

Abstract
The exocrine pancreas synthesizes all the enzymes needed for intestinal breakdown of proteins, fats, and carbohydrates in our diet. Unfortunately, the proteases needed for the digestion of the meat we eat can, if inappropriately activated inside the acinar cells, also digest the pancreas itself as well as the surrounding tissues, which is what happens in the sometimes fatal human disease acute pancreatitis. The disease is currently untreatable, but significant progress has recently been made in understanding the fundamental processes initiating the pathological changes underlying pancreatic autodigestion. It is now clear that intracellular trypsin activation—a crucial step in pathogenesis—is due to excessive release of Ca2+ from intracellular stores, principally via two types of inositol trisphosphate receptor. The unexpected recent discovery of an intrinsic protective mechanism caused by intracellular calmodulin and, specifically, the finding that this protective effect can be boosted by a membrane-permeable Ca2+-like peptide are promising.
Introduction and context
Acute pancreatitis is a human disease in which digestive proenzymes normally synthesized in the pancreatic acinar cells are activated inside the cells (rather than after they have been secreted), digesting the pancreatic tissue and its surroundings rather than food in the gut. Pancreatitis, mostly caused by gallstones or excessive alcohol intake, is found acutely in up to 100 per 100,000 people per year and causes severe disease in 20% of those with the condition. It is frequently complicated by agonizing pain, extensive pancreatic necrosis, multiple organ failure, and prolonged hospitalization. The overall mortality in patients with acute pancreatitis is about 5% [1]. It has become accepted that repeated attacks of acute pancreatitis may lead to chronic pancreatitis [1] and that chronic pancreatitis carries a markedly increased risk for development of pancreatic cancer [2], which is the fifth most common cause of death through cancer, with only about 3–4% of patients surviving beyond 5 years [3]. Heavy alcohol consumption has been known for years to be a major risk factor for the development of chronic pancreatitis, and smoking has now also been implicated as an independent risk factor [4,5]. There is currently no specific therapy for acute pancreatitis, but recently there has been progress in understanding the involvement of intracellular calcium in the initial pathobiological processes, and this may provide new opportunities for development of preventive and therapeutic measures.
There is general agreement that intracellular protease activation is the crucial initiating step and that this process depends on substantial release of Ca2+ from internal stores followed by Ca2+ entry from the extracellular space. It is also clear that the initial biochemical event, namely protease activation, occurs at the same time as—and is in some way linked to—intracellular vacuolization; that is, the transformation of zymogen granules (electron-dense proenzyme-containing secretory vesicles) into empty-looking vacuoles. Vacuolization, like protease activation, is a Ca2+-dependent process. The demonstration that trypsin activation was initiated in post-exocytotic, endocytic vacuoles was the crucial finding linking protease (trypsin) activation and vacuolization [6].
Physiological stimulus–secretion coupling and pathological stimulus–protease activation
We know that the initiation of acute pancreatitis is a Ca2+-dependent process, but so is normal protease secretion. It is therefore important to differentiate between the physiological Ca2+ signal generation resulting in normal secretion and the abnormal (toxic) Ca2+ signal generation initiating pancreatitis. Exocytotic secretion of digestive proenzymes and the crucial fluid secretion needed to wash the secreted proteins out of the duct system into the gut are controlled by the neurotransmitter acetylcholine (released from parasympathetic nerve endings) and the hormone cholecystokinin. These agonists, at physiological concentrations, generate repetitive short-lasting elevations in the concentration of cytosolic Ca2+ (Ca2+ spikes) localized in the apical (granular) region of the cells. These local Ca2+ spikes are sufficient to activate exocytosis of digestive enzymes as well as fluid secretion [7]. These findings—specifically, the link between the presence of functional cholecystokinin receptors and Ca2+ signalling—originally made in studies on mouse pancreatic acinar cells, have been confirmed in a detailed study of normal human pancreatic acinar cells [8]. High (unphysiological) concentrations of acetylcholine or cholecystokinin, as well as various pathological stimuli causing acute pancreatitis, evoke sustained global elevations of cytosolic [Ca2+]. Such signals do not result in sustained secretion of proteases, but do—in contrast to physiological stimulation—cause intracellular trypsin activation [6].
Another very important aspect to consider with regard to pathogenesis is energy production. Repetitive cytosolic Ca2+ spikes cause repeated spikes of mitochondrial [Ca2+] elevation that, in turn, activate Ca2+-dependent Krebs-cycle dehydrogenases, generating mitochondrial ATP production. In contrast, a sustained elevation of cytosolic [Ca2+] only gives rise to one initial burst of mitochondrial [Ca2+] elevation and therefore only one transient period of ATP generation [7]. As ATP is required for the secretory process, this is undoubtedly one reason for the lack of protease secretion at high (unphysiological) levels of stimulation as well as in acute pancreatitis.
Bile acids induce acute pancreatitis
A frequent cause of acute pancreatitis is gallstones, which are thought to cause disease by blocking the pancreatic duct or obstructing a common (bile–pancreatic) channel. This latter mechanism would allow reflux of bile into the pancreas and cause pancreatic injury, although the importance of this particular mechanism has been debated [9]. In any case, it has been shown that transporter-mediated bile acid uptake causes Ca2+-dependent cell death in pancreatic acinar cells in vitro [10]. The primary effect of intracellular bile acids is to release Ca2+ from both the endoplasmic reticulum and acid stores in the apical granular region through activation of inositol trisphosphate (IP3) and ryanodine receptors (intracellular calcium channels) [11], inducing either apoptosis or necrosis. The intracellular ATP level seems to be crucial in determining which type of cell death occurs. This can be demonstrated in patch clamp whole-cell recording experiments (where the cell interior is in direct contact with a large volume of pipette solution), which show that the presence of ATP in the solution leads to bile acids causing apoptosis as opposed to necrosis [12].
Alcohol: Is it dangerous for the pancreas?
Although the risk of developing pancreatitis increases with increasing alcohol intake, it is nevertheless the case that only a minority (<10%) of those drinking excessive amounts of alcohol develop pancreatitis [5]. How can this be explained? Although alcohol usually has only modest effects on cellular Ca2+ homeostasis, even in very high concentrations, results of work on isolated normal pancreatic acinar cells show that a combination of alcohol and fatty acids (fatty acid ethyl esters) causes massive intracellular Ca2+ release and acute trypsin activation. Although Ca2+ release occurs from both the endoplasmic reticulum and acid stores in the granular part of the cells, it is the Ca2+ liberation from the acid stores that is principally responsible for the intracellular trypsin activation [13]. The mechanism involves specific intracellular Ca2+ release channels (IP3 receptors of types 2 and 3). In fact, deletion of the genes for these channels prevents the toxic action of fatty acid ethyl esters [13] (Figure 1). High concentrations of long-chain fatty acids in the plasma markedly increase the risk for development of pancreatitis, and slowly increase the global cytosolic [Ca2+] [14,15]. This is mainly due to inhibition of mitochondrial energy production. The reduced level of intracellular ATP prevents full Ca2+ pump function (both in the endoplasmic reticulum membrane and in the plasma membrane), limiting the capacity for getting rid of Ca2+ accumulated in the cytosol [6]. From these studies on isolated cells, it would appear that the combination of alcohol and fat-rich meals would increase the risk of developing acute pancreatitis. It would be highly desirable to test this by conducting careful epidemiological studies.
Figure 1. Intracellular trypsin activation following stimulation with palmitoleic acid ethyl ester (POAEE) is initiated in the apical granular pole of an isolated permeabilized pancreatic acinar cell.
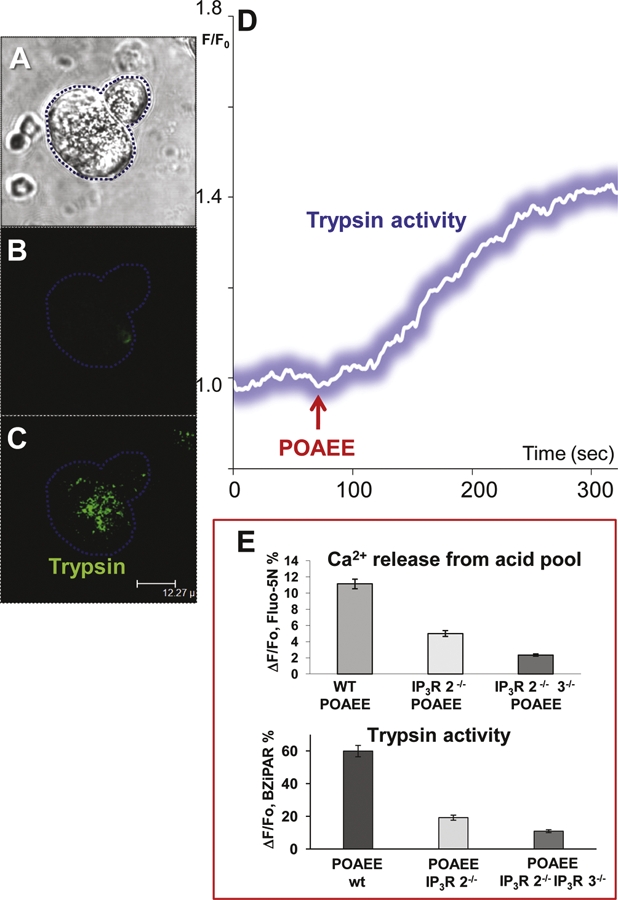
The level of trypsin activity is correlated with the degree of Ca2+ release from acid stores in the granular apical pole through inositol trisphosphate (IP3) receptors of types 2 and 3. (A) Transmitted light image showing two acinar cells. The left cell has been two-photon permeabilized. (B) and (C) Fluorescence images showing (in C) the initial localization of trypsin activity after stimulation with POAEE (the probe BZiPAR becomes fluorescent when trypsin cleaves the two oligopeptide side chains). (B) Before and (C) after start of stimulation with POAEE (100 μM). (D) The time course of the increase in intracellular trypsin activity following start of stimulation with 100 μM POAEE. (E) Results from experiments in which Ca2+ release from the acid granular pole of permeabilized cells and trypsin activation in wild-type (WT) mice were compared with results from mice in which type 2 IP3 receptors had been deleted (IP3R 2-/-) and from mice in which both types 2 and 3 IP3 receptors had been deleted (IP3R 2-/- 3-/-). Adapted from Gerasimenko et al., 2009 [13].
To summarize, although alcohol (ethanol) itself mostly has only minor acute effects on the pancreatic acinar cells, there is a minority of cells that produce large sustained Ca2+ signals when exposed to ethanol [6]. Recent data show that although ethanol has the capacity to elicit the release of substantial amounts of Ca2+ from intracellular stores, normal intact cells have an in-built protective mechanism, discussed below [16] (Figure 2).
Figure 2. The inhibitory effect of intracellular calmodulin (CaM) on ethanol-induced intracellular Ca2+ release and trypsin activation in permeabilized pancreatic acinar cells.

(A) Typical traces from two experiments in which changes in [Ca2+]store were assessed after stimulation with ethanol. In the absence of CaM, 10 mM ethanol (EtOH) evokes a clear decrease in [Ca2+]store, whereas this ethanol concentration fails to elicit any release when CaM (2.5 μM) is added to the external solution (in direct contact with the intracellular solution through the pore generated by two-photon permeabilization). A much higher ethanol concentration (100 mM) can still evoke Ca2+ release. (B) CaM inhibits ethanol-induced trypsin activation in a manner very similar to its protective effect against intracellular Ca2+ release. In addition, the lowest trace demonstrates that the Ca2+-like peptide CALP-3—in the presence of CaM—abolishes the trypsin-activating effect of even a very high ethanol concentration (100 mM). Adapted from Gerasimenko et al., 2011 [16].
Activation of the ubiquitous calcium-binding protein calmodulin protects against alcohol-induced intracellular Ca2+ release and trypsin activation
Recent insights have shed light on how alcohol-induced Ca2+ release could be prevented. It has been found that when the Ca2+-binding protein calmodulin is washed out of cells, alcohol itself has a strong and acute toxic effect on the acinar cells. Re-admission of calmodulin (at a normal intracellular concentration) has a protective effect. It is particularly exciting that recent data show that a membrane-permeable small peptide activator of calmodulin (the Ca2+-like peptide known as CALP-3), when added to the outside of isolated cells, prevents the toxic actions of even very high alcohol concentrations [16] (Figure 2), as this suggests it may have potential as a therapeutic agent to reduce alcohol-induced pancreatic damage.
Cell death pathways
Pancreatitis is characterized by cell death, but the prognosis depends a great deal on which cell death process occurs [17,18]. Apoptosis is the “physiological” cell death mechanism and occurs without losing the integrity of the plasma membrane, whereas necrosis results in loss of cell constituents to the interstitial fluid, promoting inflammation. Apoptosis requires energy in the form of ATP, so if mitochondrial function is markedly impaired by complete depolarization of the inner mitochondrial membrane, the only cell death mechanism available is necrosis (Figure 3).
Figure 3. Simplified schematic diagram indicating the two major cell death pathways employed by pancreatic acinar cells when challenged by various pathological stimuli.

The arrows at the bottom of the figure labeled +ATP and –ATP indicate that the balance between apoptosis and necrosis is to some extent dictated by the presence or absence of ATP. Δψm, electrical potential difference across inner mitochondrial membrane; CCK, cholecystokinin; ER, endoplasmic reticulum; FAEE, fatty acid ethyl ester; ROS, reactive oxygen species, ZG, zymogen granule. Adapted from Criddle et al., 2007 [17].
Potential therapeutic avenues
The experimental results demonstrating that the crucial intracellular trypsin activation is Ca2+-dependent, and promoted by excessive Ca2+ release from internal stores as well as subsequent entry of Ca2+ from the interstitial fluid, suggest that inhibition of Ca2+ release from internal stores and/or inhibition of Ca2+ entry may be helpful in limiting the damage to pathological stimuli such as alcohol and alcohol metabolites, as well as bile acids. Indeed, caffeine, which has been shown to inhibit opening of IP3 receptor channels [6,7], reduces cytosolic Ca2+ signal generation in response to fatty acid ethyl esters and has also been shown to reduce the probability of ethanol-induced pancreatitis in a clinical study [19]. However, due to the relatively low affinity of caffeine for the IP3 receptors and its activating effect on ryanodine receptors, the therapeutic potential for caffeine is limited. The recent discovery that intracellular calmodulin has an intrinsic protective effect against alcohol-induced trypsin activation and, in particular, that this protection can be boosted by CALP-induced activation of calmodulin [16] deserves further study. Inhibition of Ca2+ entry channels of the CRAC (Ca2+ release-activated Ca2+) type is another potentially interesting therapy [20] that has not yet been assessed in the pancreas, but could be powerful if Ca2+ entry through CRAC channels turns out to be the dominant pathway in the acinar cells.
Conclusions
In conclusion, the main points that have come out of the recent advances in the field are that: (a) excessive cytosolic Ca2+ loading initiates the intracellular protease activation that leads to acute pancreatitis; (b) the excessive entry of Ca2+ into the cytosol is primarily and principally due to release of Ca2+ from acid Ca2+ stores in the granular part of the acinar cells mediated via IP3 receptors of types 2 and 3; (c) an intracellular Ca2+-binding protein, calmodulin, exerts a protective effect against alcohol-related pancreatitis by reducing the probability of opening of the IP3 receptor channels; and (d) activation of calmodulin by a membrane-permeable Ca2+-like peptide boosts the protective effect of calmodulin against alcohol-induced intracellular protease activation. Hopefully by understanding more about the etiology of this disease, we will come closer to providing better preventative and therapeutic methods to relieve the suffering of those afflicted with this condition.
Acknowledgments
The experimental work in the authors’ laboratory was supported by a Programme Grant from The Medical Research Council (UK). OHP is a Medical Research Council Professor.
Abbreviations
- CALP
calcium-like peptide
- CRAC
Ca2+ release-activated Ca2+
- IP3
inositol trisphosphate
Competing interests
The authors declare that they have no competing interests.
The electronic version of this article is the complete one and can be found at: http://f1000.com/reports/m/3/15
References
- 1.Pandol SJ, Saluja AK, Imrie CW, Banks PA. Acute pancreatitis: bench to bedside. Gastroenterology. 2007;132:1127–51. doi: 10.1053/j.gastro.2007.01.055. [DOI] [PubMed] [Google Scholar]
- 2.Maisonneuve P, Lowenfels AB. Chronic pancreatitis and pancreatic cancer. Dig Dis. 2002;20:32–7. doi: 10.1159/000063165. [DOI] [PubMed] [Google Scholar]
- 3.Everhart JE, Ruhl CE. Burden of digestive disease in the United States part III: liver, biliary tract, and pancreas. Gastroenterology. 2009;136:1134–44. doi: 10.1053/j.gastro.2009.02.038. [DOI] [PubMed] [Google Scholar]
- 4.Yadav D, Whitcomb DC. The role of alcohol and smoking in pancreatitis. Nat Rev Gastroenterol Hepatol. 2010;7:131–45. doi: 10.1038/nrgastro.2010.6. [DOI] [PubMed] [Google Scholar]
- 5.Pandol SJ, Lugea A, Mareninova OA, Smoot D, Gorelick FS, Gukovskaya AS, Gukovsky I. Investigating the pathobiology of alcoholic pancreatitis. Alcohol Clin Exp Res. 2011;35:830–7. doi: 10.1111/j.1530-0277.2010.01408.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petersen OH, Tepikin AV, Gerasimenko JV, Gerasimenko OV, Sutton R, Criddle DN. Fatty acids, alcohol and fatty acid ethyl esters: toxic Ca2+ signal generation and pancreatitis. Cell Calcium. 2009;45:634–42. doi: 10.1016/j.ceca.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 7.Petersen OH, Tepikin AV. Polarized calcium signalling in exocrine gland cells. Annu Rev Physiol. 2008;70:273–99. doi: 10.1146/annurev.physiol.70.113006.100618. [DOI] [PubMed] [Google Scholar]
- 8.Murphy JA, Criddle DN, Sherwood M, Chvanov M, Mukherjee R, McLaughlin E, Booth D, Gerasimenko JV, Raraty MG, Ghaneh P, Neoptolemos JP, Gerasimenko OV, Tepikin AV, Green GM, Reeve JR, Petersen OH, Sutton R. Direct activation of cytosolic Ca2+ signaling and enzyme secretion by cholecystokinin in human pancreatic acinar cells. Gastroenterology. 2008;135:632–41. doi: 10.1053/j.gastro.2008.05.026. [DOI] [PubMed] [Google Scholar]
- 9.Lerch MM, Aghdassi AA. The role of bile acids in gallstone-induced pancreatitis. Gastroenterology. 2010;138:429–33. doi: 10.1053/j.gastro.2009.12.012. [DOI] [PubMed] [Google Scholar]
- 10.Kim JY, Kim KH, Lee JA, Namkung W, Sun AQ, Ananthanarayanan M, Suchy FJ, Shin DM, Muallem S, Lee MG. Transporter-mediated bile acid uptake causes Ca2+-dependent cell death in rat pancreatic acinar cells. Gastroenterology. 2002;122:1941–53. doi: 10.1053/gast.2002.33617. [DOI] [PubMed] [Google Scholar]; F1000 Factor 6Evaluated by Ole Petersen 21 Jul 2011
- 11.Gerasimenko JV, Flowerdew SE, Voronina SG, Sukhomlin TK, Tepikin AV, Petersen OH, Gerasimenko OV. Bile acids induce Ca2+ release from both the endoplasmic reticulum and acidic intracellular calcium stores through activation of inositol trisphosphate receptors and ryanodine receptors. J Biol Chem. 2006;281:40154–63. doi: 10.1074/jbc.M606402200. [DOI] [PubMed] [Google Scholar]; F1000 Factor 6Evaluated by Antony Galione 10 Nov 2006
- 12.Booth DM, Murphy JA, Mukherjee R, Awais M, Neoptolemos JP, Gerasimenko OV, Tepikin AV, Petersen OH, Sutton R, Criddle DN. Reactive oxygen species induced by bile acid induce apoptosis and protect against necrosis in pancreatic acinar cells. Gastroenterology. 2011;140:2116–25. doi: 10.1053/j.gastro.2011.02.054. [DOI] [PubMed] [Google Scholar]
- 13.Gerasimenko JV, Lur G, Sherwood MW, Ebisui E, Tepikin AV, Mikoshiba K, Gerasimenko OV, Petersen OH. Pancreatic protease activation by alcohol metabolite depends on Ca2+ release via acid store IP3 receptors. Proc Natl Acad Sci U S A. 2009;106:10758–63. doi: 10.1073/pnas.0904818106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y, Sternfeld L, Yang F, Rodriguez JA, Ross C, Hayden MR, Carriere F, Liu G, Hofer W, Schulz I. Enhanced susceptibility to pancreatitis in severe hypertriglyceridemic lipoprotein lipase deficient mice and agonist-like function of pancreatic lipase in pancreatic acinar cells. Gut. 2009;58:422–30. doi: 10.1136/gut.2007.146258. [DOI] [PubMed] [Google Scholar]; F1000 Factor 6Evaluated by Ole Petersen 21 Jul 2011
- 15.Yang F, Wang Y, Sternfeld L, Rodriguez JA, Ross C, Hayden MR, Carierre F, Liu G, Schulz I. The role of free fatty acids, pancreatic lipase and Ca2+ signalling in injury of isolated acinar cells and pancreatitis model in lipoprotein lipase – deficient mice. Acta Physiol. 2009;195:13–28. doi: 10.1111/j.1748-1716.2008.01933.x. [DOI] [PubMed] [Google Scholar]; F1000 Factor 6Evaluated by Ole Petersen 21 Jul 2011
- 16.Gerasimenko JV, Lur G, Ferdek P, Sherwood MW, Ebisui E, Tepikin AV, Mikoshiba K, Petersen OH, Gerasimenko OV. Calmodulin protects against alcohol-induced trypsinogen activation elicited via Ca2+ release through IP3 receptors. Proc Natl Acad Sci U S A. 2011;108:5873–8. doi: 10.1073/pnas.1016534108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Criddle DN, Gerasimenko J, Baumgartner HK, Jaffar M, Voronina SG, Sutton R, Petersen OH, Gerasimenko OV. Calcium signalling and pancreatic cell death: apoptosis or necrosis? Cell Death Differ. 2007;14:1285–94. doi: 10.1038/sj.cdd.4402150. [DOI] [PubMed] [Google Scholar]
- 18.Gukovskaya AS, Gukovsky I. Which way to die: the regulations of acinar cell death in pancreatitis by mitochondria, Ca2+, and reactive oxygen species. Gastroenterology. 2011;140:1876–80. doi: 10.1053/j.gastro.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petersen OH, Sutton R. Ca2+ signalling and pancreatitis: effects of alcohol, bile and coffee. Trends Pharmacol Sci. 2006;27:113–20. doi: 10.1016/j.tips.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 20.Di Capite JL, Bates GJ, Parekh AB. Mast cell CRAC channel as a novel therapeutic target in allergy. Curr Opin Allergy Clin Immunol. 2011;11:33–8. doi: 10.1097/ACI.0b013e32834232b0. [DOI] [PubMed] [Google Scholar]