

Abstract
Aquaporins have recently been identified as protein channels involved in water transport. These channels may play a role in the edema formation and alterations in microvascular function observed in Alzheimer disease (AD) and cerebral amyloid angiopathy (CAA). We investigated the expression of aquaporin 1 (AQP1) and aquaporin 4 (AQP 4) in 24 human autopsy brains consisting of 18 with AD and varying degrees of CAA, and 6 with no pathologic abnormalities using immunohistochemistry. In cases of AD and CAA there was enhanced AQP 4 expression compared to age- and gender-matched controls. Aquaporin 4 immunoreactivity was prominent at CSF and brain interfaces, including subpial, subependymal, pericapillary and periarteriolar spaces. Aquaporin 1 expression in AD and CAA cases was not different from that in age- and gender-matched controls. Double-labeling studies demonstrated that both AQP1 and 4 were localized to astrocytes. Both enhanced AQP4 expression and its unique staining pattern suggest that these proteins may be important in the impaired water transport observed in AD and CAA.
Keywords: Aquaporin, Alzheimer disease, Brain aging, Cerebral amyloid angiopathy
INTRODUCTION
Alzheimer disease (AD) with or without cerebral amyloid angiopathy (CAA) is a common age-related dementing disease. CAA is defined by the deposition of amyloid proteins within the walls of leptomeningeal and cortical arteries, arterioles and capillaries, sometimes leading to ischemia and hemorrhage with subsequent edema formation; it is commonly associated with AD (1-4). AD is characterized by the progressive accumulation of senile neuritic plaques, neurofibrillary tangles, neuropil threads in the brain, as well as CAA (5), and has also been associated with cerebral microvascular abnormalities. Endothelial cells of arterioles and capillaries in the brains of AD patients are often characterized by atrophy, swelling and increased pinocytic vesicles. The astrocyte end feet processes, which often extend to the abluminal aspect of brain capillaries, appear swollen (6-8). The blood-brain barrier (BBB) is abnormal in AD, showing ultrastructural evidence of “leakiness”(9). These cerebrovascular abnormalities may contribute to impaired water transport in brains of patients with AD.
We hypothesized that alterations in the expression of aquaporin protein channels may explain some of the vascular and structural abnormalities observed in CAA and AD. Aquaporins have recently been identified as protein channels that are involved in water transport in many organs, including the brain. These channels may play a role in edema formation and alterations in the microvascular physiology of the brain. Both aquaporin 1 (AQP1) and aquaporin 4 (AQP4) have been identified in human brains. Aquaporin 1 is thought to function in the production and maintenance of cerebrospinal fluid (CSF) in the choroid plexus, whereas AQP4 is involved mostly in interstitial brain fluid homeostasis, including BBB regulation (10-12).
Previous studies have suggested that aquaporins are abnormally expressed in regions of the brain affected by edema following traumatic brain injury (13-15) and cerebral ischemia (16-20). Furthermore, recent studies have demonstrated that the pathogenesis of neuromyelitis optica (NMO) is mediated by antibodies (Abs) called NMO-IgG that target AQP4. Although the exact biological role and function of NMO-IgG remain to be determined, recent reports suggest that it recognizes AQP4 expressed by astrocytes, destabilizes the BBB and contributes to astrocyte and oligodendrocyte damage (21-23). To our knowledge, aquaporin expression in neurodegenerative conditions such as AD with varying degrees of CAA have not been extensively examined. No other study to date has examined the expression of aquaporin channels in relation to CAA.
MATERIALS AND METHODS
Patients
The autopsy brains of 18 AD patients (age range, 68-90 years) (Braak stage III-VI) were examined: 12 patients with AD had severe CAA (Vonsattel grade IV) and 6 with AD did not have CAA. These specimens were obtained from the UCLA Alzheimer Disease Center Brain Bank. There were 2 sets of controls: 4 age- and gender-matched controls without significant AD pathologic changes (age range 71-86 years) and 4 young controls (age <40 years) (Table). In all cases, 3 brain regions (frontal, temporal and occipital lobes) were used for immunohistochemical study.
Table.
Demographic Data
| Case | Age at Death (years) | Sex | Diagnosis | Braak stage | PMI | Cause of death |
|---|---|---|---|---|---|---|
| 1 | 87 | M | AD+CAA | VI | - | - |
| 2 | 75 | M | AD+CAA | IV-V | - | - |
| 3 | 80 | M | AD+CAA | IV-V | - | - |
| 4 | 77 | M | AD+CAA | IV-V | - | - |
| 5 | 68 | F | AD+CAA | III | - | - |
| 6 | 90 | M | AD+CAA | IV-V | - | - |
| 7 | 86 | M | AD | VI | - | - |
| 8 | 88 | M | AD | VI | - | - |
| 9 | 74 | M | AD | VI | - | - |
| 10 | 77 | M | AD | III-IV | - | - |
| 11 | 80 | M | AD | VI | - | - |
| 12 | 79 | M | AD | III-IV | - | - |
| 13 | 78 | M | AD | V | - | - |
| 14 | 76 | M | AD | V-VI | - | - |
| 15 | 67 | F | AD | II-III | - | - |
| 16 | 68 | F | AD | IV | - | - |
| 17 | 88 | F | AD | VI | - | - |
| 18 | 95 | M | AD | V-VI | - | - |
| 19 | 86 | M | Elderly Control | N/A | 30 h | CAD |
| 20 | 76 | M | Elderly Control | N/A | 120 h | CAD/MI |
| 21 | 71 | M | Elderly Control | N/A | 20 h | CAD/MI |
| 22 | 79 | F | Elderly Control | N/A | 14 h | pancreatitis |
| 23 | 35 | M | Young Control | N/A | 15 h | malignancyΦ |
| 24 | 30 | M | Young Control | N/A | 22 h | trauma* |
| 25 | 23 | M | Young Control | N/A | 14 h | trauma** |
| 26 | 32 | F | Young Control | N/A | 16 h | malignancyΦ |
AD, Alzheimer disease; CAA, cerebral amyloid angiopathy; PMI: Post-mortem interval; CAD: Coronary artery disease.
Malignancy not involving the brain.
Abdominal trauma.
Thoracic trauma.
- not known.
N/A not applicable.
Immunohistochemistry
Immunohistochemical studies were carried out on paraffin sections. Tissues were stained with an unconjugated polyclonal Ab to AQP1 (#AB3065, Chemicon International, Temecula, CA, 1:100) or with anti-AQP4 Ab (#H-80, Santa Cruz Biotechnology, Inc, Santa Cruz, CA, 1:100). Overnight incubation of sections with primary Abs was followed by incubation with goat anti-rabbit polyclonal Ab diluted 1:200 for 1 hour at room temperature (RT). Sections were then treated with an avidin-biotin complex (ABC) for 30 minutes (#SK-4100, Vector Laboratories, Burlingame, CA). Diaminobenzidine was used as a chromogen and sections were counterstained with hematoxylin. Negative controls were performed by omitting the primary Ab. All controls gave no detectable labeling. Normal human choroid plexus was used as a positive control for AQP1 and a normal human brain sample was used for positive control for AQP4. Slides were examined and photographed using an Olympus BX 41 microscope with a microfire digital camera (Olympus America, Inc. Melville, NY).
For double-label immunohistochemistry, primary mouse anti-glial fibrillary acidic protein (GFAP) Ab (#M0761, Dako Cytomation, Glostrup, Denmark) was applied to the slides at a concentration of 1:50 overnight at 4°C. Secondary horse anti-mouse Ab (Vector Laboratories #BA-2000) was applied to the slides and incubated for 30 minutes at RT. Vector ABC solution (Vector Laboratories #PK-6100) was then applied for 30 minutes at RT. Diaminobenzidine was again used as chromogen. The primary rabbit anti-AQP 1 Ab, (1:200), Chemicon International or AQP4 Ab, (1:100), Santa Cruz Biotechnology, Inc. were applied as primary Abs overnight at 4° C followed by incubation with a secondary goat anti- rabbit (# BA-1000, Vector) Ab for 30 minutes at RT. Vector ABC (# PK-6100) was applied to the slides for 30 minutes at RT. The labeling substrate used was 3-amino-9-ethylcarbazole (#SK-4200, AEC substrate kit, Vector), resulting in a purple color. The slides were then counterstained using hematoxylin (Fisher Scientific #BM-M10), dehydrated and mounted.
All cases (parallel sections to those used for AQP/GFAP immunohistochemistry) were stained with anti-β-amyloid (Aβ) and phosphorylated tau to investigate the location of aquaporins and astrocytes relative to senile plaques and neurofibrillary tangles, respectively. Staining with these Abs was conducted using methods similar to those used for aquaporin stains, with the following exceptions: The primary rabbit anti-Aβ 1-40 or 1-42 Abs (Chemicon International) were used at a dilution of 1:500. For anti-phosphorylated tau, the primary human anti-phosphorylated tau Ab (Pierce Endogen) was used at a dilution of 1:200 while the secondary Ab used was mouse anti-human at a dilution of 1:200 (Immpress Mouse Secondary Developing Solutions, Vector).
Grading and Data Analysis
Levels of expression were graded for both gray and white matter brain regions as follow: 0, no expression; 1, trace expression; 2, moderate expression; and 3, strong expression. A Mann-Whitney U test was used to compare the expression of aquaporins among AD, CAA, and control cases. Median scores of the expression of each molecule were used for comparisons. Values of p < 0.05 were regarded as significant. All computations were performed in R version 2.4.1(24).
RESULTS
There was no difference in immunoreactivity (IR) staining intensity for AQP1 between AD and CAA cases (median = 1.00) vs. age-matched controls (median = 1.00) (p = 0.91) (Fig.1, 2A, B). There was greater AQP4 IR in AD and CAA cases (median = 3.00), vs. aged-matched controls (median = 1.00) (p = 0.009), and vs. younger controls (median =1.00) (p = 0.01) (Fig. 1, 2C, D). However, AQP1 IR was more prominent in younger controls (median = 2.00) vs. older controls (median = 1.00) (p = 0.02) and vs. AD cases (median = 1.00) (p = 0.001) (Fig. 3). There was strong interobserver reliability in assessment of staining intensities among the two authors (P.M., H.V.V.) who graded the slides (κ = 0.91, p < 0.01).
Fig. 1.

Aquaporin 1 (AQP 1) and AQP4 expression in Alzheimer disease + cerebral amyloid angiopathy (AD + CAA), (n = 6), AD only (n = 12), elderly control, (n = 4), and young control (n = 4) cases.
Fig. 2.

Immunolocalization of aquaporin 1 (AQP1) and aquaporin 4 (AQP 4) in Alzheimer disease (AD) and cerebral amyloid angiopathy (CAA), and control cases. (A, B) There is no difference in AQP1 immunoreactivity (IR) between the AD + CAA case (A) and the age- and gender-matched control (B). AQP1 IR is localized to subcortical white matter. (C, D) AQP4-IR is greater in the AD + CAA case (C) than in the control (D). AQP4 IR is present in both gray and white matter.
Fig. 3.
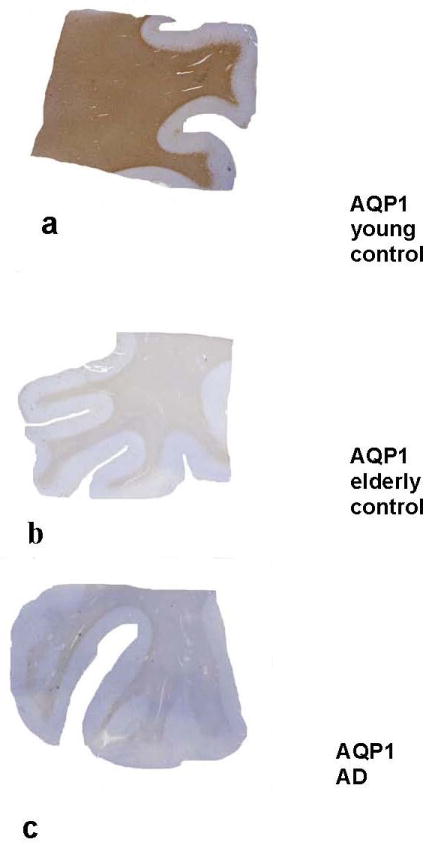
Immunolocalization of aquaporin 1 (AQP1) in young and elderly controls and in a patient with Alzheimer disease (AD). (A-C) There is prominent AQP1 immunoreactivity (IR) in the white matter of a 30-year-old control (A). Aquaporin 1 IR in the elderly control (B) and in the AD case (C) is less prominent. (Original magnification: 1x)
AQP4 IR was present in both gray and white matter (Fig.4a), and was accentuated in subpial (Fig. 5A, C, D) and subependymal regions. AQP1 IR was predominantly localized to the white matter (Fig. 4B) and was much less prominent in the gray matter, subpial (Fig. 5B), and subependymal regions when compared to AQP4 labeling intensity. Furthermore, AQP4 IR was accentuated around blood vessels of AD cases that had moderate to severe CAA (Fig. 6A, B). Blood vessels in AD cases without CAA also showed AQP4 IR; however, the IR was not as prominent or “condensed” as it was around CAA vessels (Fig. 6C). Double labeling with anti-AQP 1 or 4 and anti-GFAP Ab demonstrated that both AQP1 and AQP4 IR was abundant along the entire foot processes of astrocytes, particularly in regions corresponding to Aβ plaques. Astrocytes more distant from Aβ plaques did not have prominent aquaporin IR (Fig. 7A-C).
Fig. 4.
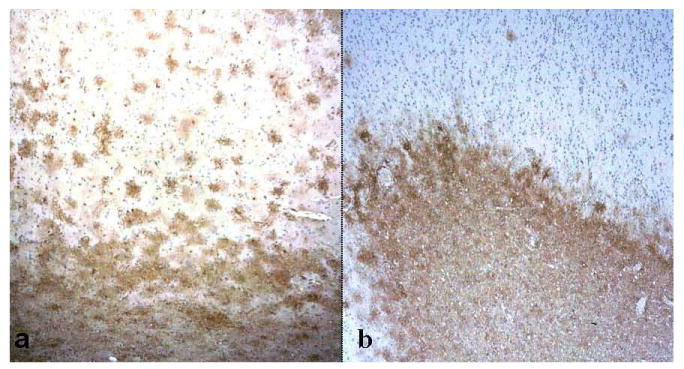
Immunolocalization of aquaporin 1 (AQP1) and aquaporin 4 (AQP 4). (A, B) AQP4 immunoreactivity (IR) is present in both gray (upper half of image) and white matter (lower half) in an Alzheimer disease + cerebral amyloid angiopathy (AD + CAA) case (A). AQP1 IR in the same patient is localized almost exclusively in subcortical white matter (lower half) (B). In both panels, the pial surface is at top of the image. (Original magnification: 4x).
Fig. 5.
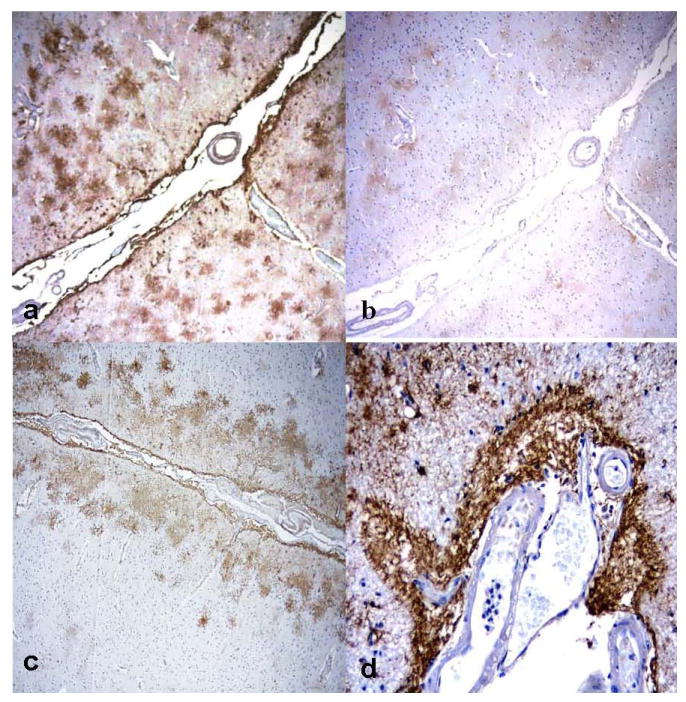
Immunolocalization of aquaporin 1 (AQP1) and aquaporin 4 (AQP 4) in subpial regions of Alzheimer disease + cerebral amyloid angiopathy (AD + CAA) cases. (A, B) There is prominent AQP4 immunoreactivity (IR) in subpial areas (A) and weak AQP1 IR in the corresponding regions from the same patient (B). (C, D) Prominent AQP4 subpial IR is noted in 2 other cases with AD + CAA. (Original magnifications: A-C, 4x; D, 20x).
Fig. 6.
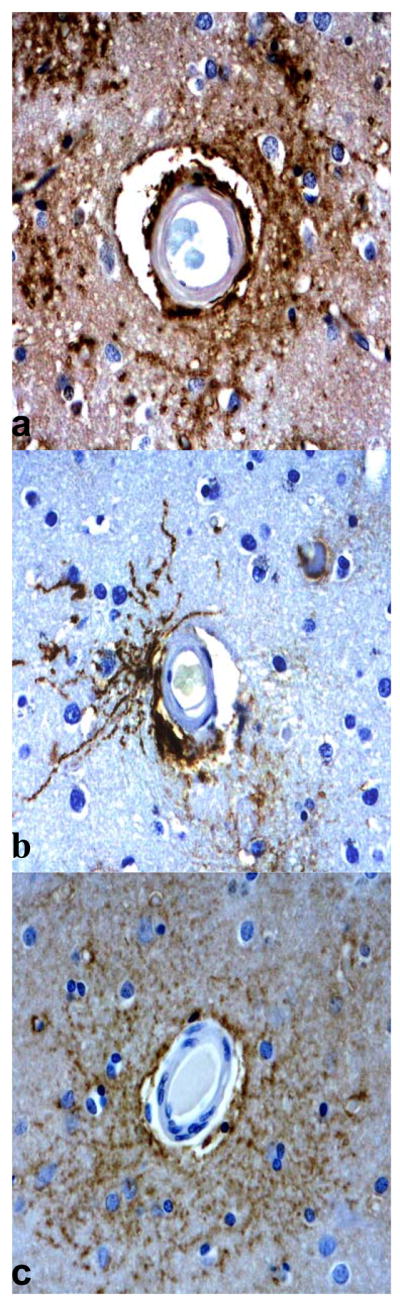
Immunolocalization of aquaporin 4 (AQP 4) around blood vessels. (A, B) There is prominent AQP4 immunoreactivity (IR) around blood vessels affected by cerebral amyloid angiopathy (CAA) in Alzheimer disease (AD) cases. (C) Although not as prominent or “condensed,” is also present around blood vessels in AD without cerebral amyloid angiopathy (CAA). (Original magnification: 40x).
Fig. 7.
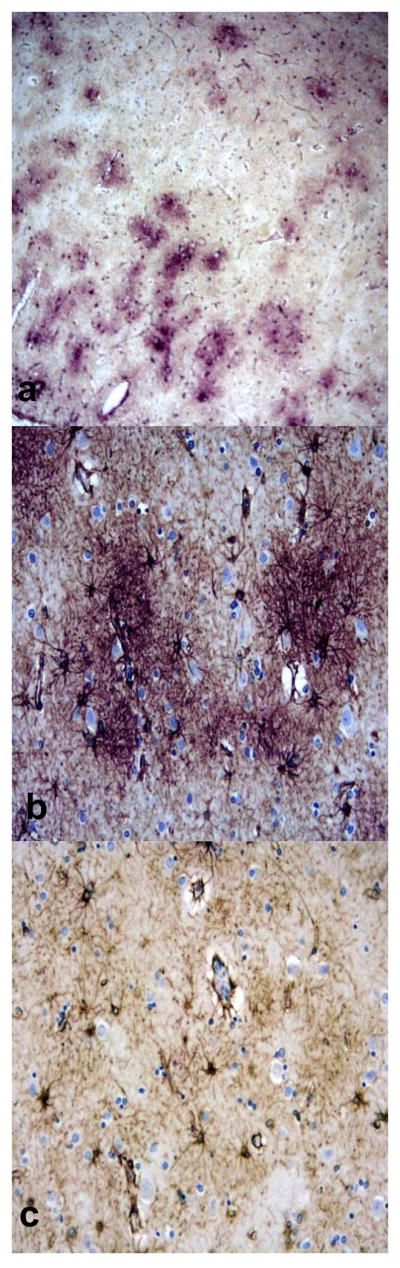
Double labeling of aquaporin 4 (AQP 4) (purple) and glial fibrillary acidic protein (GFAP) (brown) in Alzheimer disease (AD) + cerebral amyloid angiopathy (CAA) cases. (A-C) AQP4 immunoreactivity (IR) colocalizes with GFAP+ astrocytes in a plaque-like distribution, consistent with senile plaques (A, B). Not all astrocytes have associated AQP4 IR, particularly in regions with few or no associated senile plaques (C). (Original magnifications: A, C, 10x; B, 20x).
Compared to young controls, both AQP1 and AQP44 staining in the AD and CAA cases appeared to be in a “plaque” like distribution (Fig. 8A, B, D, E). When parallel sections were stained with anti-Aβ, AQP1 and AQP44 IR corresponded to regions of the brain with Aβ deposition (Fig. 8C); however, in regions where there was no plaque deposition, AQP1 and AQP4 IR were not as prominent. The older controls showed plaque-like deposition and associated AQP 1 and AQP4 IR, but the young controls did not have plaque deposition and accordingly the IR was not in a plaque-like distribution. There was no obvious relationship between aquaporins and neurofibrillary tangle IR. There was intense AQP1 and AQP4 IR in brain regions that showed microvacuolation in the AD and AD + CAA cases (Fig. 9A, B).
Fig. 8.
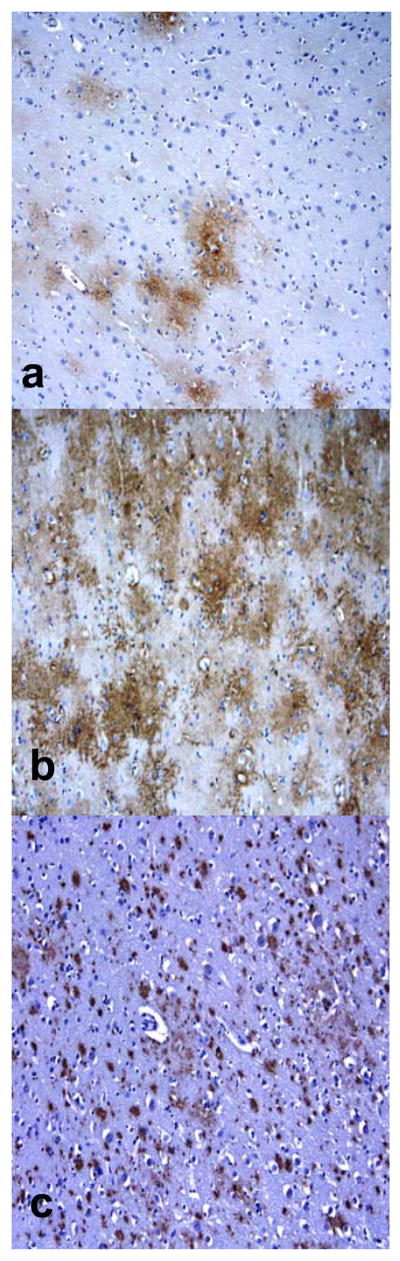

Immunolocalization of aquaporin 1 (AQP1), aquaporin 4 (AQP 4) and β-amyloid (Aβ) plaques in Alzheimer disease + cerebral amyloid angiopathy (AD + CAA). (A-C) AQP1 (A) and AQP4 (B) immunoreactivity (IR) appears to be in a plaque-like distribution that corresponds to Aβ IR (C). (D, E) AQP4 IR in a patient with AD with no CAA also shows IR in a plaque-like distribution. (Original magnifications: A, C, 10x; D, 20x; E, 40x).
Fig. 9.

A, B: Immunolocalization of aquaporin 4 (AQP 4) in areas of microvacuolation in an Alzheimer disease (AD) brain. (Original magnifications: A, 4x; B, 40x).
DISCUSSION
Aquaporins are homotetramers; each monomer consists of 6 membrane-spanning α-helical domains modeled as distinct constitutively activated water pores found in cell membranes and various barriers, including the BBB (11, 25-27). Approximately 10 types of aquaporins have been identified by homology cloning, many of them involved in a variety of human diseases. AQP1 and AQP4 are the main isoforms in the human brain.
Aquaporins have been studied in a variety of pathological conditions affecting the brain (2, 15, 16, 18, 19, 28-32). Only 1 other study focused on aquaporin expression in the frontal cortex in AD (33), but AD affects many cortical regions, such as entorhinal, parahippocampal, and temporo-parietal cortices, depending on the disease stage (34). Another study that mainly focused on transmissible spongiform encephalopathies and aquaporins examined AD patient brains, also using only frontal cortex (35). Neither of these 2 studies examined the relationship of aquaporins to CAA or Aβ plaques in AD patients.
We show that AQP4 is the main isoform demonstrating enhanced expression in AD with or without CAA. Previous studies investigating human or rat brain infarcts, human brain tumors, brain inflammatory diseases, hyponatremic rats and rat brain injuries, have all shown that AQP4 is enhanced in conditions in which water transport may be impaired (15, 16, 19, 28, 32, 37). One study has also demonstrated that in mouse models of CAA and in humans with AD and CAA, vascular amyloid deposition results in mislocalization of aquaporin 4 expression (33). The 2 previous reports on aquaporin expression in AD had conflicting results. Rodriguez et al did not observe enhanced expression of either AQP1 or AQP4 in AD (36), whereas Perez et al reported that AQP1 was enhanced in the AD cases they investigated (34).
Why we see enhanced AQP4 and not AQP1 expression in AD remains unclear. In contrast to other studies, we examined aquaporin expression patterns in relation to CAA and senile plaques. Future in-depth molecular studies that distinguish between the structure, function and regulation of these isoforms are needed to understand the true expression of aquaporins in AD and other pathological conditions in this new and complex field. Not only are there 10 aquaporin isoforms, but each of these can have different subtypes. There are 2 AQP4 subtypes (M1 and M23), and although we do not know the functional differences between them, the M23 isoform is at least 3-fold more abundant (38). Equally interesting, Dolman et al recently reported that AQP1 expression in endothelial cells is suppressed by astrocytic factors and when these astrocytic influences are absent, AQP1 expression increases (39). Clearly, aquaporins are not merely simple water channels as once thought, but rather are dynamic in their structure, function and regulation.
Our observation that there was prominent AQP4 IR in subpial and subependymal regions (in addition to perivascular localization) suggests that AQP4 may be the main isoform involved in the impaired water transport seen in AD and CAA. Regions where AQP4 is expressed include interfaces where CSF and ions are exchanged; any alterations in expression of this protein might contribute to the sequelae of edema formation (16). On the other hand, AQP1 was localized predominantly to the white matter and did not show enhanced expression in subpial or subependymal regions. Furthermore, blood vessels affected by CAA did not have prominent AQP 1 IR.
Although there was no significant change in AQP1 IR between AD cases and age-matched controls, there was a difference in AQP1 IR between young and elderly patients, regardless of the presence of AD. Based on these results, it appears that with increasing age, AQP1 expression decreases with aging in the white matter.
Both AQP1 and 4 were colocalized to astrocytes, many of which had close contact with capillaries through their foot processes. These observations suggest that the function of AQP1 is not restricted to CSF formation in the choroid plexus (17, 20, 36). Similar to AQP4, AQP1 IR within astrocytes suggests that these proteins may have an integral role in maintaining fluid balance along barriers in the brain. In AD and CAA, however, AQP4 expression appears to be more prominent than AQP1.
It is well known that in CAA, the deposition of amyloid proteins and secondary vasculopathic features (e.g. fibrinoid necrosis and vessel wall splitting) can alter the structure of affected blood vessels and make them prone to rupture (3, 4, 40, 41) The enhanced expression of AQP4 around CAA-laden blood vessels we observed suggests that these channels can alter the permeability of blood vessels, and may function to mediate CAA progression.
Our observations suggest that aquaporin expression is in a plaque-like distribution corresponding to Aβ senile plaques. The senile plaques of AD can induce both immune reactive cells and a glial response, including the upregulation of reactive astrocytes. These in turn can directly affect disease progression by modifying the degenerative plaque environment (42, 43). We found that both AQP1 IR and AQPR IR were localized to astrocytes associated with Aβ plaques. We also observed that there was prominent AQP1 and AQP4 IR in microvacuolar (usually subpial) brain areas. Microvacuolation is a histopathologic feature of AD that has been extensively observed in previous studies (44-46), but its etiology and significance remain unclear. Perhaps, these alterations in the brain tissue occur because of fluid imbalances that reflect abnormalities in aquaporin channels.
In conclusion, we demonstrate that AQP4 IR is significantly enhanced in cases of AD and CAA compared to age- and gender-matched controls. The increased expression of aquaporins may explain the fluid imbalances observed in AD and CAA (2, 8, 11, 29, 40). The finding of enhanced AQP4 IR in areas of the brain in which fluid exchange takes place (i.e. subpial, subependymal and pericapillary spaces) further supports the idea that aquaporins may be involved in the formation of edema in AD and CAA (1, 2, 47). Although AQP1 IR was not different between AD and controls, it was localized in astrocytes, which are involved in barrier regulation. Further studies focusing not only on aquaporin expression and regulation will provide a better understanding of the functional significance of our findings.
Acknowledgments
Supported in part by PHS grants P5OAG16570, PO1AG12435, SPOTRIAS NS44378, and the Sarnoff Foundation (Parham Moftakhar). Harry V. Vinters is supported in part by the Daljit S. and Elaine Sarkaria Chair in Diagnostic Medicine at the David Geffen School of Medicine, UCLA.
References
- 1.Kinnecom C, Lev MH, Wendell L, et al. Course of cerebral amyloid angiopathy-related inflammation. Neurology. 2007;68:1411–6. doi: 10.1212/01.wnl.0000260066.98681.2e. [DOI] [PubMed] [Google Scholar]
- 2.Maia LF, Mackenzie IR, Feldman HH. Clinical phenotypes of cerebral amyloid angiopathy. J Neurol Sci. 2007;257:23–30. doi: 10.1016/j.jns.2007.01.054. [DOI] [PubMed] [Google Scholar]
- 3.Vinters HV. Cerebral amyloid angiopathy. A critical review. Stroke. 1987;18:311–24. doi: 10.1161/01.str.18.2.311. [DOI] [PubMed] [Google Scholar]
- 4.Vinters HV, Wang ZZ, Secor DL. Brain parenchymal and microvascular amyloid in Alzheimer’s disease. Brain Pathol. 1996;6:179–95. doi: 10.1111/j.1750-3639.1996.tb00799.x. [DOI] [PubMed] [Google Scholar]
- 5.Duyckaerts C, Dickson DW. Neuropathology of Alzheimer’s disease. In: Dickson DW, editor. Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders. Basel: ISN Neuropath Press; 2003. pp. 47–68. [Google Scholar]
- 6.Farkas E, Luiten PGM. Cerebral microvascular pathology in aging and Alzheimer’s disease. Prog Neurobiol. 2001;64:575–611. doi: 10.1016/s0301-0082(00)00068-x. [DOI] [PubMed] [Google Scholar]
- 7.Grammas P, Yamada M, Zlokovic B. The cerebromicrovasculature: A key player in the pathogenesis of Alzheimer’s disease. J Alzheimer’s Dis. 2002;4:217–23. doi: 10.3233/jad-2002-4311. [DOI] [PubMed] [Google Scholar]
- 8.Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci. 2004;5:347–60. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- 9.Stewart PA, Hayakawa K, Akers MA, et al. A morphometric study of the blood-brain barrier in Alzheimer’s disease. Lab Invest. 1992;67:734–42. [PubMed] [Google Scholar]
- 10.Longatti PL, Basaldella L, Orvieto E, et al. Choroid plexus and aquaporin-1: A novel explanation of cerebrospinal fluid production. Pediatr Neurosurg. 2004;40:277–83. doi: 10.1159/000083740. [DOI] [PubMed] [Google Scholar]
- 11.Nielsen S, Smith BL, Christensen EI, et al. Distribution of the aquaporin CHIP in secretory and resorptive epithelia and capillary endothelia. Proc Natl Acad Sci USA. 1993;90:7275–9. doi: 10.1073/pnas.90.15.7275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oshio K, Song Y, Verkman AS, et al. Aquaporin-1 deletion reduces osmotic water permeability and cerebrospinal fluid production. Acta Neurochir Suppl. 2003;86:525–8. doi: 10.1007/978-3-7091-0651-8_107. [DOI] [PubMed] [Google Scholar]
- 13.Kiening KL, van Landeghem FK, Schreiber S, et al. Decreased hemispheric aquaporin-4 is linked to evolving brain edema following controlled cortical impact injury in rats. Neurosci Lett. 2002;324:105–8. doi: 10.1016/s0304-3940(02)00180-5. [DOI] [PubMed] [Google Scholar]
- 14.Sun MC, Honey CR, Berk C, et al. Regulation of aquaporin-4 in a traumatic brain injury model in rats. J Neurosurg. 2003;98:565–9. doi: 10.3171/jns.2003.98.3.0565. [DOI] [PubMed] [Google Scholar]
- 15.Vizuete ML, Venero JL, Vargas C, et al. Differential upregulation of aquaporin-4 mRNA expression in reactive astrocytes after brain injury: potential role in brain edema. Neurobiol Dis. 1999;4:245–58. doi: 10.1006/nbdi.1999.0246. [DOI] [PubMed] [Google Scholar]
- 16.Aoki K, Uchihara T, Tsuchiya K, et al. Enhanced expression of aquaporin 4 in human brain with infarction. Acta Neuropathol. 2003;106:121–4. doi: 10.1007/s00401-003-0709-y. [DOI] [PubMed] [Google Scholar]
- 17.Badaut J, Lasbennes F, Magistretti PJ, et al. Aquaporins in brain: distribution, physiology, and pathophysiology. J Cereb Blood Flow Metab. 2002;22:367–78. doi: 10.1097/00004647-200204000-00001. [DOI] [PubMed] [Google Scholar]
- 18.Sato S, Umenishi F, Inamasu G, et al. Expression of water channel mRNA following cerebral ischemia. Acta Neurochir Suppl. 2000;76:239–41. doi: 10.1007/978-3-7091-6346-7_48. [DOI] [PubMed] [Google Scholar]
- 19.Taniguchi M, Yamashita T, Kumura E, et al. Induction of aquaporin-4 water channel mRNA after focal cerebral ischemia in rat. Mol Brain Res. 2000;78:131–7. doi: 10.1016/s0169-328x(00)00084-x. [DOI] [PubMed] [Google Scholar]
- 20.Xiao F, Arnold TC, Zhang S, et al. Cerebral cortical aquaporin-4 expression in brain edema following cardiac arrest in rats. Acad Emerg Med. 2004;11:1001–7. doi: 10.1197/j.aem.2004.05.026. [DOI] [PubMed] [Google Scholar]
- 21.Lennon VA, Kryzer TJ, Pittock SJ, et al. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202:473–7. doi: 10.1084/jem.20050304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pittock SJ, Weinshenker BG, Lucchinetti CF, et al. Neuromyelitis optica brain lesions localized at sites of high aquaporin 4 expression. Arch Neurol. 2006;63:964–8. doi: 10.1001/archneur.63.7.964. [DOI] [PubMed] [Google Scholar]
- 23.Jarius SF, Franciotta PD, et al. Mechanisms of disease: Aquaporin-4 antibodies in neuromyelitis optica. Nat Clin Pract Neurol. 2008;4:202–14. doi: 10.1038/ncpneuro0764. [DOI] [PubMed] [Google Scholar]
- 24.R Development Core Team. A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2006. [Google Scholar]
- 25.Agre P, Brown D, Neilsen S. Aquaporin water channels: Unanswered questions and unresolved controversies. Cell Biol. 1995;7:472–483. doi: 10.1016/0955-0674(95)80003-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Borgnia M, Nielsen S, Engel A, et al. Cellular and molecular biology of aquaporin water channels. Annu Rev Biochem. 1999;68:425–58. doi: 10.1146/annurev.biochem.68.1.425. [DOI] [PubMed] [Google Scholar]
- 27.Longatti PL, Basaldella L, Orvieto E, et al. Choroid plexus and aquaporin-1: A novel explanation of cerebrospinal fluid production. Pediatr Neurosurg. 2004;40:277–83. doi: 10.1159/000083740. [DOI] [PubMed] [Google Scholar]
- 28.Aoki-Yoshina K, Uchihara T, Duyckaerts C, et al. Enhanced expression of aquaporin 4 in human brain with inflammatory diseases. Acta Neuropathol. 2005;110:281–8. doi: 10.1007/s00401-005-1052-2. [DOI] [PubMed] [Google Scholar]
- 29.Baduat J, Brunet JF, Grollimund L, et al. Aquaporin 1 and aquaporin 4 expression in human brain after subarachnoid hemorrhage and in peritumoral tissue. Acta Neurochir Suppl. 2003;86:495–8. doi: 10.1007/978-3-7091-0651-8_101. [DOI] [PubMed] [Google Scholar]
- 30.Lee TS, Eld T, Mane S, et al. Aquaporin-4 expression is increased in the sclerotic hippocampus in human temporal lobe epilepsy. Acta Neuropathol. 2004;108:493–502. doi: 10.1007/s00401-004-0910-7. [DOI] [PubMed] [Google Scholar]
- 31.Oshio K, Binder DK, Bollen A, et al. Aquaporin-1 expression in human glial tumors suggests a potential novel therapeutic target for tumor-associated edema. Acta Neurochir Suppl. 2003;86:499–502. doi: 10.1007/978-3-7091-0651-8_102. [DOI] [PubMed] [Google Scholar]
- 32.Saadoun S, Papadopoulos MC, Davies DC, et al. Aquaporin-4 expression is increased in oedematous human brain tumours. J Neurol Neurosurg Psychiatry. 2002;72:262–5. doi: 10.1136/jnnp.72.2.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilcock DM, Vitek P, Colton CA. Vascular amyloid alters astrocytic water and potassium channels in mouse models and humans with Alzheimer’s disease. Neuroscience. 2009;159:1055–69. doi: 10.1016/j.neuroscience.2009.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perez E, Barrachina M, Rodriguez A, et al. Aquaporin expression in the cerebral cortex is increased at early stages of Alzheimer’s disease. Brain Res. 2007;1128:164–74. doi: 10.1016/j.brainres.2006.09.109. [DOI] [PubMed] [Google Scholar]
- 35.Thompson PM, Mega MS, Woods RP, et al. Cortical change in Alzheimer’s disease detected with a disease-specific population-based brain atlas. Cereb Cortex. 2001;11:1–16. doi: 10.1093/cercor/11.1.1. [DOI] [PubMed] [Google Scholar]
- 36.Rodriguez A, Perez-Gracia E, Espinosa JC, et al. Increased expression of water channel aquaporin 1 and aquaporin 4 in Creutzfeldt-Jakob disease and in bovine spongiform encephalopathy-infected bovine-PrP transgenic mice. Acta Neuropathol. 2006;112:573–85. doi: 10.1007/s00401-006-0117-1. [DOI] [PubMed] [Google Scholar]
- 37.Vadja Z, Promeneur D, Doczi T, et al. Increased aquaporin-4 immunoreactivity in rat brain in response to systemic hyponatremia. Biochem Biophys Res Commun. 2000;270:495–503. doi: 10.1006/bbrc.2000.2472. [DOI] [PubMed] [Google Scholar]
- 38.Furman CS, Gorelick-Feldman DA, Davidson KG, et al. Aquaporin-4 square array assembly: opposing actions of M1 and M23 isoforms. Proc Natl Acad Sci USA. 2003;100:13609–14. doi: 10.1073/pnas.2235843100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dolman D, Drndarski S, Abbot NJ, et al. Induction of aquaporin 1 but not aquaporin 4 messenger RNA in rat primary brain microvessel endothelial cells in culture. J Neurochem. 2005;93:825–33. doi: 10.1111/j.1471-4159.2005.03111.x. [DOI] [PubMed] [Google Scholar]
- 40.Greenberg SM, Vonsattel JP, Segal AZ, et al. Association of apolipoprotein E epsilon2 and vasculopathy in cerebral amyloid angiopathy. Neurology. 1998;50:961–5. doi: 10.1212/wnl.50.4.961. [DOI] [PubMed] [Google Scholar]
- 41.McCarron MO, Nicoll JA, Stewart J, et al. The apolipoprotein E epsilon2 allele and the pathological features in cerebral amyloid angiopathy-related hemorrhage. J Neuropathol Exp Neurol. 1999;58:711–8. doi: 10.1097/00005072-199907000-00005. [DOI] [PubMed] [Google Scholar]
- 42.Pike CJ, Cummings BJ, Cotman CW. Early association of reactive astrocytes with senile plaques in Alzheimer’s disease. Exp Neurol. 1995;132:172–9. doi: 10.1016/0014-4886(95)90022-5. [DOI] [PubMed] [Google Scholar]
- 43.Vehmas AK, Kawas CH, Stewart WF, et al. Immune reactive cells in senile plaques and cognitive decline in Alzheimer’s disease. Neurobiol Aging. 2003;24:321–31. doi: 10.1016/s0197-4580(02)00090-8. [DOI] [PubMed] [Google Scholar]
- 44.Brun A, Englund E. The pattern of degeneration in Alzheimer’s disease. neuronal loss and histopathological grading. Histopathology. 1981;5:549–64. doi: 10.1111/j.1365-2559.1981.tb01818.x. [DOI] [PubMed] [Google Scholar]
- 45.Gustafson L, Abrahamson M, Grubb A, et al. Apolipoprotein-E genotyping in Alzheimer’s disease and frontotemporal dementia. Dement Geriatr Cogn Disord. 1997;8:240–3. doi: 10.1159/000106637. [DOI] [PubMed] [Google Scholar]
- 46.Sjobeck M, Englund E. Alzheimer’s disease and the cerebellum. a morphological study on neuronal and glial changes. Dement Geriatr Cogn Disord. 2001;12:211–8. doi: 10.1159/000051260. [DOI] [PubMed] [Google Scholar]
- 47.Tomimoto H, Akiguchi I, Wakita H, et al. Regressive changes of astroglia in white matter lesions in cerebrovascular disease and Alzheimer’s disease patients. Acta Neuropathol. 1997;94:146–52. doi: 10.1007/s004010050686. [DOI] [PubMed] [Google Scholar]