

Abstract
A metallo-β-lactamase-type alkylsulfatase was found to catalyze the enantioselective hydrolysis of sec-alkylsulfates with strict inversion of configuration. This catalytic event, which does not have an analog in chemocatalysis, yields homochiral (S)-configurated alcohols and nonreacted sulfate esters. The latter could be converted into (S)-sec-alcohols as the sole product in up to >99% ee via a chemoenzymatic deracemization protocol on a preparative scale.
Sulfatases are a heterogenic group of enzymes which catalyze the cleavage of the sulfate ester bond yielding the corresponding alcohol and hydrogen sulfate.1−4 In contrast to the majority of hydrolases,(5) which do not alter the stereochemistry of the substrate during catalysis, the stereochemical course of sulfate ester hydrolysis may proceed via cleavage of the S–O or the C–O bond of a sec-alkyl sulfate going in hand with retention or inversion of the stereogenic carbon atom, respectively (Scheme 1).(1)
Scheme 1. Stereochemical Course of Retaining and Inverting Alkylsulfatases.

Recently, sulfatases were reclassified based on their mode of catalysis:(6) Aryl and carbohydrate sulfatases act on sulfated carbohydrates and steroids.(4) These enzymes possess a highly conserved -C/S-X-P-X-A-X4-T-G- consensus motif,(2) which codes for a Cys or Ser residue within the active site. The latter is posttranslationally modified into a hydrated α-formylglycine moiety, which attacks the S-atom of the sulfate ester going in hand with S–O bond cleavage while the absolute configuration at C is retained.(7) On the other hand, sulfatases belonging to the Fe2+-dependent group of dioxygenases oxidatively cleave a sulfate ester at the expense of α-ketoglutarate as an electron acceptor to yield an aldehyde and inorganic sulfate, leading to destruction of the stereogenic center at C.(8) The third class of sulfatases is related to metallo-β-lactamases and (to date) harbors only a single member: Sodium dodecyl sulfatase (SdsA1).(6) Since SDS is an achiral prim-sulfate ester, the stereochemical course of SdsA1-hydrolysis is not ‘visible’. However, since the nucleophilic water molecule (W2 in the crystal structure 2cfu) strongly interacts with the sulfur atom of the substrate surrogate inhibitor 1-decanesulfonate, retention at carbon was assumed.(6)
Inverting alkylsulfatases(9) were previously studied in Pseudomonas C12B,(10)Comamonas terrigena,(11) and Rhodococcus ruber DSM 44541,(12) but due to the lack of biochemical and structural data the mechanism of inverting alkylsulfatases remained unknown. In the search for a well-characterized stable sec-alkylsulfatase, which would allow the design of a deracemization process for sec-alcohols via enantioconvergent hydrolysis of the corresponding sulfate esters, an extended whole-cell screening for sec-alkylsulfatase activity was conducted, which revealed Pseudomonas sp. DSM 6611 as the most promising candidate.(13)
Chromatographic protein purification followed by tryptic digestion and peptide mass fingerprinting allowed assignment of the obtained peptide masses to the predicted open reading frames using the full genomic sequence of the strain.(14) The protein band with the lowest mobility on SDS-PAGE (termed Pisa1, identified by de novo sequencing and peptide matching) surprisingly turned out to be a homologue of SdsA1.(15) The Pisa1 gene was amplified by PCR, cloned, and expressed in an E. coli BL21 strain with a C-terminal hexa-histidine tag. Pisa1 displayed the desired catalytic properties: (R)-2-Octyl sulfate (>99% ee) was quantitatively hydrolyzed to yield (S)-2-octanol (>99% ee) through strict inversion of configuration, whereas the (S)-enantiomer was completely unreactive. Hydrolysis of rac-2-octyl sulfate ceased at 50% conversion to furnish a homochiral product mixture of (S)-2-octanol and unreacted (S)-2-octyl sulfate, indicating perfect enantioselectivity (E >200).
Although Pisa1 shares a 44% sequence identity with SdsA1 the substrate preference of both proteins differs significantly: Based on kcat and KM values, SdsA1 has a 150-fold affinity for the prim-sulfate ester 1a, whereas Pisa1 has a pronounced (190-fold) opposite preference for the sec-sulfate ester analog 2a. Hence, Pisa1 is the first inverting sec-alkylsulfatase that is characterized on a molecular level.15,16
The stereochemical course of hydrolysis for both enzymes was investigated in detail using unlabeled 1-octyl- (1a) and rac-2-octyl sulfate (2a) in 18O-enriched buffer (label >98%, Scheme 2, part A). GC-MS analysis of the formed 1-octanol (1b) and (S)-2-octanol (2b, ee >99%) showed complete incorporation of the 18O-label in the product within analytical limits, proving C–O bond cleavage in both cases.
Scheme 2. Proof of Inversion for Pisa1 and SdsA1 via 18O-Labeling (A), Substrate Spectrum of Pisa1 (B), and Deracemization of rac-2a (C).

The nucleophilic attack of (formal) [OH–] at the chiral carbon atom of an alkyl sulfate ester as exerted by alkylsulfatases SdsA1 and Pisa1 leading to inversion of configuration at C is a remarkable catalytic event, which does not have a direct counterpart in chemical catalysis. The latter event would generate SO42–, which is a very poor leaving group.(17) Hence, inverting nucleophilic hydrolysis of sulfate esters by hydroxide, acetate, or methoxide proceeds extremely slowly and is not feasible for preparative purposes.18,19 In order to facilitate the departure of the sulfate moiety, it has to be converted into a good leaving group, i.e. HSO4– rather than SO42–.(20) Consequently, acid-catalyzed sec-sulfate ester hydrolysis is a fast process, which proceeds through retention at C.21,18 Based on the crystal structure of Pisa1,(16) an acid–base-type mechanism for the inverting enzymatic alkyl sulfate hydrolysis can be proposed (Figure 1).
Figure 1.

Schematic proposal for an acid–base mechanism for alkyl sulfate ester hydrolysis catalyzed by Pisa1 (His179/181/184/355 coordinating the Zn2+ ions were omitted for clarity).
(i) The anionic substrate is positioned by a sulfate binding site consisting of positively charged (Arg328/323) and H-bonding residues (Asn318, Thr321); (ii) a highly conserved 'nucleophile site' consisting of a binuclear Zn2+-binding cluster (Asp310/183, His179/181, Glu291) activates a water molecule to provide the nucleophile [OH–], which attacks the carbon atom bearing the sulfate ester moiety. The latter is facilitated through (iii) concomitant protonation of the liberated inorganic sulfate (presumably by Tyr417), to yield HSO4– as a good leaving group. The hydrophobic binding sites of Pisa1 and SdsA1 differ significantly with respect to the relative size of residues, which most likely accounts for the opposite substrate specificity concerning prim- versus sec-alkyl sulfate esters.
The substrate spectrum of Pisa1 proved to encompass a range of linear, branched, or cyclic ω-1 to ω-3 sec-alkyl sulfates (Table 1, Scheme 2, part B). Straight-chain (ω-1)- and (ω-2)-sulfate esters (2a–5a) were resolved with perfect enantioselectivity; the (ω-3)-analog 6a bearing two C3/C4-chains of similar size gave a respectable E-value of E = 10. Substrates bearing branched (7a), aromatic (8a, 9a), or cyclic side chains (10a) gave excellent results. In contrast to the previously employed resting whole cell preparation of Pseudomonas sp. DSM 6611(13) the reaction rates were far superior and conversion values usually reached the theoretical limit of 50%, which is required for a deracemization process.The differences in rates and selectivities are presumably due to competing sulfatases in whole cells. Most remarkably, substrates rac-11a and rac-12a, derived from propargylic alcohols bearing a synthetically useful terminal acetylene unit, were transformed with the same perfect stereoselectivity.
Table 1. Substrate Spectrum of Pisa1 Compared to Results with Whole Cells of Pseudomonas sp. DSM 6611.
|
Ps. sp. DSM 6611a |
Pisa1 |
|||||
|---|---|---|---|---|---|---|
| substrate | R1 | R2 | c [%] | Eb | c [%] | Eb |
| rac-2a | CH3 | n-C6H13 | 21 | >200 | 50 | >200 |
| rac-3a | CH3 | n-C5H11 | 17 | >200 | 50 | >200 |
| rac-4a | CH3 | n-C7H15 | 7 | >200 | 50 | >200 |
| rac-5a | C2H5 | n-C5H11 | 18 | >200 | 50 | >200 |
| rac-6a | n-C3H7 | n-C4H9 | 20 | 6 | 57 | 10 |
| rac-7a | CH3 | (CH2)2CH=CMe2 | 9 | >200 | 50 | >200 |
| rac-8a | CH3 | CH2Ph | <1 | n.d. | 30 | >200 |
| rac-9a | CH3 | (CH2)2Ph | 15 | >200 | 50 | >200 |
| rac-10a | CH3 | c-C6H11 | 5 | >200 | 10 | >200 |
| rac-11a | HC≡C | n-C4H9 | n.i. | – | 50 | >200 |
| rac-12a | HC≡C | n-C5H11 | n.i. | – | 50 | >200 |
Whole resting cells of Pseudomonas sp. DSM 6611, data from ref (13).
Enantioselectivity expressed as Enantiomeric Ratio (E-value); n.d. = not determined due to exceedingly low conversion; n.i. = not investigated.
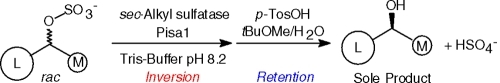
With the highly active and stereoselective inverting sec-alkylsulfatase Pisa1 in hand, the feasibility of a deracemization protocol for sec-alcohols could be demonstrated as follows (Scheme 2, part C): Treatment of rac-2-octyl sulfate (2a, 1 g) with Pisa1 in aqueous buffer gave equimolar amounts of (S)-2b and (S)-2a. The latter was hydrolyzed under acidic conditions (p-TosOH in tBuOMe/H2O/1,4-dioxane)(19) to yield enantiopure (S)-2b (>99% ee) in 87% isolated yield from the racemate (0.49 g).
Acknowledgments
This study was financed by the Austrian Science Fund (FWF, Project P18689 and DK Molecular Enzymology W9). The authors would like to express their cordial thanks to G. Rechberger, K. Zangger, K. Gruber, and G. Steinkellner (University of Graz) for their valuable support.
Supporting Information Available
Synthesis of sulfate esters, NMR-spectra, cloning, expression and purification of Pisa1 and SdsA1, H218O labeling experiments, determination of enantiomeric excess and absolute configuration, and preparative scale deracemization.This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Gadler P.; Faber K. Trends Biotechnol. 2006, 25, 83–88. [DOI] [PubMed] [Google Scholar]
- Kertesz M. A. FEMS Microbiol. Rev. 2000, 24, 135–175. [DOI] [PubMed] [Google Scholar]
- Bojarova P.; Williams S. J. Curr. Opin. Chem. Biol. 2008, 12, 573–581. [DOI] [PubMed] [Google Scholar]
- Hanson S. R.; Best M. D.; Wong C.-H. Angew. Chem., Int. Ed. 2004, 43, 5736–5763. [DOI] [PubMed] [Google Scholar]
- Bornscheuer U. T.; Kazlauskas R. J.. Hydrolases in Organic Synthesis; Wiley-VCH: Weinheim, 2005. [Google Scholar]
- Hagelueken G.; Adams T. M.; Wiehlmann L.; Widow U.; Kolmar H.; Tümmler B.; Heinz D. W.; Schubert W.-D. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 7631–7636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boltes I.; Czapinska H.; Kahnert A.; von Bülow R.; Dierks T.; Schmidt B.; von Figura K.; Kertesz M. A.; Uson I. Structure 2001, 9, 483–491. [DOI] [PubMed] [Google Scholar]
- Müller I.; Kahnert A.; Pape T.; Sheldrick G. M.; Meyer-Klaucke W.; Dierks T.; Kertesz M. A.; Uson I. Biochemistry 2004, 43, 3075–3088. [DOI] [PubMed] [Google Scholar]
- Bartholomew B.; Dodgson K. S.; Matcham G. W. J.; Shaw D. J.; White G. F. Biochem. J. 1977, 165, 575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw D. J.; Dodgson K. S.; White G. F. Biochem. J. 1980, 187, 181–196. [DOI] [PMC free article] [PubMed] [Google Scholar]; White G. F. Appl. Microbiol. Biotechnol. 1991, 35, 312–316. [DOI] [PubMed] [Google Scholar]
- Fitzgerald J. W.; Dodgson K. S.; Matcham G. W. J. Biochem. J. 1975, 149, 477–480.1180908 [Google Scholar]; Matcham G. W. J.; Dodgson K. S.; Fitzgerald J. W. Biochem. J. 1977, 167, 723–729. [DOI] [PMC free article] [PubMed] [Google Scholar]; Barrett C. H.; Dodgson K. S.; White G. F. Biochem. J. 1980, 191, 467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogorevc M.; Faber K. Appl. Environ. Microbiol. 2003, 69, 2810–2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadler P.; Faber K. Eur. J. Org. Chem. 2007, 5527–5530. [Google Scholar]
- Angelova M.; Gadler P.; Kayer H.; Krempl P.; Gülly C.; Faber K.; Macheroux P.; Thallinger G. Manuscript in preparation.
- The sequence was submitted to the European Nucleotide Archive (accession #: FR850678 – confidential until 01.07.2011). No measurable sequence similarities of Pisa1 with other sulfatases, such as the α-ketoglutarate-dependent alkylsulfatase AtsK from Pseudomonas putida, the arylsulfatase PAS from P. aeruginosa, and the human steroid sulfatases HArsA, N-acetylgalactosamin 4-sulfatase, or cerebroside-3-sulfate sulfatase, could be detected (≤10%).
- The coordinates of the crystal structure of Pisa1 (2.7 Å resolution) are available from the PDB (code 2YHE); A detailed study on the structural and mechanistic aspects of Pisa1 will be published: Knaus T.; Schober M.; Kepplinger B.; Gadler P.; Macheroux P.; Faber K.; Wagner U. G. Manuscript in preparation.
- Being the anion of the weak acid HSO4– (pKa = +1.9 to +2.7).
- Batts B. D. J. Chem. Soc. (B) 1966, 551–555. [Google Scholar]; Burwell R. L. Jr. J. Am. Chem. Soc. 1952, 74, 1462–1466. [Google Scholar]
- Wallner S. R.; Nestl B.; Faber K. Tetrahedron 2005, 61, 1517–1521. [Google Scholar]
- Based on the pKa of methyl monosulfate, which was calculated/estimated as pKa −8.4 or pKa −3.4, thus protonation of a sulfate ester requires a strong acid of pKa ca. <2; see: ; Klages F.; Jung H. A.; Hegenberg P. Chem. Ber 1966, 99, 1704–1711. [Google Scholar]; Fulford J. E.; Dupuis J. W.; March R. E. Can. J. Chem. 1978, 56, 2324–2330. [Google Scholar]
- Depending on the conditions, acid-catalyzed sulfate ester hydrolysis may be plagued by racemization,(17) which can be suppressed in the presence of 1,4-dioxane as mediator; see: Batts B. D. J. Chem. Soc. (B) 1966, 547–551. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.