogen. ZFHX4 encodes a transcription factor expressed in the adult human brain, skeletal muscle, and liver. It has been suggested as a candidate gene for congenital bilateral isolated ptosis. Our results suggest that the 8q21.11 submicroscopic deletion represents a clinically recognizable entity and that a haploinsufficient gene or genes within the minimal deletion region could underlie this syndrome.
Main Text
Historically the cytogenetic basis of microdeletion syndromes was established after the identification of similar submicroscopic deletions in a collection of individuals with a common phenotype. The introduction of whole-genome scanning technologies such as array-based comparative genomic hybridization (aCGH) enables the identification of novel imbalances in large cohorts of individuals with apparently no specific features. Once a common cytogenetic etiology is established, the clinical features of these new conditions are delineated. Recently array-based techniques have led to the discovery of several novel recurrent disease-causing microdeletions or microduplications.1
Here we report eight nonrelated individuals with an intellectual disability and an unusual phenotype who have overlapping submicroscopic deletions of 8q21.11 (0.66–13.55 Mb in size). The phenotype was proven to be recognizable in a family with five affected persons and published recently as a hitherto unreported entity,2 of whom one member is studied here. The other seven individuals are simplex cases. The phenotype is remarkably similar despite the difference in the deletion size and consisted of a round face with full cheeks, a high forehead, ptosis, cornea opacities, an underdeveloped alae, a short philtrum, a cupid's bow of the upper lip, down-turned corners of the mouth, micrognathia, low-set and prominent ears, and mild finger and toe anomalies (camptodactyly, syndactyly, and broadening of the first rays). Intellectual disability, hypotonia, decreased balance, sensorineural hearing loss, and unusual behavior were frequently observed. The clinical findings of all individuals are summarized in Table 1 and illustrated in Figure 1.
Table 1.
Clinical Data in Presently Reported Individuals With a 8q21.11 Microdeletion
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |
|---|---|---|---|---|---|---|---|---|
| Gender (M/F) | M | F | M | F | M | F | F | M |
| Age (yr) | 7 | 6 | 6 s | 17.5 | 16 | 13 | 15 | 2 |
| Growth and development | ||||||||
| Growth failure | ND | + | − | + | − | − | + | − |
| Intellectual diability | + | + | + | + | + | + (mild) | + | + |
| Face | ||||||||
| Round face | + | + | + | + | + | Square | + | + |
| High forehead | + | + | + | − | + | + | − | + |
| Eyes | Mi, SC, C, S | PR, S | Mi, SC | − | SC, S | + | D, S | |
| Hypertelorism | − | + | ND | − | − | + | − | − |
| Downslanting palpebral fissures | − | + | + | − | + | − | + | − |
| Short palpebral fissures | + | + | + | − | − | + | + | + |
| Ptosis | + | + | + | − | + | + | + | + |
| Epicanthal Folds | + | − | + | − | + | + | + | − |
| Wide nasal bridge | + | + | + | + | − | + | − | + |
| Underdeveloped alae | + | + | + | − | + | +a | − | + |
| Short philtrum | + | + | + | + | − | + | + | + |
| Cupid's bow | + | + | + | + | − | + | + | + |
| Down-turned corners mouth | + | + | + | + | + | + | − | − |
| Highly arched palate | ND | + | − | − | − | + | − | CP |
| Micrognathia | + | + | + | + | + | + | − | − |
| Prominent, low-set ears | + | + | + | + | + | + | + | + |
| Short neck | + | + | + | − | + | − | − | − |
| Neurology | ||||||||
| Hypotonia | + | + | + | − | + | − | + | − |
| Behavior | + | + | autism | Nl | Nl | unusually pleasant | Nl | Nl |
| Magnetic resonance imaging | UCC, GA | DM | UCC, GA, DM, OA | Nl | Nl | Nl | UCC | |
| Hearing | Nl | hyperacusis | hearing loss | hearing loss | Nl | Nl | Nl | hearing loss |
| Balance | Nl | Nl | Nl | impaired | Nl | Nl | Nl | impaired |
| Other | ||||||||
| Fingers | + | Nl | Ca | 4th and 5th toe abnormalities | ND | long | Ca | Sy, B, SM |
| Abnormal palmar creases | + | + | + | − | − | + | − | − |
| Various | CM, Cr, SP | AD | Cr | E | LU | DS | ||
The following abbreviations are used: M, male; F, female; ND, no data available; Mi, microphthalmia; C, cataract; SC, sclerocornea; S, strabismus; PR, pigmentary retina degeneration; D, Duane anomaly; CP, cleft palate; Nl, normal; UCC, underdeveloped corpus callosum; GA, gyration abnormalities; DM, decreased myelination; Ca, camptodactyly; Sy, syndactyly 3–4 finger; B, broad first ray; SM, short metacarpals; CM, capillary malformation; Cr, cryptorchidism; SP, small penis; AD, abnormal dentition; E, eczema; LU, lozenge shaped umbilicus; DS, decreased sense of smell.
This individual also had agenesis nasal cartilage; which had been surgically corrected before the picture was taken.
Figure 1.
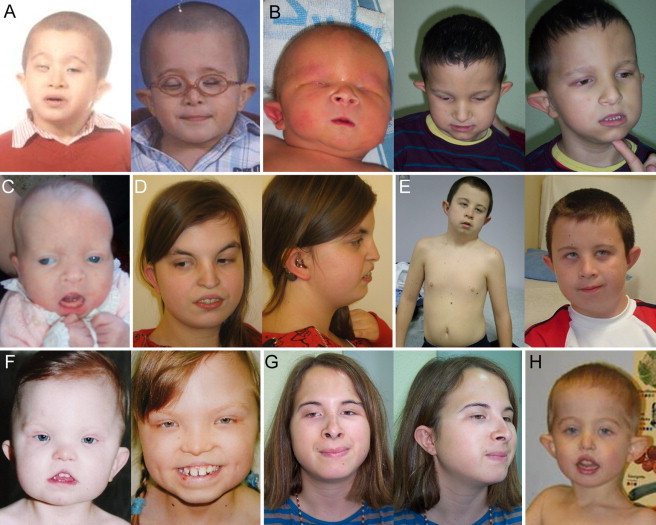
Facial Views of Individuals Included in the Present Study
(A) Individual 1 at age 4 (left) and 7 years (right).
(B) Individual 3 at age 1 month (left) and 6 years (middle and right).
(C) Individual 2 at age 6 months.
(D) Individual 4 at age 17 years (left and right).
(E) Individual 5 at age 12 years (left and right).
(F) Individual 6 at age 20 months (left) and 9 years (right).
(G) Individual 7 at age 15 years (left and right) and (H) individual 8 at age 3 years.
Individuals 1–3 and 7 were referred for genetic assessment because of intellectual disability and an unusual phenotype. They were studied by classical cytogenetics and array comparative genomic hybridization (aCGH). Individuals 1 and 2 had de novo apparently balanced chromosome translocations involving 8q21; individual 3 was found to have a de novo Robertsonian translocation of chromosome 21; and individual 7 had a normal karyotype (Table 2). Individual 4 was recruited through the DECIPHER database3 (ID 2121). Samples from individuals 5 and 6, who also had de novo 8q21 microdeletions, were obtained from colleagues in Ireland and Poland. One of them (individual 6) had been published.4 Individual 8 was recently published as part of a four-generation family suggested to have a hitherto unrecognized entity characterized by intellectual disability and an unusual phenotype2. He had a previous normal aCGH result at a 0.7 Mb resolution. The resemblance to individuals 1–7 was recognized and samples obtained. The study received ethical approval from the ethical committee of the University Hospital La Paz and, after a complete description of the study to the participants, written informed consent was obtained. In individuals 1–7, overlapping 8q21 deletions were ascertained by aCGH with the Agilent Kit 105A (Figure 2). Individual 8, who had a previous normal result with a commercial aCGH platform, was directly analyzed with a high-density custom oligonucleotide array for the 8q21 region. The deletions were confirmed by fluorescent in situ hybridization (FISH), multiplex ligation-dependent probe amplification, or microsatellite analysis (Figure 3). The 8q21.11 deletion region has not been reported to show copy number variation (CNV) in normal individuals. No other potentially pathogenic copy number alterations were observed elsewhere in the genome (data not shown).
Table 2.
Results of aCGH and Classical Cytogenetic Studies of Eight 8q21 Microdeletions
| Deletion | Size (Mb) | Start | End | Nomenclature | Karyotype | |
|---|---|---|---|---|---|---|
| 1 | 8q21.11q21.3 8q21.2q21.3 | 9.79 1.50 | 74915253 86536728 | 84715130 88037242 | arr 8q21.11q21.3 (74915253-84715130)x1 dn arr 8q21.2q21.3 (86536728-88037242)x1 dn |
46,XY,t(4;8;7)(q13;q13;q11.2)dn. ish del (8)(q21.11)(RP11-48D4-) |
| 2 | 8q21.11q21.2 | 9.80 | 74362438 | 84172022 | arr 8q21.11q21.2(74362438-84172022)x1 dn | 46,XX,t(8;15)(q13;q15)dn. ish del(8) (q21.1)(RP11-48D4-) |
| 3 | 8q21.11q21.13 | 4.74 | 77179233 | 81924253 | arr 8q21.11q21.13 (77179233-81924253)x1 dn | 45,XY,der(21;21)(q10,q10)dn. ish del(8)(q21.11)(RP11-48D4-) |
| 4 | 8q21.11 | 3.58 | 74487194 | 78073108 | arr 8q21.11 (74487194-78073108)x1 dn | 46, XX |
| 5 | 8q21.11q21.3 | 13.55 | 77220253 | 90774646 | arr 8q21.11q21.3 (77220253-90774646)x1 dn | 46, XY |
| 64 | 8q21.11q21.13 | 5.69 | 74600271 | 80291446 | arr 8q21.11q21.13 (74600271-80291446)x1 dn | 46, XX |
| 7 | 8q21.11q21.13 | 5.11 | 77226464 | 82338741 | arr 8q21.11q21.13 (77226464-82338741)x1 dn | 46, XX, ish del(8) (q21.1)(RP11-48D4-) |
| 82 | 8q21.11 | 0.66 | 77098877 | 77766239 | arr 8q21.11(77098877-77766239)x1 | 46, XY |
Data are from the UCSC Genome Browser (February 2009 assembly: hg19, NCBI Build 37).
Figure 2.
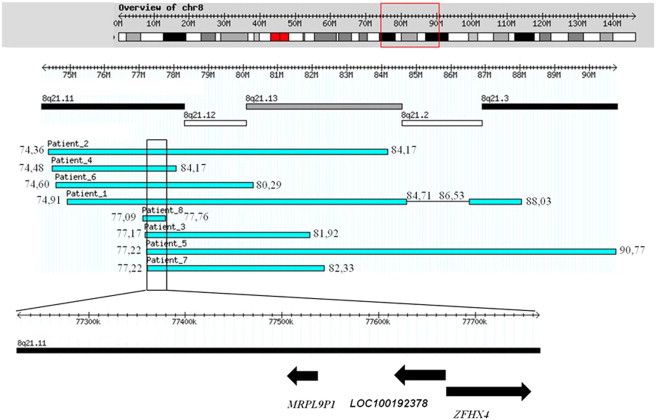
Graphical Representation of the Deletions in All Individuals and Genes within the Minimal Region of Overlap
The minimal region of overlap shown includes hg 19, chromosome 8: 77226464-77766239.
Figure 3.
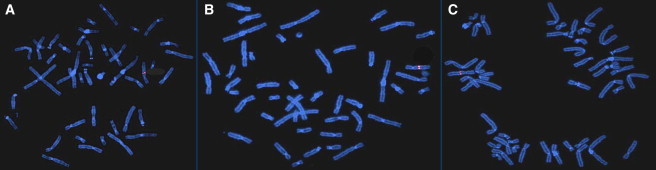
FISH Confirmation of the Heterozygous Deletion of 8q21 in Individuals
Individuals 1 (A), 2 (B), and 3 (C) are shown with BAC probe RP11-48D4 mapping to 8q21.11 (red).
In order to define more precisely the breakpoint interval of the deletions, we designed a high-density targeted oligonucleotide array with a 140 bp average probe spacing spanning a 20 Mb interval of 8q13.3-q21.3 (AMADID 029896, Agilent Technologies, Santa Clara, CA, USA). Results demonstrated that all eight individuals carried overlapping deletions defining a minimal region of 539.77 kb (hg 19, chromosome 8: 77226464–77766239) (Table 2, illustrated in Figure 2 and Figure S1, available online). In addition individual 1 showed a complex genomic rearrangement with a pattern consistent with two deletions close to one another and involving the 8q21 region (Figure 2 and Figure S1).
Syndrome-associated nonrecurrent rearrangements usually share a common genomic region of overlap (the smallest region of overlap [SRO]) encompassing the locus associated with the genomic disorder. We analyzed the present individuals with 8q21 rearrangements by using high-density arrays to narrow the SRO to a 539.77 kb interval. In the minimal region of overlap only one gene, Zinc Finger Homeobox 4 (ZFHX4 [MIM 606940]); one microRNA of unknown function (LOC100192378); and one nonfunctional pseudogen (Mitochondrial Ribosomal Protein L9 Pseudogene 1 [MRPL9P1]) were mapped (Figure 2). ZFHX4 belongs to the family of zinc-finger homeodomain transcription factors. This gene has been suggested to regulate neural and mesenchymal cell differentiation.5 Hemmi et al.6 demonstrated that ZFHX4 is expressed in the human adult brain, muscle, and liver and that Zfhx4 is expressed in the brain and in developing muscle during embryogenesis of the mouse. Because ZFHX4 is 90% homologous to mouse Zfhx4 in the genomic structure, amino acid sequence, and expression profile, the authors suggest that ZFHX4 plays a role in human neuronal and muscle differentiation comparable to that in mice. Levels of the ZFHX4 protein were high in the brainstem at embryonic and neonatal periods and in the midbrain and diencephalon in the neonatal rat brain. An immunolocalization study showed that postmitotic neurons in the brainstem were the major site of ZFHX4 localization, and the levels of protein varied depending on age and anatomical sites. Although ZFHX4 levels decreased after birth, the protein was detected in the mature neurons. Thus, it has been postulated that ZFHX4 participates in the regulation of neural cell maturation or region-specific differentiation of the brain.5 Further support for this is provided by the 52% homology of ZFHX4 with Zinc Finger Homeobox 3 (ZFHX3 [MIM 104155]), involved in early development of the central nervous system, maintenance of the undifferentiated state of myoblasts, and regulation of various genes important for cell cycle and growth.6 ZFHX4 was found to be disrupted in an individual with congenital bilateral isolated ptosis (MIM 178300) and a de novo balanced translocation t(1;8)(p34.3;q21.12).7 The breakpoint occurred in intron 4 of ZFHX4 and resulted in truncation of this gene and its protein product. This person did not have an intellectual disability or any other unusual morphological characteristic. Levels of the protein Zfhx4 were high in the midbrain, including the oculomotor nuclei, during embryogenesis in mice,5 and the oculomotor nerves innervate the levator palpebrae superioris that regulate the elevation of superior eyelid. Abnormal protein levels of ZFHX4 might cause congenital bilateral isolated ptosis.7 Linkage studies did show linkage to 8q21 in several families, but subsequent sequencing failed to show a mutation and no abnormal methylation of ZFHX4 was found either.8 The findings in the present individuals fit in well with the expression pattern of ZFHX4: the intellectual disability, disturbed eye movements, hearing and balance problems, and decreased sense of smell fit in with an altered ZFHX4 function in brain development, and the hypotonia fits in with ZFHX4 expression in muscle. Detailed muscular studies have not been performed in the present series of individuals and could show interesting results. The absence of major malformations (except for the eye findings; see below) elsewhere in the anatomy fits in with the expression pattern as well. However, the fact that the individual reported by McMullan et al.7 showed no intellectual disability or physical abnormalities other than the congenital bilateral isolated ptosis makes it difficult to establish a direct association between the haploinsufficiency for this gene and the cause of the intellectual disability observed in these individuals. Moreover a CNV that partially affects ZFHX4 and the microRNA located in the SRO has been reported in one apparently normal Yoruba individual from the HapMap Project.9 The significance of this observation is unclear, and the identification of the genomic cause underlying this clinically recognizable entity is difficult at this stage.
The deletions presented here show different sizes and proximal and distal breakpoints in all cases. Breakpoint junction sequencing analyses in individuals 3, 4, and 5 provided base pair resolution and showed that the breakpoints were located in unique sequences with no apparent homology (Figure S2). Thus nonallelic homologous recombination between clusters of flanking low-copy repeats or repetitive elements, the prevailing mechanism for recurrent rearrangements causing genomic disorders, seems not to be the rearrangement mechanism underlying 8q21 microdeletions.
In addition to the common 8q21 microdeletion phenotype, individuals 1 and 3 presented with severe eye abnormalities: severe microphthalmia, aphakia, cataracts, atrophy of optic nerves, corneal opacity, and sclerocornea. The individual reported by Taysi et al.10 who had an interstitial 8q12-q22 deletion also showed severe bilateral microphthalmia. One might hypothesize that individuals with 8q21 microdeletions have a contiguous gene syndrome because a gene for eye development might be present in the deleted region. Because individuals 2 and 5–7 have a deletion that overlaps with the deleted regions in individuals 1 and 3, this seems less likely. The 8q21 microdeletion might have a variable presentation, and the extreme end of the spectrum might present with eye development defects. Lastly, the 8q21 microdeletion might unmask recessive mutations on the nondeleted allele. It is of interest that two genes involved in eye morphogenesis map close to 8q21 deletions. Eyes Absent 1 (EYA1 [MIM 601653]), mapping 2 Mb upstream from the most proximal deletion breakpoint, encodes a member of the eyes-absent family of proteins playing a role in the developing kidneys, branchial arches, eyes, and ears. Mutations of this gene have been associated with sporadic cases of congenital cataracts and ocular anterior segment anomalies. The Growth Differentiation Factor 6 (GDF6 [MIM 601147]) is located 6 Mb downstream from the most distal breakpoint and encodes a member of the bone morphogenetic protein family and the TGF-beta superfamily of secreted signaling molecules. It is required for normal formation of some bones and joints in the limbs, skull, and axial skeleton. Mutations in this gene also result in congenital abnormalities in ocular development. Neither gene is directly disrupted or deleted by 8q21 rearrangements, but one cannot completely exclude a position effect through the loss of regulatory sequences because enhancer elements can regulate the expression of a gene at large distance, even with other uninvolved genes located within the genomic region separating the enhancer and the regulated gene.
Three of the eight 8q21.11 deletion individuals had corneal opacities. Seven of them had heterozygous deletions of Peroxisomal Biogenesis Factor 2 (PEX2 [MIM 170993]) in addition to ZFHX4 deletions. PEX2 encodes an integral peroxisomal membrane protein required for peroxisome biogenesis. Mutations in this gene result in Zellweger syndrome (ZS [MIM 214100]). Although carriers of heterozygous mutations in PEX2 are normal, phenotypic effects of heterozygous mutations in parents of four infants who had ZS have been reported,11 describing curvilinear, cortical lens opacities. One cannot exclude a contribution of PEX2 haploinsufficiency to the phenotype in some of the present individuals.
Parent-of-origin studies were carried out by segregation analysis of 18 sequence-tagged sites in samples from available parents of individuals 1–4, 6, and 7. Individual 8 belongs to a family in whom five members in four generations are affected, including his mother. Samples from other family members were not available. These studies revealed that the deletion occurred on the paternal chromosome in five individuals (1–4 and 6), and on the maternal chromosome in individual 7; individual 8 very likely inherited the deletion from his mother. This could suggest the existence of a parent-of-origin bias indicating that the mechanisms that lead to this rearrangement occur preferentially in the paternal germline, but the difference could also well be coincidence. Parent-of-origin effects in the phenotype were not observed; ZFHX4 was found not to be imprinted,8 and no other imprinted genes have been described within the regions deleted in our individuals.
Large interstitial deletions that also involve chromosome region 8q21 have been reported infrequently in the literature.10,12–14 Persons with these deletions showed a phenotype, similar to the that in the 8q21.11 microdeletion reported here, that included a high forehead, a wide nasal bridge, hypertelorism, down-turned corners of the mouth, micrognathia, abnormally shaped ears, a short neck, hypotonia, growth retardation, and intellectual disability. At the time of description, these deletions could be characterized only at the cytogenetic level, which hampers using the individuals for further delineation of the phenotype.
In summary, we have observed a characteristic common phenotype in eight individuals with a microdeletion of 8q21.11, suggesting that this could potentially represent a clinically recognizable entity, at least in retrospect. We could narrow down the SRO to a 539.77 kb interval encompassing ZFHX4, one microRNA of unknown function, and one nonfunctional pseudogen. Our observations suggest that the core phenotype of this microdeletion could be due to a haploinsufficient gene or genes within the minimal deletion. We cannot discard the existence within the SRO of a hitherto not-annotated gene. Characterization of additional 8q21 deletion patients might help refine this critical region, potentially allowing the identification of the underlying gene(s) by mutation analysis in phenotypically similar patients without a detectable deletion.
Acknowledgments
We would like to thank all the individuals and families for their participation in this study. This work was supported by a grant from the Fondo de Investigaciones Sanitarias (PI081187) of the Instituto de Salud Carlos III, Ministerio de Ciencia e Innovación of Spain.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Agilent eArray, https://earray.chem.agilent.com/earray/
Database of Genomic Variants, http://projects.tcag.ca/cgi-bin/variation/gbrowse/hg19/
Database of Genomic Variants Archive, http://www.ebi.ac.uk/dgva/
Database of Genomic Structural Variation (dbVAR), http://www.ncbi.nlm.nih.gov/dbvar/
DECIPHER, http://decipher.sanger.ac.uk
Genomic Imprint, http://www.geneimprint.com
UCSC Genome Browser (build 37), http://genome.ucsc.edu
International Mouse Strain Resource, http://www.findmice.org/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
References
- 1.Slavotinek A.M. Novel microdeletion syndromes detected by chromosome microarrays. Hum. Genet. 2008;124:1–17. doi: 10.1007/s00439-008-0513-9. [DOI] [PubMed] [Google Scholar]
- 2.Belligni E.F., Hennekam R.C. Familial occurrence of ptosis, nasal speech, prominent ears, hand anomalies and learning problems. Eur. J. Med. Genet. 2010;53:192–196. doi: 10.1016/j.ejmg.2010.03.009. [DOI] [PubMed] [Google Scholar]
- 3.Firth H.V., Richards S.M., Bevan A.P., Clayton S., Corpas M., Rajan D., Van Vooren S., Moreau Y., Pettett R.M., Carter N.P. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 2009;84:524–533. doi: 10.1016/j.ajhg.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nowakowska B., Stankiewicz P., Obersztyn E., Ou Z., Li J., Chinault A.C., Smyk M., Borg K., Mazurczak T., Cheung S.W. Application of metaphase HR-CGH and targeted Chromosomal Microarray Analyses to genomic characterization of 116 patients with mental retardation and dysmorphic features. Am. J. Med. Genet. A. 2008;146A:2361–2369. doi: 10.1002/ajmg.a.32475. [DOI] [PubMed] [Google Scholar]
- 5.Nogami S., Ishii Y., Kawaguchi M., Sakata N., Oya T., Takagawa K., Kanamori M., Sabit H., Obata T., Kimura T. ZFH4 protein is expressed in many neurons of developing rat brain. J. Comp. Neurol. 2005;482:33–49. doi: 10.1002/cne.20382. [DOI] [PubMed] [Google Scholar]
- 6.Hemmi K., Ma D., Miura Y., Kawaguchi M., Sasahara M., Hashimoto-Tamaoki T., Tamaoki T., Sakata N., Tsuchiya K. A homeodomain-zinc finger protein, ZFHX4, is expressed in neuronal differentiation manner and suppressed in muscle differentiation manner. Biol. Pharm. Bull. 2006;29:1830–1835. doi: 10.1248/bpb.29.1830. [DOI] [PubMed] [Google Scholar]
- 7.McMullan T.W., Crolla J.A., Gregory S.G., Carter N.P., Cooper R.A., Howell G.R., Robinson D.O. A candidate gene for congenital bilateral isolated ptosis identified by molecular analysis of a de novo balanced translocation. Hum. Genet. 2002;110:244–250. doi: 10.1007/s00439-002-0679-5. [DOI] [PubMed] [Google Scholar]
- 8.Nakashima M., Nakano M., Hirano A., Kishino T., Kondoh S., Miwa N., Niikawa N., Yoshiura K. Genome-wide linkage analysis and mutation analysis of hereditary congenital blepharoptosis in a Japanese family. J. Hum. Genet. 2008;53:34–41. doi: 10.1007/s10038-007-0214-6. [DOI] [PubMed] [Google Scholar]
- 9.Matsuzaki H., Wang P.H., Hu J., Rava R., Fu G.K. High resolution discovery and confirmation of copy number variants in 90 Yoruba Nigerians. Genome Biol. 2009;10:R125. doi: 10.1186/gb-2009-10-11-r125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taysi K., Noetzel M.J., Strauss A.W. Presumptive long arm deletion of chromosome 8: A new syndrome? Hum. Genet. 1979;51:49–53. doi: 10.1007/BF00278291. [DOI] [PubMed] [Google Scholar]
- 11.Hittner H.M., Kretzer F.L., Mehta R.S. Zellweger syndrome. Lenticular opacities indicating carrier status and lens abnormalities characteristic of homozygotes. Arch. Ophthalmol. 1981;99:1977–1982. doi: 10.1001/archopht.1981.03930020853008. [DOI] [PubMed] [Google Scholar]
- 12.Dallapiccola B., Santoro L., Trabace S., Ramenghi M., Mastroiacovo P., Gandini E. Deletion of the long arm of chromosome 8 resulting from a de novo translocation t(4;8) (q13;q213) Hum. Genet. 1977;38:125–130. doi: 10.1007/BF00527393. [DOI] [PubMed] [Google Scholar]
- 13.Donahue M.L., Ryan R.M. Interstitial deletion of 8q21—>22 associated with minor anomalies, congenital heart defect, and Dandy-Walker variant. Am. J. Med. Genet. 1995;56:97–100. doi: 10.1002/ajmg.1320560122. [DOI] [PubMed] [Google Scholar]
- 14.Fryburg J.S., Golden W.L. Interstitial deletion of 8q13.3—>22.1 associated with craniosynostosis. Am. J. Med. Genet. 1993;45:638–641. doi: 10.1002/ajmg.1320450524. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.