

Abstract
Autophagy is an evolutionarily conserved process whereby cytoplasm and cellular organelles are degraded in lysosomes for amino acid and energy recycling. Autophagy is a survival pathway activated in response to nutrient deprivation and other stressful stimuli, such as metabolic stress and exposure to anticancer drugs. However, autophagy may also result in cell death, if it proceeds to completion. Defective autophagy is implicated in tumorigenesis, as the essential autophagy regulator beclin 1 is monoallelically deleted in human breast, ovarian and prostate cancers, and beclin 1+/− mice are tumor-prone. How autophagy suppresses tumorigenesis is under intense investigation. Cell-autonomous mechanisms, involving protection of genome integrity and stability, and a non-cell-autonomous mechanism, involving suppression of necrosis and inflammation, have been discovered so far. The role of autophagy in treatment responsiveness is also complex. Autophagy inhibition concurrently with chemotherapy or radiotherapy has emerged as a novel approach in cancer treatment, as autophagy-competent tumor cells depend on autophagy for survival under drug- and radiation-induced stress. Alternatively, autophagy stimulation and preservation of cellular fitness bymaintenance of protein and organelle quality control, suppression of DNA damage and genomic instability, and limitation of necrosis-associated inflammation may play a critical role in cancer prevention.
Keywords: Autophagy, Tumorigenesis, Tumor suppression, Autophagy regulation, Cancer therapy, Autophagy modulation
1. Introduction
Macroautophagy, hereafter referred to as autophagy, is a cellular self-consumption process characterized by sequestration of bulk cytoplasm, long-lived proteins and cellular organelles in double-membrane vesicles called autophagosomes, which are delivered to and degraded in lysosomes [1]. Basal autophagy plays an important role in cellular homeostasis by degrading excessive, damaged and/or aged proteins and organelles, and thus maintaining quality control of essential cellular components [2,3]. Defective autophagy has been implicated in the pathogenesis of diverse disease states, such as myopathy [4], neuronal degeneration [5], microbial infection [6], inflammatory bowel disease [7,8], aging [9] and cancer [10]. In addition to its basal function, autophagy is readily induced in response to nutrient deprivation [11–14], metabolic stress [15–17], endoplasmic reticulum (ER)-stress [18,19], radiation [20] and anticancer drugs [21–24]. The role of autophagy as an alternate energy source, and thus as a temporary survival mechanism, under stressful conditions is well recognized [25]. The presence of autophagosomes in dying cells raises the possibility that autophagymay also play an active role in cell death [26]. However, in most occasions, it is unclear whether this is the case or autophagy is just a bystander merely representing the cell’s desperate attempt to sustain survival upon severe stress and/or injury.
The mechanism by which defective autophagy contributes to tumorigenesis is under intense investigation. Inactivation of apoptosis, and thus deregulation of cell death, is a frequent occurrence in tumor cells [27], indicating that aberrant cell survival and cell death drive cancer progression. Loss of a survival pathway, such as autophagy, might have then been expected to undermine tumorigenesis; however, the recognition of the essential autophagy regulator beclin 1 as a haploinsufficient tumor suppressor [28,29] argues against this simplistic scenario.
In this publication, the role of autophagy in tumorigenesis and the possible implications of the functional status of autophagy on treatment responsiveness will be reviewed. What is currently known on the regulation of autophagy in tumor cells will also be presented, together with a discussion on how pharmacologic modulation of autophagy may lead to improved combinatorial anticancer regimens.
2. beclin 1 as a haploinsufficient tumor suppressor
The autophagy-related (atg) genes play essential roles at different stages of the autophagic process, including induction, vesicle formation, and autophagosome degradation, and were first identified and characterized in yeast [30]. Autophagy is evolutionarily conserved, and many yeast Atg proteins have homologues in higher eukaryotes. beclin 1 is the mammalian ortholog of the Saccharomyces cerevisiae atg6/VPS30 gene, which is required for both autophagy and sorting of the vacuole resident hydrolase carboxypeptidase Y through the Vps pathway [31]. The possibility that defective autophagy may play a role in cancer was first recognized through studies focused on Beclin 1, as summarized below.
beclin 1 was originally discovered during the positional cloning of BRCA1 and was entered in GenBank as a gene of unknown function [32]. Beclin 1 was independently rediscovered in a yeast two-hybrid screen of an adult mouse brain library as a Bcl-2 interacting protein [33]. beclin 1 maps to a centromeric region of BRCA1 on chromosome 17q21 that is commonly deleted in 75, 50 and 40% of ovarian, breast, and prostate cancers, respectively [34–38]. In addition to beclin 1, this commonly deleted region contains at least 11 more genes, 6 genes of known function and not considered cancer-related, and 5 completely novel genes. FISH analysis of human breast cancer cell lines using the beclin 1-containing PAC 452O8 as a probe revealed that 9 out of 22 cell lines had allelic beclin 1 deletions [39]. The cell lines examined were mostly aneuploid with 3–7 copies of chromosome 17 and showed only a “partial” beclin 1 deletion, still retaining 2–3 copies of beclin 1. Sequencing of the retained beclin 1 alleles did not reveal any mutations, and, interestingly, Beclin 1 mRNA levels were comparable in all cell lines, independently of beclin 1 copy number [39]. The autophagy potential of these cell lines has been only marginally characterized so far [40,41].
Ectopic expression of Beclin 1 restores full autophagy potential in MCF-7 cells, which are tetraploid, but have three beclin 1 copies, and slows cell proliferation in vitro and in xenograft tumors in vivo [41]. This finding together with the frequent allelic deletion of beclin 1 in human breast cancer cell lines raised the possibility that beclin 1 may be a tumor suppressor, a hypothesis subsequently confirmed by knockout mouse technology. beclin 1 heterozygous mice develop lymphomas, liver and lung cancers as they age, as well as hyperproliferative, preneoplastic mammary lesions [29,42]. The second beclin 1 allele is retained in all tumors developing in beclin 1+/− mice, and is neither mutated nor silenced [28], indicating that beclin 1 is a haploinsufficient tumor suppressor. Monoallelic beclin 1 loss also accelerates the development of hepatitis B virus-induced premalignant liver lesions [28]. Furthermore, beclin 1+/− immortalized baby mouse kidney (iBMK) cells [15,17] and immortalized mouse mammary epithelial cells (iMMECs) [16], which exhibit compromised autophagy under metabolic stress, display accelerated tumorigenesis in nude mouse allografts.
Lower Beclin 1 protein expression, as compared to Beclin 1 levels in normal adjacent breast tissue, was confirmed in a small series of human breast tumors [41], but any correlation between allelic beclin 1 loss, and thus defective autophagy, and clinical outcome in breast cancer remains to be investigated. Decreased Beclin 1 mRNA and protein expression was also demonstrated in glioblastoma multiforme (GBM) and other high-grade brain tumors [43]. In contrast, higher expression of Beclin 1 was detected in the majority of colorectal (95%) and gastric (83%) carcinomas examined as compared to the normal stomach and colon mucosa, which show very low or undetectable Beclin 1 levels [44]. In this case, Beclin 1 expression did not show any correlation with pathological and clinical characteristics, such as stage, invasion, and metastasis [44]. It is, therefore, conceivable that the tumor suppressive function of Beclin 1 may be tissue-specific, which is certainly worthy of further investigation.
3. The Beclin 1 protein network
As mentioned earlier, Beclin 1 was originally identified as a Bcl-2 interacting protein in a yeast two-hybrid system [33]. This interaction is not very surprising, given that Beclin 1 possesses a putative Bcl-2-homology-3 (BH3) amphipathic alpha-helix (amino acids 112–123), as demonstrated by X-ray crystallography [45], NMR spectroscopy [46], and mutational analysis [47], which can interact with the BH3 receptor domain (hydrophobic grove) of the anti-apoptotic proteins Bcl-2, Bcl-xL, Bcl-w, and to a lesser extent Mcl-1 [48]. Binding of Beclin 1 to Bcl-2 or Bcl-xL is much weaker than that of proapoptotic BH3-only proteins, such as Bid, Bad, Bak, and Bim [46]. Upon nutrient deprivation, which is the most potent autophagy inducer, BH3-only proteins induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2 or Bcl-xL [49]. At this point, it is unclear whether the autophagy-inhibitory Beclin 1–Bcl-2 or Beclin 1–Bcl-xL interaction plays any role in the tumor promoting function of the anti-apoptotic Bcl-proteins, especially since only endoplasmic reticulum (ER)-targeted Bcl-2 binds Beclin 1 and the interaction between these two proteins is disrupted under conditions of metabolic stress, which is a frequent occurrence in tumors in vivo.
The role of autophagy in tumor suppression has further been established by the identification and characterization of Beclin 1-interacting proteins. Co-immunoprecipitation studies have identified class III phosphoinositide-3-kinase (PI3KIII)/Vps34 as a major physiological partner of Beclin 1 in a complex required for autophagy initiation [50] and tumor suppression [51]. The UV irradiation resistance-associated gene protein, UVRAG, interacts with Beclin 1 and PI3KIII to promote autophagosome formation, autophagy activation and inhibition of human colon cancer cell proliferation and tumorigenicity [52]. UVRAG was recently shown to disrupt an in vitro and in vivo observed Beclin 1-dimer interface, normally stabilized by Bcl-2-like proteins, to induce autophagy [53]. Similarly to beclin 1, UVRAG is monoallelically deleted in human colon cancers [52]. Furthermore, a polyadenine tract in the UVRAG gene (A10 in exon 8) is a target for frameshift mutations decreasing the autophagy potential of colon [54] and gastric [55] cancers with microsatellite instability (MSI).
Bif-1, also known as SH3GLB1 or Endophilin B1, was originally discovered as a Bax-binding protein [56,57], and was subsequently shown to associate with membranes of intracellular organelles, such as the Golgi apparatus [58,59] and mitochondria [60,61], and participate in vesicle formation and membrane dynamics. More recently, Bif-1 was identified as a Beclin 1-interacting protein through UVRAG, a PI3KIII activator, and thus a regulator of autophagosome formation, and a novel tumor suppressor, as bif-1−/− mice develop lymphomas and solid tumors at about 12 months of age [62].
4. Other autophagy-related genes as tumor suppressors
Whereas beclin 1−/− mice die early in embryogenesis [28,29], atg5−/− and atg7−/− mice are born normally, but die soon after birth [11,12]. Also, in contrast to aging beclin 1+/− mice which are tumor-prone, older atg5+/− and atg7+/− mice do not develop malignancies, and neither tumorigenesis nor enhanced cell proliferation is observed in atg7-deficient liver, which is abnormally large mostly due to hepatocyte swelling [11]. These studies indicate that Beclin 1 may play a more important role in embryonic development and tumor suppression than Atg5 and Atg7 or, alternatively, that autophagy-independent properties of Beclin 1 may be responsible for these functions. More recently, atg5−/− iBMK cells were found to be more tumorigenic than atg5+/− and atg5+/+ iBMK cells in nude mouse allografts [17], suggesting that autophagy defects indeed play a role in tumorigenesis. This was further confirmed by the finding that mice deficient in atg4C/autophagin-3, a cysteine protease involved in Atg8/LC3 processing required for autophagy execution [63], show increased susceptibility to chemical carcinogen-induced fibrosarcoma development [64].
5. How does autophagy suppress tumorigenesis?
The mechanism by which autophagy defects lead to accelerated tumorigenesis is not readily apparent, especially given the well-documented prosurvival function of autophagy, which prolongs both normal and tumor cell survival under metabolic stress [25,65]. How loss of a survival pathway promotes tumorigenesis presents an intriguing paradox. Recent studies have revealed involvement of both non-cell-autonomous and cell-autonomous mechanisms [65], and have provided insight into the role of autophagy in tumor suppression.
5.1. Non-cell-autonomous mechanism
While functional autophagy acts as a metabolic stress buffer, the combined impairment of both apoptosis and autophagy promotes necrotic cell death in vitro and in tumors in vivo [15]. Thus, autophagy defects impair survival of apoptosis-defective tumor cells upon nutrient and oxygen limitation, leading to cell death by necrosis, which in turn is associated with inflammatory cell recruitment, cytokine production and nuclear factor κB (NFκB) activation, which has been linked to accelerated tumor growth [66]. Thus, autophagy may function in tumor suppression by mitigating metabolic stress and, in concert with apoptosis, by preventing tumor cell death by necrosis. However, the specific interactions between necrotic tumors, their microenvironment and the immune system, as well as the specific molecular pathways implicated in tumorigenesis under these conditions, have yet to be elucidated.
5.2. Cell-autonomous mechanism
An alternative, and likely complementary, mechanism by which autophagy suppresses tumorigenesis involves its unique role in preservation of cellular fitness and genome integrity. Autophagy defects are associated with accumulation of 1) ubiquitin-positive protein aggregates in neurons and liver [67], 2) deformed, and likely dysfunctional, cellular organelles, such as mitochondria [11] and peroxisomes [68], and 3) DNA damage and genomic instability in tumor cells with inactivated cell cycle checkpoints [16,65]. Although the exact mechanism by which autophagy limits genome damage has not yet been determined, several hypotheses are being actively investigated, including maintenance of energy balance and/or prevention of oxidative stress, potentially caused by defective organelles and accumulated unfolded proteins in autophagy-deficient cells.
6. Regulation of autophagy in cancer
In normal cells, the mammalian target of rapamycin (mTOR) kinase, which is downstream of the nutrient-sensor PI3K, primarily regulates autophagy. In response to nutrient and growth factor availability, the PI3K/AKT/mTOR axis is activated leading to suppression of autophagy and stimulation of cell proliferation. To the contrary, starvation suppresses the PI3K pathway and de-represses autophagy, which can now takeover as an alternate process for energy and amino acid generation to sustain cell survival, at least temporarily. Regulation of autophagy in tumors is governed by similar principles, only in a much more complicated manner, given the frequently observed abnormal PI3K activation in cancer and the multitude of interactions between the PI3K/AKT/mTOR pathway and other cell signaling cascades, often also deregulated in tumor cells.
6.1. The PI3K/AKT/mTOR pathway
Constitutive activation of the PI3K/AKT/mTOR axis is a prototypic survival mechanism commonly encountered in human cancer [69]. Diverse cellular events, such as loss of the tumor suppressors phosphatase and tensin-homolog deleted on chromosome 10 (PTEN) and tuberous sclerosis complex (TSC) 1 and TSC2, amplification or mutation of class I PI3K, overexpression of AKT, constitutive activation of tyrosine kinase growth factor receptors and exposure to carcinogens, can all result in abnormal activation of this pathway [70–73] and, ultimately, in autophagy suppression [74,75] (Fig. 1). To the contrary, G-protein coupled receptor (GPCR) antagonists to growth factor receptors (GFR), class I PI3K inhibitors such as lithium and carbamazepine, AKT inhibitors such as perifostine and AKT/PKB signaling inhibitor-2 (API-2), and mTOR inhibitors such as rapamycin, RAD-001 and CCI-779, result in autophagy induction and provide multiple means for autophagy modulation in cancer treatment (Fig.1).
Fig. 1.
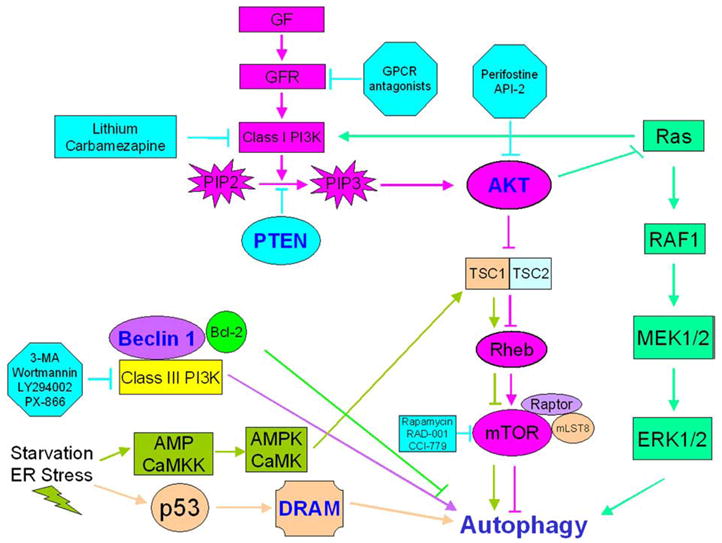
Autophagy regulation. Growth factor signaling activates the PI3K/AKT/mTOR axis resulting in autophagy inhibition. Consequently, G-protein coupled receptor (GPCR) antagonists inhibiting growth factor receptors, lithium and carbamazepine negatively regulating class I PI3K, perifostine and AKT/PKB signaling inhibitor-2 (API-2) downregulating AKT activation, and rapamycin, RAD001 and CCI-779 inhibiting mTOR play pivotal roles in autophagy upregulation. In contrast, class III PI3K activates autophagy, and thus class III PI3K inhibitors such as 3-MA, wortmannin, LY294002 or PX-866 suppress autophagy. At least two pathways are responsible for autophagy activation in response to starvation and ER stress: one mediated by AMPK and CaMKK, and the other involving p53 and damage-regulated autophagy modulator (DRAM) activation. In this complex regulatory network, Ras exhibits dual function as both an autophagy inhibitor (via class I PI3K activation) and an autophagy activator (via the RAF1/MEK1/2/ERK1/2 pathway).
How much, if any, of the tumorigenicity conferred by the activated class I PI3K pathway is due to autophagy inhibition is not currently known. A deregulated PI3K/AKT/mTOR axis not only suppresses autophagy, but also induces protein translation, cell growth and proliferation, a major driving force in tumorigenesis. Tumors with high metabolic demands, such as those with constitutively active PI3K mutations, PTEN loss or AKT activation, would be expected to be dependent on autophagy for energy homeostasis and survival. Thus, suppression of autophagy by the PI3K signaling cascade presents a disadvantage that these rapidly proliferating tumor cells may have to overcome to remain viable, and leads to the prediction that compensatory mechanisms, such as deregulated apoptosis and/or metabolism, may be concurrently activated to counteract the negative implications of defective autophagy on tumor cell survival. Two more predictions pertain to the nature of the resultant apoptosis-and-autophagy doubly-defective tumors: first, necrosis and inflammation may be prominent features of these tumors, a hypothesis already confirmed in tumors generated by apoptosis-deficient iBMK cells expressing activated AKT [15], and second, tumors resulting from PI3K pathway activation may exhibit higher levels of DNA damage and genomic instability, as autophagy defects compromise genome integrity and stability, especially when apoptosis and cell-cycle checkpoints have also been inactivated [16,17], a common finding in PTEN-deficient tumors [76], but, to this point in time, it is unclear if this is autophagy-related.
In contrast to class I PI3K, class III PI3K induces autophagy in a complex with Beclin 1, and thus class III PI3K inhibitors, such as methyl-adenine (3-MA), wortmannin, LY294002 and PX-866, inhibit autophagy (Fig. 1).
6.2. Role of p53
The relationship between p53 and autophagy is complex, as p53 appears to have a dual role in autophagy regulation [77]. The tumor suppressor p53 is a critical checkpoint protein in mammalian cells [78] which is activated under genotoxic stress conditions, including DNA damage, hypoxia and oncogene activation, and responds by initiating tumor suppression mechanisms, such as cell cycle arrest, senescence and apoptosis. Under these conditions, p53 has been shown to transactivate autophagy-inducing genes and stimulate autophagy by inhibiting mTOR in an AMP-activated protein kinase (AMPK)- and TSC1/TSC2-dependent manner [79,80]. This is, in turn, accomplished by the LKB1-mediated activation of AMPK by an increased AMP/ATP ratio [81,82]. p53 also induces autophagy via its direct target damage-regulated autophagy modulator (DRAM) [83] (Fig. 1). At the same time, however, genetic or pharmacologic inactivation of cytoplasmic p53 also triggers autophagy [84], indicating that the non-nuclear p53 pool is a potent autophagy repressor. Thus, autophagy is activated as a stress-mitigating mechanism by both stress-mediated p53 induction and stress-exacerbating p53 loss. The circumstances and the molecular pathways involved in the decision to use p53 for autophagy activation versus inhibition in cancer cells have not yet been determined. Plausibly, p53 loss, and thus autophagy induction, or negative regulation of autophagy inhibition, may be one of the compensatory mechanisms that tumor cells use to counter-balance the survival-undermining effects of autophagy suppression by an activated PI3K/AKT/mTOR axis. Interestingly, germline mutations in lkb1, tsc2 and pten tumor suppressor genes, all expected to activate mTOR and suppress autophagy, result in hamartomatous syndromes with common tumor biological characteristics [85]. At present, it is unclear whether defective autophagy plays any role in the pathogenesis and high malignancy potential of conditions, such as the Peutz–Jeghers syndrome [86], tuberous sclerosis [87] and Cowden disease [88], and, if yes, whether pharmacologic upregulation of autophagy may play a disease- and cancer-preventative role for affected patients.
6.3. Role of apoptosis
The potential for crosstalk between apoptosis and autophagy was first recognized when Beclin 1 was initially identified as a Bcl-2-interacting protein [33]. Apoptosis and autophagy share similarities in that both are self-degradative cellular pathways activated under conditions of stress. Apoptosis is a tightly regulated cell death mechanism (type I cell death) activated to eliminate unwanted or irreparably damaged cells, whereas autophagy involves cellular “self-digestion”, as a stress adaptive response to prolong cell survival. Similarly to apoptosis, autophagy can potentially lead to cell death, if allowed to proceed to completion (hence, autophagic or type II cell death). Regulators of apoptosis, such as Bcl-2/Bcl-xL and the BH3-only proteins, interact with Beclin 1 and can modulate autophagy (Fig. 1). The anti-apoptotic protein Bcl-2 binds to Beclin 1 under non-stress conditions and inhibits autophagy in the ER [89], whereas the BH3-only protein Bad [47], BNIP3 [90], and BH3 mimetics, such as ABT737 [47], competitively inhibit the interaction between Beclin 1 and Bcl-2/ Bcl-xL and stimulate autophagy. Thus, positive regulators of apoptosis also induce autophagy, which is not very surprising given that both pathways are activated under similar stress conditions. The functional relationship between autophagy and apoptosis is complicated by the fact that there is an apparent hierarchy in the activation and execution of these two stress-responsive pathways. Apoptosis-competent cells tend to undergo a rapid, “clean” apoptotic cell death under severe stress with hardly any signs of autophagy activation, whereas cells with apoptosis defects can sustain prolonged stress by upregulating autophagy [13,15–17]. On the other hand, when autophagy is blocked, apoptosis is accelerated [40,91] or apoptosis-defective cells undergo metabolic catastrophe and die by necrosis [15,92]. Thus, cell fate in response to metabolic stress is determined by the functional status and the interaction between the stress-mitigating pathways of apoptosis and autophagy [65].
6.4. Calcium signaling
Calcium (Ca2+) is a pluripotent intracellular signaling ion and an ubiquitous second messenger that plays an essential role in many processes, including muscle contraction, motility, metabolism, gene expression, enzyme activity, cell proliferation and cell death [93,94]. Ca2+ signaling pathways are often remodeled or deregulated in cancer, but whether these changes are required for tumorigenesis remains to be determined [95]. An increase in free cytosolic Ca2+ concentration ([Ca2+]c) induces autophagy via activation of the Ca2+/calmodulin-dependent kinase kinase-β (CaMKKβ), which then activates AMPK that ultimately inhibits mTOR [96] (Fig. 1). Increased [Ca2+]c also stimulates death-associated protein kinases (DAPKs) and calpains, which are enzyme families linked to both autophagy [97–99] (Fig. 1) and cancer [100,101] regulation. The implications of calcium-mediated autophagy for tumorigenesis are not yet known.
6.5. Role of endoplasmic reticulum (ER)-stress
The ER is responsible for the folding and quality control of proteins destined to be used in cell membranes, such as transmembrane receptors and other integral membrane proteins, or to be exocytosed. Accumulation of misfolded or unfolded proteins in the ER (ER-stress) in response to diverse cellular insults has recently been shown to upregulate autophagy via protein kinase-like endoplasmic reticulum kinase (PERK), inositol-requiring kinase 1 (IRE1) and increased [Ca2+]c-dependent mechanisms [18,19,21,96,102,103]. Conditions inducing ER-stress and the resultant unfolded protein response (UPR), such as hypoxia and glucose deprivation, are frequently encountered in rapidly growing tumors. In general, ER-stress and UPR activation can be considered double-edge swords in cancer progression: they may initiate apoptosis and promote tumor cell death, but they may also upregulate tumor cell adaptive responses, rendering tumors more aggressive by increasing their resistance to metabolic stress, and possibly to chemotherapeutic agents, and enabling them to metastasize more efficiently. Autophagy induction in tumors under conditions of ER-stress, though not yet documented, likely occurs as such an adaptive mechanism and potentially represents a novel target that can be exploited for therapeutic benefit.
6.6. Ras/RAF1/MEK1/2/ERK1/2 pathway
Ras is frequently mutated and constitutively activated in human cancer, more frequently in pancreatic, colon, lung and thyroid tumors [104]. Ras appears to regulate autophagy in a dual manner: it inhibits autophagy by activating class I PI3K and the AKT/mTOR pathway [105–107], and at the same time, it may induce autophagy via the RAF1/MEK1/2/ERK1/2 pathway [108] (Fig. 1). The impact of Ras activation on the functional status of autophagy in tumors has not yet been determined. Also, whether autophagy modulation by Ras plays any role in the tumorigenicity conferred by this oncogene remains to be determined. Valid arguments can be made for either scenario: autophagy inhibition may accelerate Ras-mediated tumorigenesis by the cell-autonomous and/or non-cell-autonomous mechanisms described earlier, whereas autophagy stimulation may provide Ras-transformed tumor cells with a survival benefit under rapid growth conditions or treatment-induced metabolic stress.
7. Autophagy as a therapeutic target in cancer
The role and regulation of autophagy in cancer is apparently quite complex, making autophagy a challenging, but potentially very important, target for cancer prevention and treatment. In tumors, autophagy is activated in areas of hypoxia and sustains tumor cell survival under metabolic stress conditions [15,16]. Thus, autophagy induction seen in response to anticancer agents mostly represents a survival mechanism activated to counteract the deleterious effects of endogenous metabolic stress and possibly also treatment on tumor cells. On the other hand, when autophagy proceeds to completion, cell death ensues raising the possibility that excessive autophagy induced by drugs can potentially result in tumor cell elimination. Regarding cancer prevention, the function of autophagy as a protector of cellular homeostasis and genome integrity may be particularly important.
7.1. Modulation of autophagy for cancer treatment
7.1.1. Autophagy as a treatment resistance mechanism
7.1.1.1. Treatment of autophagy-competent tumors
Autophagy-competent tumors may activate autophagy as an adaptive response to anticancer agents, in which case autophagy may act as a treatment resistance mechanism prolonging tumor cell survival. In this case, concurrent inhibition of autophagy is expected to deprive cancer cells of an essential coping mechanism, and, thus, enhance the efficacy of anticancer drugs. Given that apoptosis-defective cancer cells rely on autophagy for survival under metabolic stress, in contrast to tumors with intact apoptosis which may undergo rapid apoptotic cell death when stressed, it is also expected that autophagy inhibition will likely be therapeutically more beneficial in the treatment of tumors with apoptosis defects, but functional autophagy. Furthermore, the higher metabolic demands of rapidly proliferating cancer cells may render these cells “addicted” to autophagy for survival, and, consequently, more vulnerable to autophagy inhibition than normal cells, a concept that can be exploited for preferential tumor cell killing and reduction of undesired treatment side-effects. Autophagy inhibition as a means to sensitize cancer cells to treatment has been validated in several studies: inhibition of autophagy by chloroquine, a lysosomotropic agent that raises intralysosomal pH and interferes with autophagosome degradation within lysosomes, was shown to enhance the anticancer activity of the alkylating agent cyclophosphamide in a myc-induced lymphoma model [109] and to induce p53-dependent cell death and tumor suppression in different myc-induced and atm-deficiency lymphoma models [110]; both chloroquine and 3-methyladenine (3-MA), a class III PI3K inhibitor, synergistically augmented the proapoptotic effects and overall anticancer activity of the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) in chronic myelogenous leukemia (CML) cells [111]; targeting autophagy was used to circumvent TRAIL-resistance in tumors with apoptosis defects [112]; knockdown of autophagy, in combination with tamoxifen or 4-hydroxy-tamoxifen (4-OH-T), resulted in decreased cell viability of estrogen receptor-positive MCF-7 and T-47D cells [113,114]; inhibition of autophagy along with irradiation lead to enhanced cytotoxicity of radiotherapy in resistant cancer cells [115]. The translational testing of this paradigm has already begun in a few US institutions. Table 1 from the National Cancer Institute (NCI) website (http://clinicaltrials.gov) lists trials actively accruing patients in the USA that utilize hydroxychloroquine (HCQ) to modulate autophagy for therapeutic benefit in cancer.
Table 1.
Clinical trials utilizing hydrochloroquine to modulate autophagy in cancer therapy
| Clinical trials | Principal investigator, lead institution |
|---|---|
| HCQ and bortezomib in relapsed or refractory multiple myeloma (NCT00568880) | Stadtmauer, University of Pennsylvania (UPenn) |
| HCQ, RT and temozolomide in patients with glioblastoma multiforme (NCT00486603) | Rosenfeld, NCI/UPenn/Multiple Institutes |
| Ixabepilone and HCQ in metastatic breast cancer (NCT00765765) | Karantza-Wadsworth, Cancer Institute of New Jersey (CINJ) |
| HCQ, carboplatin, paclitaxel and bevacizumab in advanced lung cancer (NCT00728845) | Aisner, CINJ |
| HCQ in patients with PSA progression after local therapy for prostate cancer (NCT00726596) | Stein, CINJ |
7.1.1.2. Treatment of autophagy-deficient tumors
Chronically autophagy-deficient tumors likely adjust to their autophagy-defective status over time and acquire compensatory cell survival mechanisms. Thus, cancer cells with autophagy defects are not expected to depend on autophagy for cytoprotection during chemotherapy and radiotherapy, in which case autophagy inhibition may not augment the cytotoxicity conferred by anticancer drugs and irradiation. On the other hand, autophagy-deficient tumor cells likely exhibit increased susceptibility to metabolic stress, high levels of DNA damage and propensity toward genomic instability [16,17], all properties with distinct implications for treatment responsiveness. Though not yet documented, autophagy-defective tumors may be particularly sensitive to metabolic stress-inducing regimens, such as anti-angiogenic drugs, growth factor receptor inhibitors and glucose deprivation, and to DNA damage-inducing agents, including platinum compounds and topoisomerase inhibitors. Defective autophagy may not only sensitize tumor cells to certain drugs, but it may also confer resistance to agents inducing gene amplification as a resistance mechanism. In the latter case, methotrexate may prove ineffective for the treatment of autophagy-deficient tumors, a hypothesis that remains to be tested. Elucidation of the molecular pathways compensating for tumor cell survival under conditions of inactivated autophagy and determination of the mechanisms responsible for the genomic instability exhibited by autophagy-deficient tumors will be critical for identifying novel therapeutic targets in the treatment of such malignancies.
7.1.2. Autophagy as a cell death mechanism
Accumulation of autophagosomes in dying cells has, in many occasions, been described as an active cell death mechanism. Along the same thinking, autophagy induction by anticancer agents has been considered a mediator of cytotoxicity, rather than drug resistance as described in the previous section. Although “self-eating” by autophagy can potentially lead to cell death when cytoplasmic material and cellular organelles are consumed beyond a critical-for-cell-survival point, in most cases, it is unclear whether autophagy represents an active dying mode or the cell’s desperate, and often exhausted, attempt to survive. Complicating matters further, autophagy is usually not observed in cells with intact apoptosis, as these cells undergo rapid, apoptotic cell death under stress. As explained earlier, autophagy becomes apparent as a survival mechanism mostly in an apoptosis-defective background [13,15,16], thus tumor cells exhibiting signs of treatment-induced autophagy likely have inherent apoptotic defects and cannot undergo cell death characterized by classic signs of apoptosis. In this case, non-apoptotic cell death occurs via alternative death pathways, including necrosis [15,16] and possibly autophagy [116]. The active role of autophagy as a cell death mechanism can be in principle validated by experiments documenting prolongation of cell survival upon autophagy downregulation. This has been successfully demonstrated in a few occasions [117–119]. However, in most cases, autophagy inhibition under conditions of stress reduces cell viability [13,15,120], and changes the mode of cell death to either apoptosis [40,91] or necrosis [15,16], making a strong argument against this hypothesis.
7.2. Modulation of autophagy for cancer prevention
Basal autophagy plays a critical role in cellular homeostasis, as it is responsible for the degradation of excessive or malfunctioning organelles and damaged or misfolded proteins [2,3]. Accumulation of protein aggregates due to defective autophagy plays a central role in neurodegeneration, including Alzheimer’s, Parkinson’s and polyglutamine diseases, and in hepatic dysfunction, but whether it also contributes to cancer development and progression is not yet clear. A reasonable hypothesis is that protein aggregation in autophagy-defective tumor cells is a likely source of genotoxic stress, including ER- and oxidative-stress, in turn contributing to the genomic damage and instability, and thus increased tumorigenicity, associated with autophagy defects. The prediction would then be that autophagy induction, and thus maintenance of cellular fitness, may be used for cancer prevention, similarly to the rapidly evolving use of autophagy stimulators for prevention of neurodegeneration [121].
8. Concluding remarks
There is no doubt that the role of autophagy in cancer is complex and that much work is still needed to determine the molecular details of autophagy regulation in tumor cells, define how the functional status of autophagy in tumors impacts cancer progression and response to treatment, and elucidate how to best modulate autophagy for cancer prevention and therapeutic benefit. Autophagy defects are associated with susceptibility to metabolic stress, DNA damage accumulation, genomic instability, and accelerated tumorigenicity, and these observations lead to the predictions that autophagy stimulation may preserve cellular fitness and genome integrity, and thus prevent cancer, and that tumors with chronic autophagy deficiency may be particularly sensitive to certain anticancer agents, such as DNA-damaging and anti-angiogenic drugs. Tumors with intact autophagy likely utilize and potentially rely on this pathway for survival under metabolic stress conditions, such as during rapid tumor growth, metastasis and chemo- or radiotherapy. In this case, inhibition of autophagy concurrently with treatment may augment the anti-tumor activity, and thus the efficacy, of radiation and/or anticancer drugs. Ultimately, pharmacologic manipulation of autophagy for cancer prevention and treatment will depend on our ability to successfully recognize the functional status of autophagy in tumors and on the availability of specific autophagy modulators.
References
- 1.Klionsky DJ. Autophagy revisited: a conversation with Christian de Duve. Autophagy. 2008;4:740–743. doi: 10.4161/auto.6398. [DOI] [PubMed] [Google Scholar]
- 2.Eskelinen EL, Saftig P. Autophagy: a lysosomal degradation pathway with a central role in health and disease. Biochim Biophys Acta. 2008 doi: 10.1016/j.bbamcr.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 3.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malicdan MC, Noguchi S, Nonaka I, Saftig P, Nishino I. Lysosomal myopathies: an excessive build-up in autophagosomes is too much to handle. Neuromuscul Disord. 2008;18:521–529. doi: 10.1016/j.nmd.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 5.Winslow AR, Rubinsztein DC. Autophagy in neurodegeneration and development. Biochim Biophys Acta. 2008 doi: 10.1016/j.bbadis.2008.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Orvedahl A, Levine B. Eating the enemy within: autophagy in infectious diseases. Cell Death Differ. 2008 doi: 10.1038/cdd.2008.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, Kishi C, Kc W, Carrero JA, Hunt S, Stone CD, Brunt EM, Xavier RJ, Sleckman BP, Li E, Mizushima N, Stappenbeck TS, Virgin HWt. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259–263. doi: 10.1038/nature07416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, Tanaka K, Kawai T, Tsujimura T, Takeuchi O, Yoshimori T, Akira S. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264–268. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 9.Yen WL, Klionsky DJ. How to live long and prosper: autophagy, mitochondria, and aging. Physiology (Bethesda) 2008;23:248–262. doi: 10.1152/physiol.00013.2008. [DOI] [PubMed] [Google Scholar]
- 10.Jin S, White E. Tumor suppression by autophagy through the management of metabolic stress. Autophagy. 2008;4:563–566. [PMC free article] [PubMed] [Google Scholar]
- 11.Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, Kominami E, Tanaka K, Chiba T. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 13.Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–248. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 14.Onodera J, Ohsumi Y. Autophagy is required for maintenance of amino acid levels and protein synthesis under nitrogen starvation. J Biol Chem. 2005;280:31582–31586. doi: 10.1074/jbc.M506736200. [DOI] [PubMed] [Google Scholar]
- 15.Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gelinas C, Fan Y, Nelson DA, Jin S, White E. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer cell. 2006;10:51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S, White E. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 2007;21:1621–1635. doi: 10.1101/gad.1565707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, Chen G, Jin S, White E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007;21:1367–1381. doi: 10.1101/gad.1545107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sakaki K, Wu J, Kaufman RJ. Protein kinase Ctheta is required for autophagy in response to stress in the endoplasmic reticulum. J Biol Chem. 2008;283:15370–15380. doi: 10.1074/jbc.M710209200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yorimitsu T, Nair U, Yang Z, Klionsky DJ. Endoplasmic reticulum stress triggers autophagy. J Biol Chem. 2006;281:30299–30304. doi: 10.1074/jbc.M607007200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ito H, Daido S, Kanzawa T, Kondo S, Kondo Y. Radiation-induced autophagy is associated with LC3 and its inhibition sensitizes malignant glioma cells. Int J Oncol. 2005;26:1401–1410. [PubMed] [Google Scholar]
- 21.Gills JJ, Lopiccolo J, Tsurutani J, Shoemaker RH, Best CJ, Abu-Asab MS, Borojerdi J, Warfel NA, Gardner ER, Danish M, Hollander MC, Kawabata S, Tsokos M, Figg WD, Steeg PS, Dennis PA. Nelfinavir, a lead HIV protease inhibitor, is a broad-spectrum, anticancer agent that induces endoplasmic reticulum stress, autophagy, and apoptosis in vitro and in vivo. Clin Cancer Res. 2007;13:5183–5194. doi: 10.1158/1078-0432.CCR-07-0161. [DOI] [PubMed] [Google Scholar]
- 22.Katayama M, Kawaguchi T, Berger MS, Pieper RO. DNA damaging agent-induced autophagy produces a cytoprotective adenosine triphosphate surge in malignant glioma cells. Cell Death Differ. 2007;14:548–558. doi: 10.1038/sj.cdd.4402030. [DOI] [PubMed] [Google Scholar]
- 23.Pan J, Chen B, Su CH, Xu ZX, Sun L, Lee MH, Yeung SC. Autophagy induced by farnesyltransferase inhibitors in cancer cells. Cancer Biol Ther. 2008;7 doi: 10.4161/cbt.7.10.6661. [DOI] [PubMed] [Google Scholar]
- 24.Park MA, Zhang G, Martin AP, Hamed H, Mitchell C, Hylemon PB, Graf M, Rahmani M, Ryan K, Liu X, Spiegel S, Norris J, Fisher PB, Grant S, Dent P. Vorinostat and sorafenib increase ER stress, autophagy and apoptosis via ceramide-dependent CD95 and PERK activation. Cancer Biol Ther. 2008;7 doi: 10.4161/cbt.7.10.6623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 26.Baehrecke EH. Autophagy: dual roles in life and death? Nat Rev. 2005;6:505–510. doi: 10.1038/nrm1666. [DOI] [PubMed] [Google Scholar]
- 27.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 28.Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, Cattoretti G, Levine B. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–1820. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100:15077–15082. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klionsky DJ, Cregg JM, Dunn WA, Emr SD, Sakai Y, Sandoval IV, Sibirny A, Subramani S, Thumm M, Veenhuis M, Ohsumi Y. A unified nomenclature for yeast autophagy-related genes. Dev Cell. 2003;5:539–545. doi: 10.1016/s1534-5807(03)00296-x. [DOI] [PubMed] [Google Scholar]
- 31.Kametaka S, Okano T, Ohsumi M, Ohsumi Y. Apg14p and Apg6/Vps30p form a protein complex essential for autophagy in the yeast, Saccharomyces cerevisiae. J Biol Chem. 1998;273:22284–22291. doi: 10.1074/jbc.273.35.22284. [DOI] [PubMed] [Google Scholar]
- 32.Friedman LS, Ostermeyer EA, Lynch ED, Welcsh P, Szabo CI, Meza JE, Anderson LA, Dowd P, Lee MK, Rowell SE, et al. 22 genes from chromosome 17q21: cloning, sequencing, and characterization of mutations in breast cancer families and tumors. Genomics. 1995;25:256–263. doi: 10.1016/0888-7543(95)80133-7. [DOI] [PubMed] [Google Scholar]
- 33.Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, Herman B, Levine B. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol. 1998;72:8586–8596. doi: 10.1128/jvi.72.11.8586-8596.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eccles DM, Cranston G, Steel CM, Nakamura Y, Leonard RC. Allele losses on chromosome 17 in human epithelial ovarian carcinoma. Oncogene. 1990;5:1599–1601. [PubMed] [Google Scholar]
- 35.Futreal PA, Soderkvist P, Marks JR, Iglehart JD, Cochran C, Barrett JC, Wiseman RW. Detection of frequent allelic loss on proximal chromosome 17q in sporadic breast carcinoma using microsatellite length polymorphisms. Cancer Res. 1992;52:2624–2627. [PubMed] [Google Scholar]
- 36.Gao X, Zacharek A, Salkowski A, Grignon DJ, Sakr W, Porter AT, Honn KV. Loss of heterozygosity of the BRCA1 and other loci on chromosome 17q in human prostate cancer. Cancer Res. 1995;55:1002–1005. [PubMed] [Google Scholar]
- 37.Russell SE, Hickey GI, Lowry WS, White P, Atkinson RJ. Allele loss from chromosome 17 in ovarian cancer. Oncogene. 1990;5:1581–1583. [PubMed] [Google Scholar]
- 38.Saito H, Inazawa J, Saito S, Kasumi F, Koi S, Sagae S, Kudo R, Saito J, Noda K, Nakamura Y. Detailed deletion mapping of chromosome 17q in ovarian and breast cancers: 2-cM region on 17q21.3 often and commonly deleted in tumors. Cancer Res. 1993;53:3382–3385. [PubMed] [Google Scholar]
- 39.Aita VM, Liang XH, Murty VV, Pincus DL, Yu W, Cayanis E, Kalachikov S, Gilliam TC, Levine B. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics. 1999;59:59–65. doi: 10.1006/geno.1999.5851. [DOI] [PubMed] [Google Scholar]
- 40.Abedin MJ, Wang D, McDonnell MA, Lehmann U, Kelekar A. Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ. 2007;14:500–510. doi: 10.1038/sj.cdd.4402039. [DOI] [PubMed] [Google Scholar]
- 41.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 42.Kanzawa T, Kondo Y, Ito H, Kondo S, Germano I. Induction of autophagic cell death in malignant glioma cells by arsenic trioxide. Cancer Res. 2003;63:2103–2108. [PubMed] [Google Scholar]
- 43.Miracco C, Cosci E, Oliveri G, Luzi P, Pacenti L, Monciatti I, Mannucci S, De Nisi MC, Toscano M, Malagnino V, Falzarano SM, Pirtoli L, Tosi P. Protein and mRNA expression of autophagy gene Beclin 1 in human brain tumours. Int J Oncol. 2007;30:429–436. [PubMed] [Google Scholar]
- 44.Ahn CH, Jeong EG, Lee JW, Kim MS, Kim SH, Kim SS, Yoo NJ, Lee SH. Expression of beclin-1, an autophagy-related protein, in gastric and colorectal cancers. APMIS. 2007;115:1344–1349. doi: 10.1111/j.1600-0463.2007.00858.x. [DOI] [PubMed] [Google Scholar]
- 45.Oberstein A, Jeffrey PD, Shi Y. Crystal structure of the Bcl-XL-Beclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J Biol Chem. 2007;282:13123–13132. doi: 10.1074/jbc.M700492200. [DOI] [PubMed] [Google Scholar]
- 46.Feng W, Huang S, Wu H, Zhang M. Molecular basis of Bcl-xL’s target recognition versatility revealed by the structure of Bcl-xL in complex with the BH3 domain of Beclin-1. J Mol Biol. 2007;372:223–235. doi: 10.1016/j.jmb.2007.06.069. [DOI] [PubMed] [Google Scholar]
- 47.Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K, Tavernarakis N, Hickman JA, Geneste O, Kroemer G. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007;26:2527–2539. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Erlich S, Mizrachy L, Segev O, Lindenboim L, Zmira O, Adi-Harel S, Hirsch JA, Stein R, Pinkas-Kramarski R. Differential interactions between Beclin 1 and Bcl-2 family members. Autophagy. 2007;3:561–568. doi: 10.4161/auto.4713. [DOI] [PubMed] [Google Scholar]
- 49.Maiuri MC, Criollo A, Tasdemir E, Vicencio JM, Tajeddine N, Hickman JA, Geneste O, Kroemer G. BH3-only proteins and BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-X(L) Autophagy. 2007;3:374–376. doi: 10.4161/auto.4237. [DOI] [PubMed] [Google Scholar]
- 50.Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem. 2000;275:992–998. doi: 10.1074/jbc.275.2.992. [DOI] [PubMed] [Google Scholar]
- 51.Furuya N, Yu J, Byfield M, Pattingre S, Levine B. The evolutionarily conserved domain of Beclin 1 is required for Vps34 binding, autophagy and tumor suppressor function. Autophagy. 2005;1:46–52. doi: 10.4161/auto.1.1.1542. [DOI] [PubMed] [Google Scholar]
- 52.Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, Jung JU. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol. 2006;8:688–699. doi: 10.1038/ncb1426. [DOI] [PubMed] [Google Scholar]
- 53.Noble CG, Dong JM, Manser E, Song H. Bcl-xL and UVRAG cause a monomer–dimer switch in Beclin1. J Biol Chem. 2008;283:26274–26282. doi: 10.1074/jbc.M804723200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ionov Y, Nowak N, Perucho M, Markowitz S, Cowell JK. Manipulation of nonsense mediated decay identifies gene mutations in colon cancer cells with microsatellite instability. Oncogene. 2004;23:639–645. doi: 10.1038/sj.onc.1207178. [DOI] [PubMed] [Google Scholar]
- 55.Kim MS, Jeong EG, Ahn CH, Kim SS, Lee SH, Yoo NJ. Frameshift mutation of UVRAG, an autophagy-related gene, in gastric carcinomas with microsatellite instability. Hum Pathol. 2008;39:1059–1063. doi: 10.1016/j.humpath.2007.11.013. [DOI] [PubMed] [Google Scholar]
- 56.Cuddeback SM, Yamaguchi H, Komatsu K, Miyashita T, Yamada M, Wu C, Singh S, Wang HG. Molecular cloning and characterization of Bif-1. A novel Src homology 3 domain-containing protein that associates with Bax. J Biol Chem. 2001;276:20559–20565. doi: 10.1074/jbc.M101527200. [DOI] [PubMed] [Google Scholar]
- 57.Pierrat B, Simonen M, Cueto M, Mestan J, Ferrigno P, Heim J. SH3GLB, a new endophilin-related protein family featuring an SH3 domain. Genomics. 2001;71:222–234. doi: 10.1006/geno.2000.6378. [DOI] [PubMed] [Google Scholar]
- 58.Farsad K, Ringstad N, Takei K, Floyd SR, Rose K, De Camilli P. Generation of high curvature membranes mediated by direct endophilin bilayer interactions. J Cell Biol. 2001;155:193–200. doi: 10.1083/jcb.200107075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang JS, Zhang L, Lee SY, Gad H, Luini A, Hsu VW. Key components of the fission machinery are interchangeable. Nat Cell Biol. 2006;8:1376–1382. doi: 10.1038/ncb1503. [DOI] [PubMed] [Google Scholar]
- 60.Karbowski M, Jeong SY, Youle RJ. Endophilin B1 is required for the maintenance of mitochondrial morphology. J Cell Biol. 2004;166:1027–1039. doi: 10.1083/jcb.200407046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Takahashi Y, Karbowski M, Yamaguchi H, Kazi A, Wu J, Sebti SM, Youle RJ, Wang HG. Loss of Bif-1 suppresses Bax/Bak conformational change and mitochondrial apoptosis. Mol Cell Biol. 2005;25:9369–9382. doi: 10.1128/MCB.25.21.9369-9382.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mul JJ, Pledger WJ, Wang HG. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol. 2007;9:1142–1151. doi: 10.1038/ncb1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Marino G, Uria JA, Puente XS, Quesada V, Bordallo J, Lopez-Otin C. Human autophagins, a family of cysteine proteinases potentially implicated in cell degradation by autophagy. J Biol Chem. 2003;278:3671–3678. doi: 10.1074/jbc.M208247200. [DOI] [PubMed] [Google Scholar]
- 64.Marino G, Salvador-Montoliu N, Fueyo A, Knecht E, Mizushima N, Lopez-Otin C. Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. J Biol Chem. 2007;282:18573–18583. doi: 10.1074/jbc.M701194200. [DOI] [PubMed] [Google Scholar]
- 65.Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev, Cancer. 2007;7:961–967. doi: 10.1038/nrc2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 67.Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, Hamazaki J, Nishito Y, Iemura S, Natsume T, Yanagawa T, Uwayama J, Warabi E, Yoshida H, Ishii T, Kobayashi A, Yamamoto M, Yue Z, Uchiyama Y, Kominami E, Tanaka K. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 68.Sakai Y, Oku M, van der Klei IJ, Kiel JA. Pexophagy: autophagic degradation of peroxisomes. Biochim Biophys Acta. 2006;1763:1767–1775. doi: 10.1016/j.bbamcr.2006.08.023. [DOI] [PubMed] [Google Scholar]
- 69.LoPiccolo J, Blumenthal GM, Bernstein WB, Dennis PA. Targeting the PI3K/Akt/ mTOR pathway: effective combinations and clinical considerations. Drug Resist Updat. 2008;11:32–50. doi: 10.1016/j.drup.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Steelman LS, Abrams SL, Whelan J, Bertrand FE, Ludwig DE, Basecke J, Libra M, Stivala F, Milella M, Tafuri A, Lunghi P, Bonati A, Martelli AM, McCubrey JA. Contributions of the Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways to leukemia. Leukemia. 2008;22:686–707. doi: 10.1038/leu.2008.26. [DOI] [PubMed] [Google Scholar]
- 71.Hart S, Fischer OM, Prenzel N, Zwick-Wallasch E, Schneider M, Hennighausen L, Ullrich A. GPCR-induced migration of breast carcinoma cells depends on both EGFR signal transactivation and EGFR-independent pathways. Biol Chem. 2005;386:845–855. doi: 10.1515/BC.2005.099. [DOI] [PubMed] [Google Scholar]
- 72.Hii CS, Moghadammi N, Dunbar A, Ferrante A. Activation of the phosphatidylinositol 3-kinase-Akt/protein kinase B signaling pathway in arachidonic acid-stimulated human myeloid and endothelial cells: involvement of the ErbB receptor family. J Biol Chem. 2001;276:27246–27255. doi: 10.1074/jbc.M103250200. [DOI] [PubMed] [Google Scholar]
- 73.Povsic TJ, Kohout TA, Lefkowitz RJ. Beta-arrestin1 mediates insulin-like growth factor 1 (IGF-1) activation of phosphatidylinositol 3-kinase (PI3K) and anti-apoptosis. J Biol Chem. 2003;278:51334–51339. doi: 10.1074/jbc.M309968200. [DOI] [PubMed] [Google Scholar]
- 74.Arico S, Petiot A, Bauvy C, Dubbelhuis PF, Meijer AJ, Codogno P, Ogier-Denis E. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol Chem. 2001;276:35243–35246. doi: 10.1074/jbc.C100319200. [DOI] [PubMed] [Google Scholar]
- 75.Ueno T, Sato W, Horie Y, Komatsu M, Tanida I, Yoshida M, Ohshima S, Mak TW, Watanabe S, Kominami E. Loss of Pten, a tumor suppressor, causes the strong inhibition of autophagy without affecting LC3 lipidation. Autophagy. 2008;4:692–700. doi: 10.4161/auto.6085. [DOI] [PubMed] [Google Scholar]
- 76.Yin Y, Shen WH. PTEN: a new guardian of the genome. Oncogene. 2008;27:5443–5453. doi: 10.1038/onc.2008.241. [DOI] [PubMed] [Google Scholar]
- 77.Tasdemir E, Chiara Maiuri M, Morselli E, Criollo A, D’Amelio M, Djavaheri-Mergny M, Cecconi F, Tavernarakis N, Kroemer G. A dual role of p53 in the control of autophagy. Autophagy. 2008;4:810–814. doi: 10.4161/auto.6486. [DOI] [PubMed] [Google Scholar]
- 78.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 79.Feng Z, Hu W, de Stanchina E, Teresky AK, Jin S, Lowe S, Levine AJ. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007;67:3043–3053. doi: 10.1158/0008-5472.CAN-06-4149. [DOI] [PubMed] [Google Scholar]
- 80.Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A. 2005;102:8204–8209. doi: 10.1073/pnas.0502857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA, Cantley LC. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell. 2004;6:91–99. doi: 10.1016/j.ccr.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 82.Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A. 2004;101:3329–3335. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–134. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 84.Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D’Amelio M, Criollo A, Morselli E, Zhu C, Harper F, Nannmark U, Samara C, Pinton P, Vicencio JM, Carnuccio R, Moll UM, Madeo F, Paterlini-Brechot P, Rizzuto R, Szabadkai G, Pierron G, Blomgren K, Tavernarakis N, Codogno P, Cecconi F, Kroemer G. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10:676–687. doi: 10.1038/ncb1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zbuk KM, Eng C. Hamartomatous polyposis syndromes. Nat Clin Pract Gastroenterol Hepatol. 2007;4:492–502. doi: 10.1038/ncpgasthep0902. [DOI] [PubMed] [Google Scholar]
- 86.Giardiello FM, Trimbath JD. Peutz–Jeghers syndrome and management recommendations. Clin Gastroenterol Hepatol. 2006;4:408–415. doi: 10.1016/j.cgh.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 87.Curatolo P, Bombardieri R, Jozwiak S. Tuberous sclerosis. Lancet. 2008;372:657–668. doi: 10.1016/S0140-6736(08)61279-9. [DOI] [PubMed] [Google Scholar]
- 88.Gustafson S, Zbuk KM, Scacheri C, Eng C. Cowden syndrome. Semin Oncol. 2007;34:428–434. doi: 10.1053/j.seminoncol.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 89.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 90.Tracy K, Dibling BC, Spike BT, Knabb JR, Schumacker P, Macleod KF. BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Mol Cell Biol. 2007;27:6229–6242. doi: 10.1128/MCB.02246-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, Metivier D, Meley D, Souquere S, Yoshimori T, Pierron G, Codogno P, Kroemer G. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25:1025–1040. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jin S, DiPaola RS, Mathew R, White E. Metabolic catastrophe as a means to cancer cell death. J Cell Sci. 2007;120:379–383. doi: 10.1242/jcs.03349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- 94.Clapham DE. Calcium signaling. Cell. 1995;80:259–268. doi: 10.1016/0092-8674(95)90408-5. [DOI] [PubMed] [Google Scholar]
- 95.Roderick HL, Cook SJ. Ca2+ signalling checkpoints in cancer: remodelling Ca2+ for cancer cell proliferation and survival, Nat. Rev. Cancer. 2008;8:361–375. doi: 10.1038/nrc2374. [DOI] [PubMed] [Google Scholar]
- 96.Hoyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N, Elling F, Rizzuto R, Mathiasen IS, Jaattela M. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell. 2007;25:193–205. doi: 10.1016/j.molcel.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 97.Demarchi F, Bertoli C, Copetti T, Tanida I, Brancolini C, Eskelinen EL, Schneider C. Calpain is required for macroautophagy in mammalian cells. J Cell Biol. 2006;175:595–605. doi: 10.1083/jcb.200601024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Inbal B, Bialik S, Sabanay I, Shani G, Kimchi A. DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. J Cell Biol. 2002;157:455–468. doi: 10.1083/jcb.200109094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, Brunner T, Simon HU. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. 2006;8:1124–1132. doi: 10.1038/ncb1482. [DOI] [PubMed] [Google Scholar]
- 100.Demarchi F, Schneider C. Cell Cycle. Vol. 6. Georgetown, Tex: 2007. The calpain system as a modulator of stress/damage response; pp. 136–138. [DOI] [PubMed] [Google Scholar]
- 101.Gozuacik D, Kimchi A. DAPk protein family and cancer. Autophagy. 2006;2:74–79. doi: 10.4161/auto.2.2.2459. [DOI] [PubMed] [Google Scholar]
- 102.Criollo A, Maiuri MC, Tasdemir E, Vitale I, Fiebig AA, Andrews D, Molgo J, Diaz J, Lavandero S, Harper F, Pierron G, di Stefano D, Rizzuto R, Szabadkai G, Kroemer G. Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Differ. 2007;14:1029–1039. doi: 10.1038/sj.cdd.4402099. [DOI] [PubMed] [Google Scholar]
- 103.Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, Yin XM. Linking of autophagy to ubiquitin–proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol. 2007;171:513–524. doi: 10.2353/ajpath.2007.070188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev. 2008;9:517–531. doi: 10.1038/nrm2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Budovskaya YV, Stephan JS, Reggiori F, Klionsky DJ, Herman PK. The Ras/cAMP-dependent protein kinase signaling pathway regulates an early step of the autophagy process in Saccharomyces cerevisiae. J Biol Chem. 2004;279:20663–20671. doi: 10.1074/jbc.M400272200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Furuta S, Hidaka E, Ogata A, Yokota S, Kamata T. Ras is involved in the negative control of autophagy through the class I PI3-kinase. Oncogene. 2004;23:3898–3904. doi: 10.1038/sj.onc.1207539. [DOI] [PubMed] [Google Scholar]
- 107.Li D, Cui Q, Chen SG, Wu LJ, Tashiro S, Onodera S, Ikejima T. Inactivation of ras and changes of mitochondrial membrane potential contribute to oridonin-induced autophagy in a431 cells. J Pharmacol Sci. 2007;105:22–33. doi: 10.1254/jphs.fpj06022x. [DOI] [PubMed] [Google Scholar]
- 108.Pattingre S, Bauvy C, Codogno P. Amino acids interfere with the ERK1/2- dependent control of macroautophagy by controlling the activation of Raf-1 in human colon cancer HT-29 cells. J Biol Chem. 2003;278:16667–16674. doi: 10.1074/jbc.M210998200. [DOI] [PubMed] [Google Scholar]
- 109.Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest. 2007;117:326–336. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Maclean KH, Dorsey FC, Cleveland JL, Kastan MB. Targeting lysosomal degradation induces p53-dependent cell death and prevents cancer in mouse models of lymphomagenesis. J Clin Invest. 2008;118:79–88. doi: 10.1172/JCI33700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Carew JS, Nawrocki ST, Kahue CN, Zhang H, Yang C, Chung L, Houghton JA, Huang P, Giles FJ, Cleveland JL. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood. 2007;110:313–322. doi: 10.1182/blood-2006-10-050260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Han J, Hou W, Goldstein LA, Lu C, Stolz DB, Yin XM, Rabinowich H. Involvement of protective autophagy in TRAIL resistance of apoptosis-defective tumor cells. J Biol Chem. 2008;283:19665–19677. doi: 10.1074/jbc.M710169200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Qadir MA, Kwok B, Dragowska WH, To KH, Le D, Bally MB, Gorski SM. Macroautophagy inhibition sensitizes tamoxifen-resistant breast cancer cells and enhances mitochondrial depolarization. Breast Cancer Res Treat. 2008 doi: 10.1007/s10549-007-9873-4. [DOI] [PubMed] [Google Scholar]
- 114.Samaddar JS, Gaddy VT, Duplantier J, Thandavan SP, Shah M, Smith MJ, Browning D, Rawson J, Smith SB, Barrett JT, Schoenlein PV. A role for macroautophagy in protection against 4-hydroxytamoxifen-induced cell death and the development of antiestrogen resistance. Mol Cancer Ther. 2008;7:2977–2987. doi: 10.1158/1535-7163.MCT-08-0447. [DOI] [PubMed] [Google Scholar]
- 115.Apel A, Herr I, Schwarz H, Rodemann HP, Mayer A. Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res. 2008;68:1485–1494. doi: 10.1158/0008-5472.CAN-07-0562. [DOI] [PubMed] [Google Scholar]
- 116.Berry DL, Baehrecke EH. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell. 2007;131:1137–1148. doi: 10.1016/j.cell.2007.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Samara C, Syntichaki P, Tavernarakis N. Autophagy is required for necrotic cell death in Caenorhabditis elegans. Cell Death Differ. 2008;15:105–112. doi: 10.1038/sj.cdd.4402231. [DOI] [PubMed] [Google Scholar]
- 118.Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, Tsujimoto Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6:1221–1228. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- 119.Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH, Lenardo MJ. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science (New York, NY. 2004;304:1500–1502. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- 120.Melendez A, Talloczy Z, Seaman M, Eskelinen EL, Hall DH, Levine B. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science (New York, NY. 2003;301:1387–1391. doi: 10.1126/science.1087782. [DOI] [PubMed] [Google Scholar]
- 121.Sarkar S, Rubinsztein DC. Small molecule enhancers of autophagy for neurodegenerative diseases. Mol Biosyst. 2008;4:895–901. doi: 10.1039/b804606a. [DOI] [PubMed] [Google Scholar]